Glyoxalase System as a Therapeutic Target against Diabetic Retinopathy

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
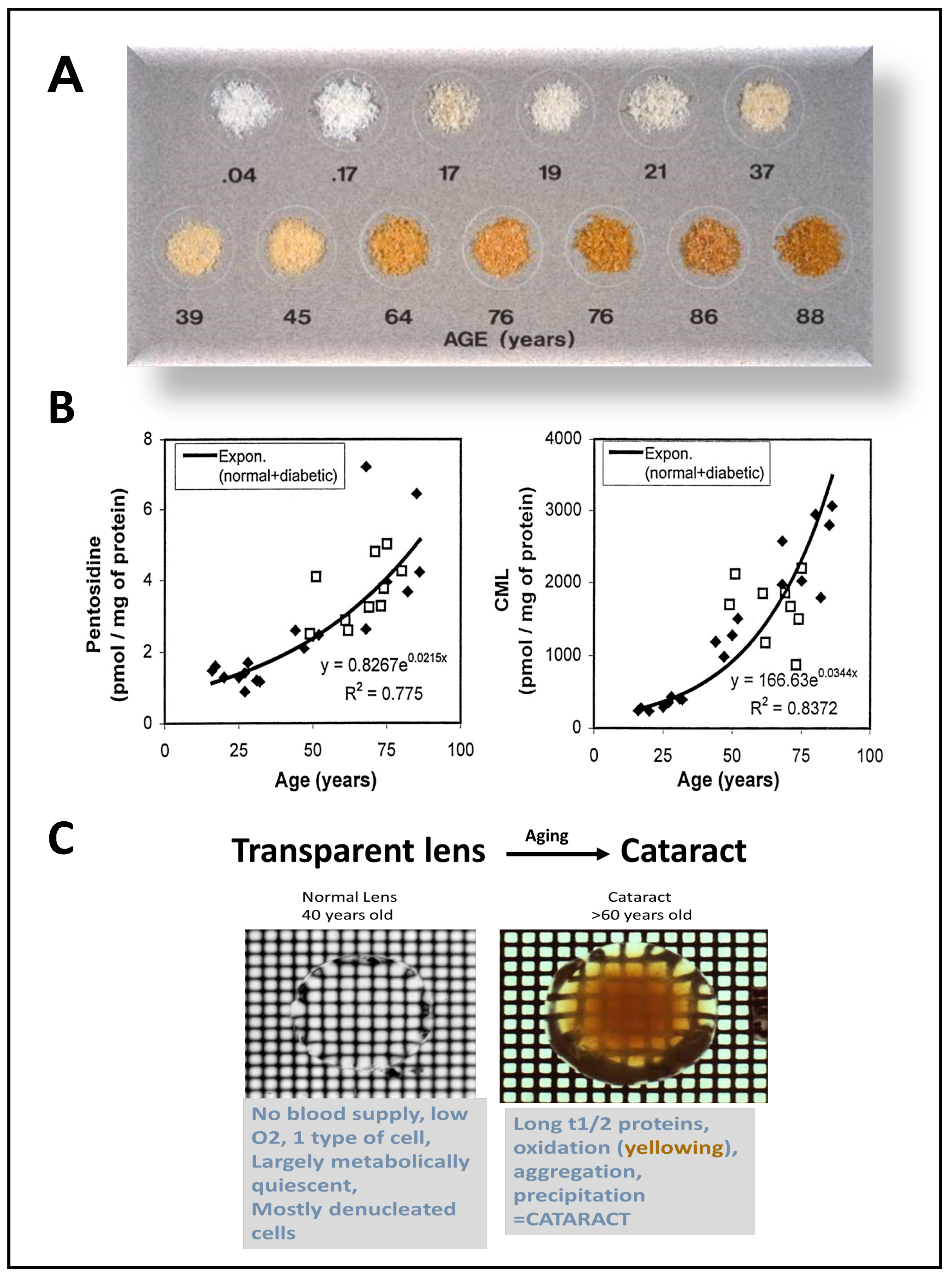
1. Oxidative Stress is Related to Many Age-Related Eye Diseases
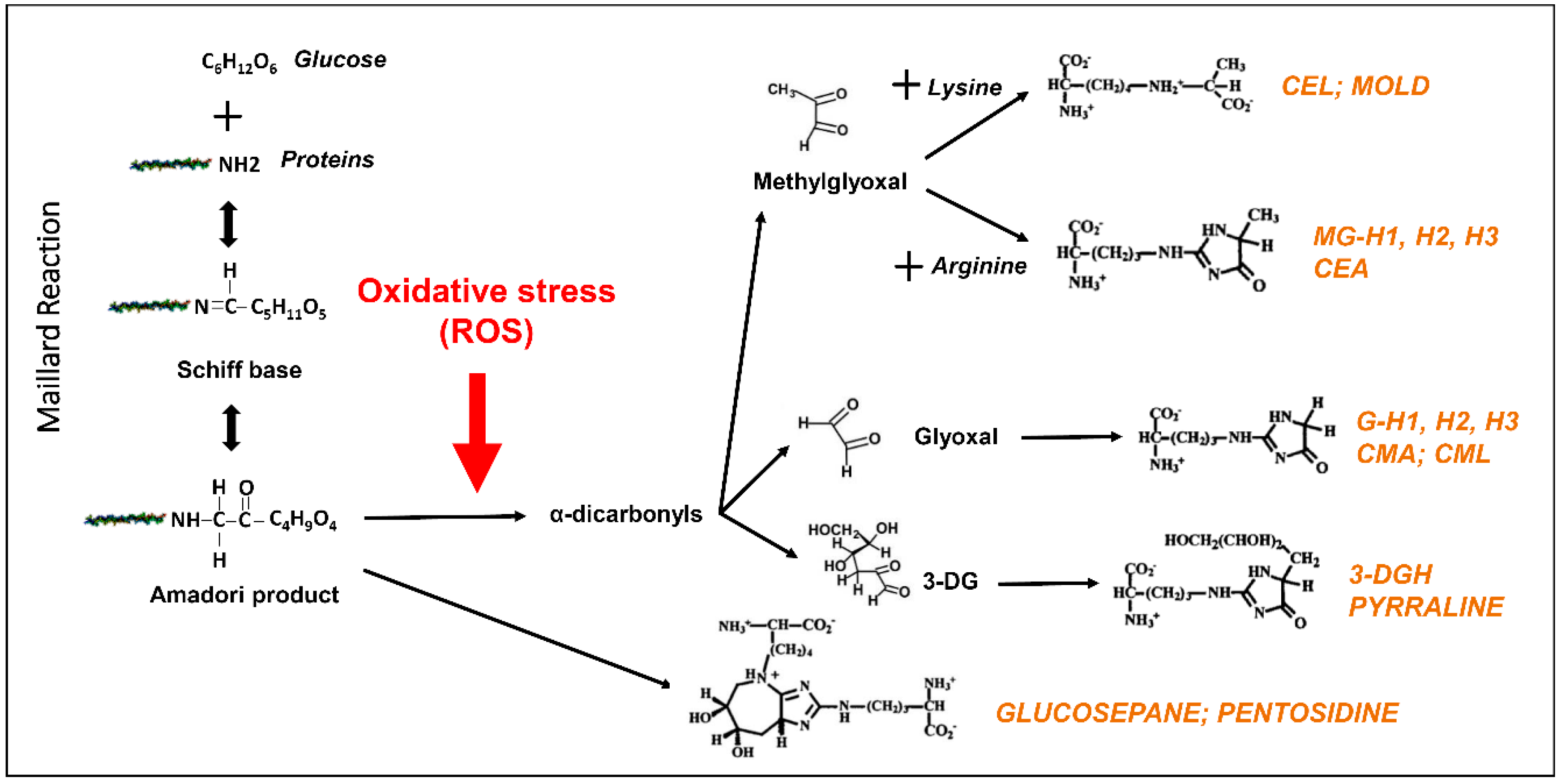
2. Advanced Glycation End Products: A Special Case of Oxidative Stress Found in Aged Eye Tissues and Throughout the Body
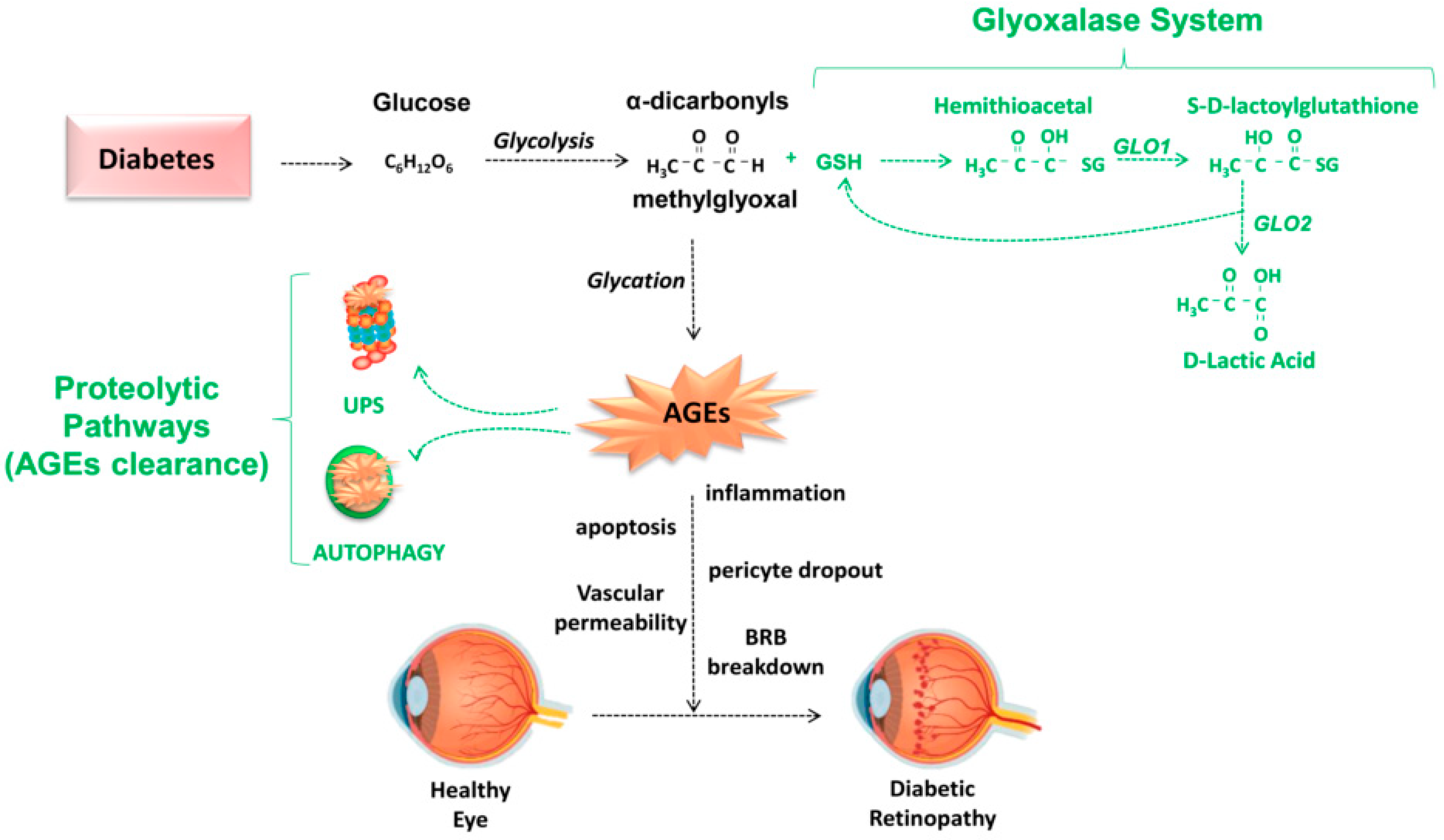
3. Role of Advanced Glycation End Products in the Pathogenesis of DR
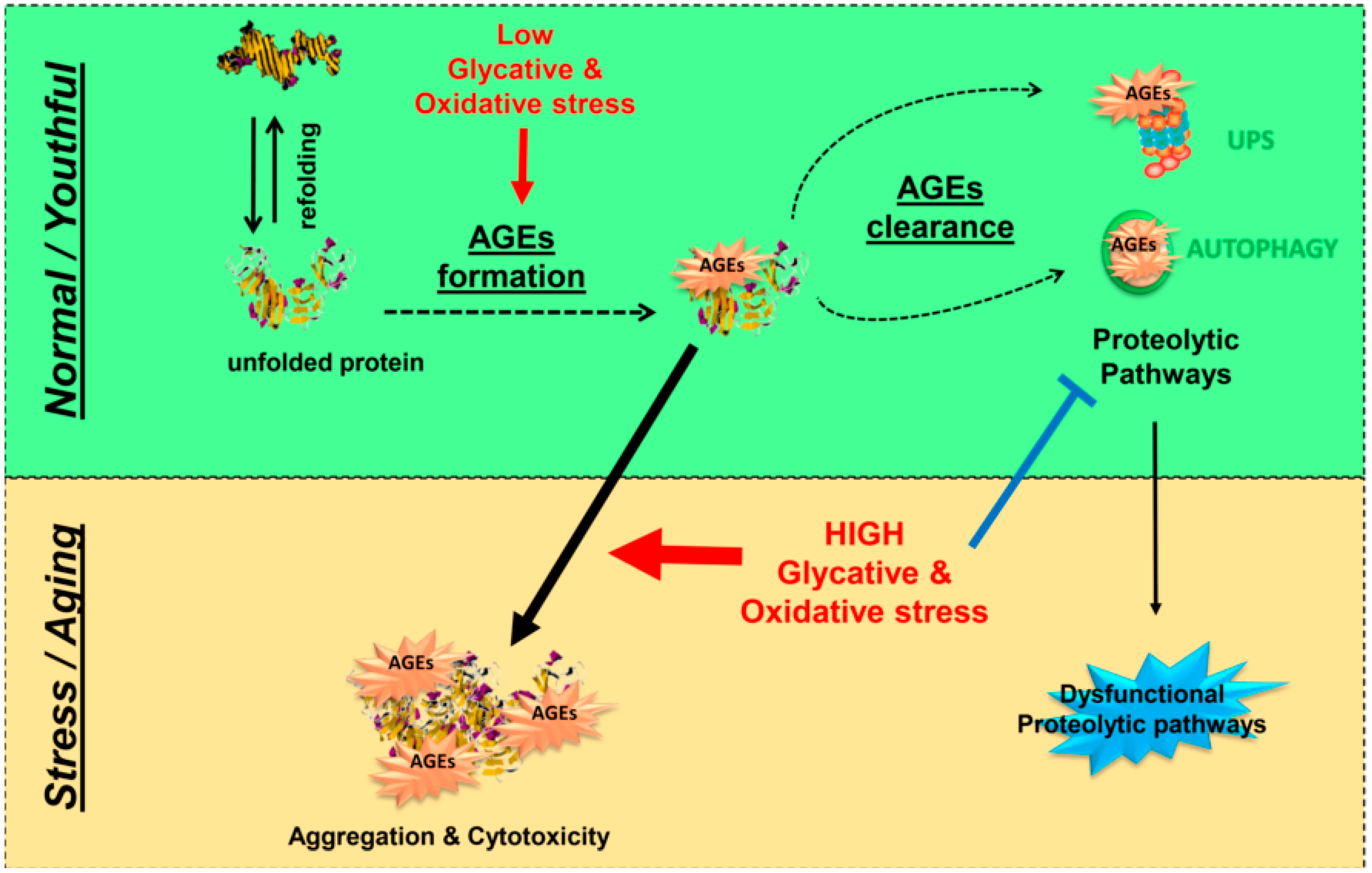
4. Protective Mechanisms against Glycation-Derived Damage
4.1. Antioxidant Enzymes, Antioxidants, and Signaling
4.2. Detoxifying Mechanisms against AGEs: Glyoxalase System, Aldehyde Dehydrogenases (ALDH), Aldoketoreductases (AKR), DJ-1/Park7, and Aldol Condensations
4.3. Proteolytic Pathways: The Last Line of Defense against Glycation-Derived Damage
4.4. Protective Role of NRF2 against Glycation-Derived Damage and Modulation of GLO1
5. The Use of Glyoxalase 1 in Animal Models
5.1. The Decline of Glyoxalase 1 Activity with Age
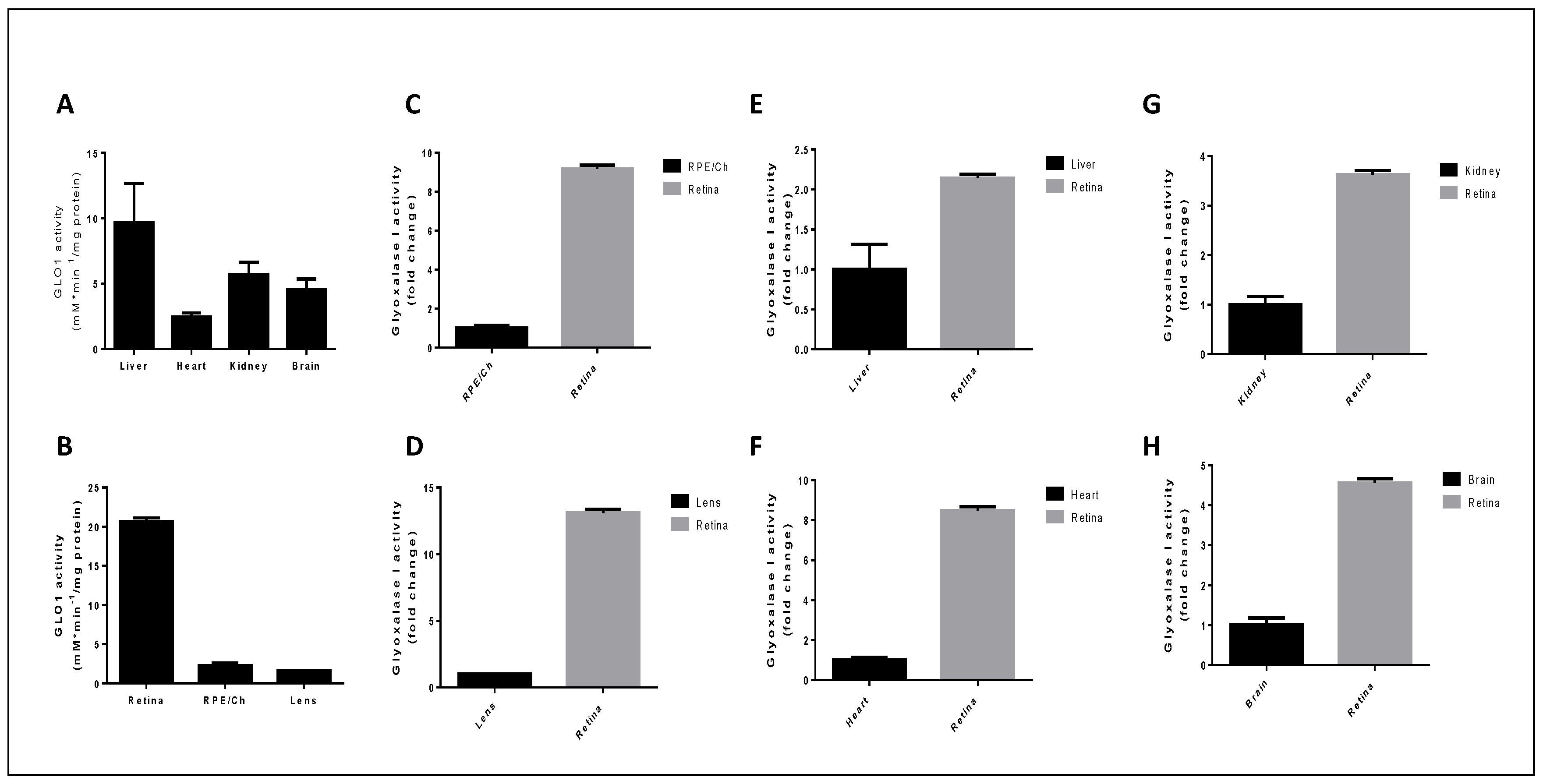
5.2. Glyoxalase 1 Activity in Ocular Tissues
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stevens, G.A.; White, R.A.; Flaxman, S.R.; Price, H.; Jonas, J.B.; Keeffe, J.; Leasher, J.; Naidoo, K.; Pesudovs, K.; Resnikoff, S.; et al. Global prevalence of vision impairment and blindness: Magnitude and temporal trends, 1990–2010. Ophthalmology 2013, 120, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Eckert, K.A.; Carter, M.J.; Lansingh, V.C.; Wilson, D.A.; Furtado, J.M.; Frick, K.D.; Resnikoff, S. A Simple Method for Estimating the Economic Cost of Productivity Loss Due to Blindness and Moderate to Severe Visual Impairment. Ophthalmic Epidemiol. 2015, 22, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Ramrattan, R.S.; Wolfs, R.C.; Panda-Jonas, S.; Jonas, J.B.; Bakker, D.; Pols, H.A.; Hofman, A.; de Jong, P.T. Prevalence and causes of visual field loss in the elderly and associations with impairment in daily functioning: The Rotterdam Study. Arch. Ophthalmol. 2001, 119, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Köberlein, J.; Beifus, K.; Schaffert, C.; Finger, R.P. The economic burden of visual impairment and blindness: A systematic review. BMJ Open 2013, 3, e003471. [Google Scholar] [CrossRef]
- WHO. Draft action plan for the prevention of avoidable blindness and visual impairment 2014–2019. In Universal Eye Health: A Global Action Plan 2014–2019; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Bourne, R.R.A.; Flaxman, S.R.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; Leasher, J.; Limburg, H.; et al. Magnitude, temporal trends, and projections of the global prevalence of blindness and distance and near vision impairment: A systematic review and meta-analysis. Lancet. Glob. Health 2017, 5, e888–e897. [Google Scholar] [CrossRef]
- Williams, D.L. Oxidative stress and the eye. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Domènech, E.B.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Bungau, S.; Abdel-Daim, M.M.; Tit, D.M.; Ghanem, E.; Sato, S.; Maruyama-Inoue, M.; Yamane, S.; Kadonosono, K. Health Benefits of Polyphenols and Carotenoids in Age-Related Eye Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9783429. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; El-Tawil, O.S.; Bungau, S.G.; Atanasov, A.G. Applications of Antioxidants in Metabolic Disorders and Degenerative Diseases: Mechanistic Approach. Oxidative Med. Cell. Longev. 2019, 2019, 4179676. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Chan, P.S. Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 43603. [Google Scholar] [CrossRef]
- Cabrera, M.P.; Chihuailaf, R.H. Antioxidants and the integrity of ocular tissues. Vet. Med. Int. 2011, 2011, 905153. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.N.; Conley, S.M.; Naash, M.I. Therapeutic Approach of Nanotechnology for Oxidative Stress Induced Ocular Neurodegenerative Diseases. Adv. Exp. Med. Biol. 2016, 854, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E. Role of Protein Carbonylation in Skeletal Muscle Mass Loss Associated with Chronic Conditions. Proteomes 2016, 4, 18. [Google Scholar] [CrossRef]
- Jahngen-Hodge, J.; Cyr, D.; Laxman, E.; Taylor, A. Ubiquitin and ubiquitin conjugates in human lens. Exp. Eye Res. 1992, 55, 897–902. [Google Scholar] [CrossRef]
- Dudek, E.J.; Shang, F.; Valverde, P.; Liu, Q.; Hobbs, M.; Taylor, A. Selectivity of the ubiquitin pathway for oxidatively modified proteins: Relevance to protein precipitation diseases. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1707–1709. [Google Scholar] [CrossRef] [PubMed]
- Obin, M.; Shang, F.; Gong, X.; Handelman, G.; Blumberg, J.; Taylor, A. Redox regulation of ubiquitin-conjugating enzymes: Mechanistic insights using the thiol-specific oxidant diamide. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1998, 12, 561–569. [Google Scholar] [CrossRef]
- Jahngen-Hodge, J.; Obin, M.S.; Gong, X.; Shang, F.; Nowell, T.R., Jr.; Gong, J.; Abasi, H.; Blumberg, J.; Taylor, A. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J. Biol. Chem. 1997, 272, 28218–28226. [Google Scholar] [CrossRef]
- Trpkovic, A.; Resanovic, I.; Stanimirovic, J.; Radak, D.; Mousa, S.A.; Cenic-Milosevic, D.; Jevremovic, D.; Isenovic, E.R. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit. Rev. Clin. Lab. Sci. 2015, 52, 70–85. [Google Scholar] [CrossRef]
- Jacob, K.D.; Noren Hooten, N.; Trzeciak, A.R.; Evans, M.K. Markers of oxidant stress that are clinically relevant in aging and age-related disease. Mech. Ageing Dev. 2013, 134, 139–157. [Google Scholar] [CrossRef]
- Chen, J.; Patil, S.; Seal, S.; McGinnis, J.F. Rare earth nanoparticles prevent retinal degeneration induced by intracellular peroxides. Nat. Nanotechnol. 2006, 1, 142–150. [Google Scholar] [CrossRef]
- Martínez-Fernández de la Cámara, C.; Salom, D.; Sequedo, M.D.; Hervás, D.; Marín-Lambíes, C.; Aller, E.; Jaijo, T.; Díaz-Llopis, M.; Millán, J.M.; Rodrigo, R. Altered antioxidant-oxidant status in the aqueous humor and peripheral blood of patients with retinitis pigmentosa. PLoS ONE 2013, 8, e74223. [Google Scholar] [CrossRef]
- Liu, Q.; Ju, W.K.; Crowston, J.G.; Xie, F.; Perry, G.; Smith, M.A.; Lindsey, J.D.; Weinreb, R.N. Oxidative stress is an early event in hydrostatic pressure induced retinal ganglion cell damage. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4580–4589. [Google Scholar] [CrossRef]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef]
- Serebryany, E.; Yu, S.; Trauger, S.A.; Budnik, B.; Shakhnovich, E.I. Dynamic disulfide exchange in a crystallin protein in the human eye lens promotes cataract-associated aggregation. J. Biol. Chem. 2018, 293, 17997–18009. [Google Scholar] [CrossRef]
- Weikel, K.A.; Chiu, C.J.; Taylor, A. Nutritional modulation of age-related macular degeneration. Mol. Asp. Med. 2012, 33, 318–375. [Google Scholar] [CrossRef] [PubMed]
- Saccà, S.C.; Corazza, P.; Gandolfi, S.; Ferrari, D.; Sukkar, S.; Iorio, E.L.; Traverso, C.E. Substances of Interest That Support Glaucoma Therapy. Nutrients 2019, 11, 239. [Google Scholar] [CrossRef]
- Perez, C.I.; Singh, K.; Lin, S. Relationship of lifestyle, exercise, and nutrition with glaucoma. Curr. Opin. Ophthalmol. 2019, 30, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Weikel, K.A.; Garber, C.; Baburins, A.; Taylor, A. Nutritional modulation of cataract. Nutr. Rev. 2014, 72, 30–47. [Google Scholar] [CrossRef]
- Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: The Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA 2013, 309, 2005–2015. [CrossRef]
- Thornalley, P.J.; Langborg, A.; Minhas, H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344, 109–116. [Google Scholar]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl proteome and genome damage in metabolic and vascular disease. Biochem. Soc. Trans. 2014, 42, 425–432. [Google Scholar] [CrossRef]
- Bejarano, E.; Taylor, A. Too sweet: Problems of protein glycation in the eye. Exp. Eye Res. 2019, 178, 255–262. [Google Scholar] [CrossRef]
- Kandarakis, S.A.; Piperi, C.; Topouzis, F.; Papavassiliou, A.G. Emerging role of advanced glycation-end products (AGEs) in the pathobiology of eye diseases. Prog. Retin. Eye Res. 2014, 42, 85–102. [Google Scholar] [CrossRef]
- Lapolla, A.; Flamini, R.; Dalla Vedova, A.; Senesi, A.; Reitano, R.; Fedele, D.; Basso, E.; Seraglia, R.; Traldi, P. Glyoxal and methylglyoxal levels in diabetic patients: Quantitative determination by a new GC/MS method. Clin. Chem. Lab. Med. 2003, 41, 1166–1173. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef]
- Miyata, S.; Monnier, V. Immunohistochemical detection of advanced glycosylation end products in diabetic tissues using monoclonal antibody to pyrraline. J. Clin. Investig. 1992, 89, 1102–1112. [Google Scholar] [CrossRef]
- Sell, D.R.; Nagaraj, R.H.; Grandhee, S.K.; Odetti, P.; Lapolla, A.; Fogarty, J.; Monnier, V.M. Pentosidine: A molecular marker for the cumulative damage to proteins in diabetes, aging, and uremia. Diabetes Metab. Rev. 1991, 7, 239–251. [Google Scholar] [CrossRef]
- Schleicher, E.D.; Wagner, E.; Nerlich, A.G. Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J. Clin. Investig. 1997, 99, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Moore, J.E.; Sharkey, J.A.; Murphy, G.; Simpson, D.A.; Bucala, R.; Vlassara, H.; Archer, D.B. Advanced glycation end products in vitreous: Structural and functional implications for diabetic vitreopathy. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2517–2523. [Google Scholar]
- Dyer, D.G.; Blackledge, J.A.; Katz, B.M.; Hull, C.J.; Adkisson, H.D.; Thorpe, S.R.; Lyons, T.J.; Baynes, J.W. The Maillard reaction in vivo. Z. Fur Ernahr. 1991, 30, 29–45. [Google Scholar] [CrossRef]
- Pescosolido, N.; Barbato, A.; Giannotti, R.; Komaiha, C.; Lenarduzzi, F. Age-related changes in the kinetics of human lenses: Prevention of the cataract. Int. J. Ophthalmol. 2016, 9, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Tessier, F.; Obrenovich, M.; Monnier, V.M. Structure and mechanism of formation of human lens fluorophore LM-1. Relationship to vesperlysine A and the advanced Maillard reaction in aging, diabetes, and cataractogenesis. J. Biol. Chem. 1999, 274, 20796–20804. [Google Scholar] [CrossRef]
- Xu, J.; Chen, L.J.; Yu, J.; Wang, H.J.; Zhang, F.; Liu, Q.; Wu, J. Involvement of Advanced Glycation End Products in the Pathogenesis of Diabetic Retinopathy. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 48, 705–717. [Google Scholar] [CrossRef]
- Okamoto, T.; Yamagishi, S.; Inagaki, Y.; Amano, S.; Koga, K.; Abe, R.; Takeuchi, M.; Ohno, S.; Yoshimura, A.; Makita, Z. Angiogenesis induced by advanced glycation end products and its prevention by cerivastatin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1928–1930. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef]
- Frey, T.; Antonetti, D.A. Alterations to the blood-retinal barrier in diabetes: Cytokines and reactive oxygen species. Antioxid Redox Signal 2011, 15, 1271–1284. [Google Scholar] [CrossRef]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef]
- Nojima, H.; Watanabe, H.; Yamane, K.; Kitahara, Y.; Sekikawa, K.; Yamamoto, H.; Yokoyama, A.; Inamizu, T.; Asahara, T.; Kohno, N. Effect of aerobic exercise training on oxidative stress in patients with type 2 diabetes mellitus. Metab. Clin. Exp. 2008, 57, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Rodiño-Janeiro, B.K.; González-Peteiro, M.; Ucieda-Somoza, R.; González-Juanatey, J.R.; Alvarez, E. Glycated albumin, a precursor of advanced glycation end-products, up-regulates NADPH oxidase and enhances oxidative stress in human endothelial cells: Molecular correlate of diabetic vasculopathy. Diabetes Metab. Res. Rev. 2010, 26, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kanwar, M. Effects of curcumin on retinal oxidative stress and inflammation in diabetes. Nutr. Metab. 2007, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.L.; Pérez, S.; Mena-Mollá, S.; Desco, M.C.; Ortega, Á.L. Oxidative Stress and Microvascular Alterations in Diabetic Retinopathy: Future Therapies. Oxidative Med. Cell. Longev. 2019, 2019, 4940825. [Google Scholar] [CrossRef]
- Tang, J.; Zhu, X.W.; Lust, W.D.; Kern, T.S. Retina accumulates more glucose than does the embryologically similar cerebral cortex in diabetic rats. Diabetologia 2000, 43, 1417–1423. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef]
- Sugasawa, K.; Deguchi, J.; Okami, T.; Yamamoto, A.; Omori, K.; Uyama, M.; Tashiro, Y. Immunocytochemical analyses of distributions of Na, K-ATPase and GLUT1, insulin and transferrin receptors in the developing retinal pigment epithelial cells. Cell Struct. Funct. 1994, 19, 21–28. [Google Scholar] [CrossRef]
- You, Z.-P.; Zhang, Y.-L.; Shi, K.; Shi, L.; Zhang, Y.-Z.; Zhou, Y.; Wang, C.-y. Suppression of diabetic retinopathy with GLUT1 siRNA. Sci. Rep. 2017, 7, 7437. [Google Scholar] [CrossRef]
- Ulas, M.; Orhan, C.; Tuzcu, M.; Ozercan, I.H.; Sahin, N.; Gencoglu, H.; Komorowski, J.R.; Sahin, K. Anti-diabetic potential of chromium histidinate in diabetic retinopathy rats. BMC Complement. Altern. Med. 2015, 15, 16. [Google Scholar] [CrossRef]
- Badr, G.A.; Tang, J.; Ismail-Beigi, F.; Kern, T.S. Diabetes downregulates GLUT1 expression in the retina and its microvessels but not in the cerebral cortex or its microvessels. Diabetes 2000, 49, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Rajah, T.T.; Olson, A.L.; Grammas, P. Differential glucose uptake in retina- and brain-derived endothelial cells. Microvasc. Res. 2001, 62, 236–242. [Google Scholar] [CrossRef]
- Nass, N.; Bartling, B.; Navarrete Santos, A.; Scheubel, R.J.; Börgermann, J.; Silber, R.E.; Simm, A. Advanced glycation end products, diabetes and ageing. Z. Fur. Gerontol. Und Geriatr. 2007, 40, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Li, Y.M.; Gardiner, T.A.; Bucala, R.; Archer, D.B.; Vlassara, H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am. J. Pathol. 1997, 150, 523–531. [Google Scholar]
- Chen, M.; Curtis, T.M.; Stitt, A.W. Advanced glycation end products and diabetic retinopathy. Curr. Med. Chem. 2013, 20, 3234–3240. [Google Scholar] [CrossRef]
- McVicar, C.M.; Ward, M.; Colhoun, L.M.; Guduric-Fuchs, J.; Bierhaus, A.; Fleming, T.; Schlotterer, A.; Kolibabka, M.; Hammes, H.P.; Chen, M.; et al. Role of the receptor for advanced glycation endproducts (RAGE) in retinal vasodegenerative pathology during diabetes in mice. Diabetologia 2015, 58, 1129–1137. [Google Scholar] [CrossRef]
- Genuth, S.; Sun, W.; Cleary, P.; Sell, D.R.; Dahms, W.; Malone, J.; Sivitz, W.; Monnier, V.M. Glycation and carboxymethyllysine levels in skin collagen predict the risk of future 10-year progression of diabetic retinopathy and nephropathy in the diabetes control and complications trial and epidemiology of diabetes interventions and complications participants with type 1 diabetes. Diabetes 2005, 54, 3103–3111. [Google Scholar] [CrossRef]
- Genuth, S.; Sun, W.; Cleary, P.; Gao, X.; Sell, D.R.; Lachin, J.; Monnier, V.M. Skin advanced glycation end products glucosepane and methylglyoxal hydroimidazolone are independently associated with long-term microvascular complication progression of type 1 diabetes. Diabetes 2015, 64, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Inagaki, Y.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Matsuura, T.; Narama, I.; Motomiya, Y.; et al. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species-mediated vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 20213–20220. [Google Scholar] [CrossRef]
- Borg, D.J.; Forbes, J.M. Targeting advanced glycation with pharmaceutical agents: Where are we now? Glycoconj. J. 2016, 33, 653–670. [Google Scholar] [CrossRef]
- Gardiner, T.A.; Anderson, H.R.; Stitt, A.W. Inhibition of advanced glycation end-products protects against retinal capillary basement membrane expansion during long-term diabetes. J. Pathol. 2003, 201, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Martin, S.; Federlin, K.; Geisen, K.; Brownlee, M. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc. Natl. Acad. Sci. USA 1991, 88, 11555–11558. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Brownlee, M.; Edelstein, D.; Saleck, M.; Martin, S.; Federlin, K. Aminoguanidine inhibits the development of accelerated diabetic retinopathy in the spontaneous hypertensive rat. Diabetologia 1994, 37, 32–35. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hammes, H.P.; Strödter, D.; Weiss, A.; Bretzel, R.G.; Federlin, K.; Brownlee, M. Secondary intervention with aminoguanidine retards the progression of diabetic retinopathy in the rat model. Diabetologia 1995, 38, 656–660. [Google Scholar] [CrossRef]
- Freedman, B.I.; Wuerth, J.P.; Cartwright, K.; Bain, R.P.; Dippe, S.; Hershon, K.; Mooradian, A.D.; Spinowitz, B.S. Design and baseline characteristics for the aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTION II). Control. Clin. Trials 1999, 20, 493–510. [Google Scholar] [CrossRef]
- Bolton, W.K.; Cattran, D.C.; Williams, M.E.; Adler, S.G.; Appel, G.B.; Cartwright, K.; Foiles, P.G.; Freedman, B.I.; Raskin, P.; Ratner, R.E.; et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am. J. Nephrol. 2004, 24, 32–40. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Berger, J.; Shephard, D.; Morrow, F.; Sadowski, J.; Haire, T.; Taylor, A. Reduced and total ascorbate in guinea pig eye tissues in response to dietary intake. Curr. Eye Res. 1988, 7, 681–686. [Google Scholar] [CrossRef]
- Berger, J.; Shepard, D.; Morrow, F.; Taylor, A. Relationship between dietary intake and tissue levels of reduced and total vitamin C in the nonscorbutic guinea pig. J. Nutr. 1989, 119, 734–740. [Google Scholar] [CrossRef]
- Taylor, A.; Jacques, P.F.; Nowell, T.; Perrone, G.; Blumberg, J.; Handelman, G.; Jozwiak, B.; Nadler, D. Vitamin C in human and guinea pig aqueous, lens and plasma in relation to intake. Curr. Eye Res. 1997, 16, 857–864. [Google Scholar] [CrossRef]
- Yeum, K.J.; Shang, F.M.; Schalch, W.M.; Russell, R.M.; Taylor, A. Fat-soluble nutrient concentrations in different layers of human cataractous lens. Curr. Eye Res. 1999, 19, 502–505. [Google Scholar] [CrossRef]
- Sajithlal, G.B.; Chithra, P.; Chandrakasan, G. Effect of curcumin on the advanced glycation and cross-linking of collagen in diabetic rats. Biochem. Pharmacol. 1998, 56, 1607–1614. [Google Scholar] [CrossRef]
- Li, X.; Zheng, T.; Sang, S.; Lv, L. Quercetin inhibits advanced glycation end product formation by trapping methylglyoxal and glyoxal. J. Agric. Food Chem. 2014, 62, 12152–12158. [Google Scholar] [CrossRef] [PubMed]
- Sampath, C.; Rashid, M.R.; Sang, S.; Ahmedna, M. Green tea epigallocatechin 3-gallate alleviates hyperglycemia and reduces advanced glycation end products via nrf2 pathway in mice with high fat diet-induced obesity. Biomed. Pharm. 2017, 87, 73–81. [Google Scholar] [CrossRef]
- Kishore, L.; Kaur, N.; Singh, R. Effect of Kaempferol isolated from seeds of Eruca sativa on changes of pain sensitivity in Streptozotocin-induced diabetic neuropathy. Inflammopharmacology 2018, 26, 993–1003. [Google Scholar] [CrossRef]
- Navarro, M.; Morales, F.J.; Ramos, S. Olive leaf extract concentrated in hydroxytyrosol attenuates protein carbonylation and the formation of advanced glycation end products in a hepatic cell line (HepG2). Food Funct. 2017, 8, 944–953. [Google Scholar] [CrossRef]
- Hajizadeh-Sharafabad, F.; Sahebkar, A.; Zabetian-Targhi, F.; Maleki, V. The impact of resveratrol on toxicity and related complications of advanced glycation end products: A systematic review. Biofactors 2019, 45, 651–665. [Google Scholar] [CrossRef]
- Basta, G.; Lazzerini, G.; Del Turco, S.; Ratto, G.M.; Schmidt, A.M.; De Caterina, R. At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1401–1407. [Google Scholar] [CrossRef]
- Nam, M.H.; Son, W.R.; Lee, Y.S.; Lee, K.W. Glycolaldehyde-derived advanced glycation end products (glycol-AGEs)-induced vascular smooth muscle cell dysfunction is regulated by the AGES-receptor (RAGE) axis in endothelium. Cell Commun. Adhes. 2015, 22, 67–78. [Google Scholar] [CrossRef]
- Dobi, A.; Bravo, S.B.; Veeren, B.; Paradela-Dobarro, B.; Álvarez, E.; Meilhac, O.; Viranaicken, W.; Baret, P.; Devin, A.; Rondeau, P. Advanced glycation end-products disrupt human endothelial cells redox homeostasis: New insights into reactive oxygen species production. Free Radic. Res. 2019, 53, 150–169. [Google Scholar] [CrossRef]
- Wang, X.L.; Yu, T.; Yan, Q.C.; Wang, W.; Meng, N.; Li, X.J.; Luo, Y.H. AGEs Promote Oxidative Stress and Induce Apoptosis in Retinal Pigmented Epithelium Cells RAGE-dependently. J. Mol. Neurosci. 2015, 56, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Torreggiani, M.; Zhu, L.; Chen, X.; He, J.C.; Striker, G.E.; Vlassara, H. AGER1 regulates endothelial cell NADPH oxidase-dependent oxidant stress via PKC-delta: Implications for vascular disease. Am. J. Physiol. Cell Physiol. 2010, 298, C624–C634. [Google Scholar] [CrossRef]
- Vlassara, H.; Cai, W.; Goodman, S.; Pyzik, R.; Yong, A.; Chen, X.; Zhu, L.; Neade, T.; Beeri, M.; Silverman, J.M.; et al. Protection against loss of innate defenses in adulthood by low advanced glycation end products (AGE) intake: Role of the antiinflammatory AGE receptor-1. J. Clin. Endocrinol. Metab. 2009, 94, 4483–4491. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: Potential role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A. Mechanistically linking age-related diseases and dietary carbohydrate via autophagy and the ubiquitin proteolytic systems. Autophagy 2012, 8, 1404–1406. [Google Scholar] [CrossRef][Green Version]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef]
- Antognelli, C.; Talesa, V.N. Glyoxalases in Urological Malignancies. Int. J. Mol. Sci. 2018, 19, 415. [Google Scholar] [CrossRef]
- Thornalley, P.J. The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 1990, 269, 1–11. [Google Scholar] [CrossRef]
- Abordo, E.A.; Minhas, H.S.; Thornalley, P.J. Accumulation of alpha-oxoaldehydes during oxidative stress: A role in cytotoxicity. Biochem. Pharmacol. 1999, 58, 641–648. [Google Scholar] [CrossRef]
- Clelland, J.D.; Thornalley, P.J. S-2-hydroxyacylglutathione-derivatives: Enzymatic preparation, purification and characterisation. J. Chem. Soc. Perkin Trans. 1 1991, 3009–3015. [Google Scholar] [CrossRef]
- Shinohara, M.; Thornalley, P.J.; Giardino, I.; Beisswenger, P.; Thorpe, S.R.; Onorato, J.; Brownlee, M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Investig. 1998, 101, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- McCann, V.J.; Davis, R.E.; Welborn, T.A.; Constable, I.J.; Beale, D.G. Glyoxalase phenotypes in patients with diabetes mellitus. Aust. N. Z. J. Med. 1981, 11, 380–382. [Google Scholar] [CrossRef]
- Kalousová, M.; Germanová, A.; Jáchymová, M.; Mestek, O.; Tesar, V.; Zima, T. A419C (E111A) polymorphism of the glyoxalase I gene and vascular complications in chronic hemodialysis patients. Ann. N. Y. Acad. Sci. 2008, 1126, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S.; Ciaccio, P.J.; Walsh, E.S.; Tew, K.D. Genomic sequence of human glyoxalase-I: Analysis of promoter activity and its regulation. Gene 1999, 240, 149–155. [Google Scholar] [CrossRef]
- Conboy, C.M.; Spyrou, C.; Thorne, N.P.; Wade, E.J.; Barbosa-Morais, N.L.; Wilson, M.D.; Bhattacharjee, A.; Young, R.A.; Tavaré, S.; Lees, J.A.; et al. Cell cycle genes are the evolutionarily conserved targets of the E2F4 transcription factor. PLoS ONE 2007, 2, e1061. [Google Scholar] [CrossRef] [PubMed]
- Orso, F.; Corà, D.; Ubezio, B.; Provero, P.; Caselle, M.; Taverna, D. Identification of functional TFAP2A and SP1 binding sites in new TFAP2A-modulated genes. Bmc Genom. 2010, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef]
- Kold-Christensen, R.; Johannsen, M. Methylglyoxal Metabolism and Aging-Related Disease: Moving from Correlation toward Causation. Trends Endocrinol. Metab. 2020, 31, 81–92. [Google Scholar] [CrossRef]
- Li, D.; Ferrari, M.; Ellis, E.M. Human aldo-keto reductase AKR7A2 protects against the cytotoxicity and mutagenicity of reactive aldehydes and lowers intracellular reactive oxygen species in hamster V79-4 cells. Chem. Biol. Interact. 2012, 195, 25–34. [Google Scholar] [CrossRef]
- Li, D.; Ellis, E.M. Aldo-keto reductase 7A5 (AKR7A5) attenuates oxidative stress and reactive aldehyde toxicity in V79-4 cells. Toxicol. Vitr. Int. J. Publ. Assoc. Bibra 2014, 28, 707–714. [Google Scholar] [CrossRef]
- Baba, S.P.; Barski, O.A.; Ahmed, Y.; O’Toole, T.E.; Conklin, D.J.; Bhatnagar, A.; Srivastava, S. Reductive metabolism of AGE precursors: A metabolic route for preventing AGE accumulation in cardiovascular tissue. Diabetes 2009, 58, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Vander Jagt, D.L.; Hunsaker, L.A. Methylglyoxal metabolism and diabetic complications: Roles of aldose reductase, glyoxalase-I, betaine aldehyde dehydrogenase and 2-oxoaldehyde dehydrogenase. Chem. Biol. Interact. 2003, 143-144, 341–351. [Google Scholar] [CrossRef]
- Morgenstern, J.; Fleming, T.; Schumacher, D.; Eckstein, V.; Freichel, M.; Herzig, S.; Nawroth, P. Loss of Glyoxalase 1 Induces Compensatory Mechanism to Achieve Dicarbonyl Detoxification in Mammalian Schwann Cells. J. Biol. Chem. 2017, 292, 3224–3238. [Google Scholar] [CrossRef]
- Lodd, E.; Wiggenhauser, L.M.; Morgenstern, J.; Fleming, T.H.; Poschet, G.; Büttner, M.; Tabler, C.T.; Wohlfart, D.P.; Nawroth, P.P.; Kroll, J. The combination of loss of glyoxalase1 and obesity results in hyperglycemia. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Morgenstern, J.; Oguchi, Y.; Volk, N.; Kopf, S.; Groener, J.B.; Nawroth, P.P.; Fleming, T.; Freichel, M. Compensatory mechanisms for methylglyoxal detoxification in experimental & clinical diabetes. Mol. Metab. 2018, 18, 143–152. [Google Scholar] [CrossRef]
- Fujii, E.; Iwase, H.; Ishii-Karakasa, I.; Yajima, Y.; Hotta, K. The presence of 2-keto-3-deoxygluconic acid and oxoaldehyde dehydrogenase activity in human erythrocytes. Biochem. Biophys. Res. Commun. 1995, 210, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Collard, F.; Vertommen, D.; Fortpied, J.; Duester, G.; Van Schaftingen, E. Identification of 3-deoxyglucosone dehydrogenase as aldehyde dehydrogenase 1A1 (retinaldehyde dehydrogenase 1). Biochimie 2007, 89, 369–373. [Google Scholar] [CrossRef]
- McDowell, R.E.; McGahon, M.K.; Augustine, J.; Chen, M.; McGeown, J.G.; Curtis, T.M. Diabetes Impairs the Aldehyde Detoxifying Capacity of the Retina. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4762–4771. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Lev, N.; Roncevic, D.; Ickowicz, D.; Melamed, E.; Offen, D. Role of DJ-1 in Parkinson’s disease. J. Mol. Neurosci. 2006, 29, 215–225. [Google Scholar] [CrossRef]
- Lee, J.Y.; Song, J.; Kwon, K.; Jang, S.; Kim, C.; Baek, K.; Kim, J.; Park, C. Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet. 2012, 21, 3215–3225. [Google Scholar] [CrossRef]
- Richarme, G.; Mihoub, M.; Dairou, J.; Bui, L.C.; Leger, T.; Lamouri, A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 2015, 290, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, D.H.; Fleming, T.; Nawroth, P.; Teleman, A.A. Evidence Against a Role for the Parkinsonism-associated Protein DJ-1 in Methylglyoxal Detoxification. J. Biol. Chem. 2017, 292, 685–690. [Google Scholar] [CrossRef]
- Richarme, G.; Liu, C.; Mihoub, M.; Abdallah, J.; Leger, T.; Joly, N.; Liebart, J.C.; Jurkunas, U.V.; Nadal, M.; Bouloc, P.; et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science 2017, 357, 208–211. [Google Scholar] [CrossRef]
- Galligan, J.J.; Wepy, J.A.; Streeter, M.D.; Kingsley, P.J.; Mitchener, M.M.; Wauchope, O.R.; Beavers, W.N.; Rose, K.L.; Wang, T.; Spiegel, D.A.; et al. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. USA 2018, 115, 9228–9233. [Google Scholar] [CrossRef]
- Zheng, Q.; Omans, N.D.; Leicher, R.; Osunsade, A.; Agustinus, A.S.; Finkin-Groner, E.; D’Ambrosio, H.; Liu, B.; Chandarlapaty, S.; Liu, S.; et al. Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat. Commun. 2019, 10, 1289. [Google Scholar] [CrossRef]
- Kalapos, M.P. On the mammalian acetone metabolism: From chemistry to clinical implications. Biochim. Biophys. Acta 2003, 1621, 122–139. [Google Scholar] [CrossRef]
- Salomón, T.; Sibbersen, C.; Hansen, J.; Britz, D.; Svart, M.V.; Voss, T.S.; Møller, N.; Gregersen, N.; Jørgensen, K.A.; Palmfeldt, J.; et al. Ketone Body Acetoacetate Buffers Methylglyoxal via a Non-enzymatic Conversion during Diabetic and Dietary Ketosis. Cell Chem. Biol. 2017, 24, 935–943.e937. [Google Scholar] [CrossRef]
- Barnosky, A.R.; Hoddy, K.K.; Unterman, T.G.; Varady, K.A. Intermittent fasting vs daily calorie restriction for type 2 diabetes prevention: A review of human findings. Transl. Res. J. Lab. Clin. Med. 2014, 164, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Takabatake, Y.; Kimura, T.; Maejima, I.; Namba, T.; Yamamoto, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; et al. Autophagy Inhibits the Accumulation of Advanced Glycation End Products by Promoting Lysosomal Biogenesis and Function in the Kidney Proximal Tubules. Diabetes 2017, 66, 1359–1372. [Google Scholar] [CrossRef]
- Uchiki, T.; Weikel, K.A.; Jiao, W.; Shang, F.; Caceres, A.; Pawlak, D.; Handa, J.T.; Brownlee, M.; Nagaraj, R.; Taylor, A. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics). Aging Cell 2012, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cross, S.D.; Stanton, J.B.; Marmorstein, A.D.; Le, Y.Z.; Marmorstein, L.Y. Early AMD-like defects in the RPE and retinal degeneration in aged mice with RPE-specific deletion of Atg5 or Atg7. Mol. Vis. 2017, 23, 228–241. [Google Scholar] [PubMed]
- Chai, P.; Ni, H.; Zhang, H.; Fan, X. The Evolving Functions of Autophagy in Ocular Health: A Double-edged Sword. Int. J. Biol. Sci. 2016, 12, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Rosa, M.D.; Distefano, G.; Gagliano, C.; Rusciano, D.; Malaguarnera, L. Autophagy in Diabetic Retinopathy. Curr. Neuropharmacol. 2016, 14, 810–825. [Google Scholar] [CrossRef]
- Grimm, S.; Ernst, L.; Grötzinger, N.; Höhn, A.; Breusing, N.; Reinheckel, T.; Grune, T. Cathepsin D is one of the major enzymes involved in intracellular degradation of AGE-modified proteins. Free Radic. Res. 2010, 44, 1013–1026. [Google Scholar] [CrossRef]
- Bulteau, A.L.; Verbeke, P.; Petropoulos, I.; Chaffotte, A.F.; Friguet, B. Proteasome inhibition in glyoxal-treated fibroblasts and resistance of glycated glucose-6-phosphate dehydrogenase to 20 S proteasome degradation in vitro. J. Biol. Chem. 2001, 276, 45662–45668. [Google Scholar] [CrossRef]
- Raupbach, J.; Ott, C.; Koenig, J.; Grune, T. Proteasomal degradation of glycated proteins depends on substrate unfolding: Preferred degradation of moderately modified myoglobin. Free Radic. Biol. Med. 2020, 152, 516–524. [Google Scholar] [CrossRef]
- Gavilán, E.; Pintado, C.; Gavilan, M.P.; Daza, P.; Sánchez-Aguayo, I.; Castaño, A.; Ruano, D. Age-related dysfunctions of the autophagy lysosomal pathway in hippocampal pyramidal neurons under proteasome stress. Neurobiol. Aging 2015, 36, 1953–1963. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef]
- Shang, F.; Taylor, A. Ubiquitin Conjugates: A Sensitive Marker of Oxidative Stress. In Biomarkers for Antioxidant Defense and Oxidative Damage: Principles and Practical Applications; Wiley: Hoboken, NJ, USA, 2010; pp. 219–228. [Google Scholar] [CrossRef]
- Shang, F.; Taylor, A. Roles for the ubiquitin-proteasome pathway in protein quality control and signaling in the retina: Implications in the pathogenesis of age-related macular degeneration. Mol. Asp. Med. 2012, 33, 446–466. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Ishii, T.; Wakabayashi, N.; Yamamoto, M. Regulatory mechanisms of cellular response to oxidative stress. Free Radic. Res. 1999, 31, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Itoh, K.; Suzuki, T.; Osanai, H.; Nishikawa, K.; Katoh, Y.; Takagi, Y.; Yamamoto, M. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells Devoted Mol. Cell. Mech. 2002, 7, 807–820. [Google Scholar] [CrossRef]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells Devoted Mol. Cell. Mech. 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Fornelli, C.; Retta, F.; Cassoni, P.; Talesa, V.N.; Retta, S.F. Data in support of sustained upregulation of adaptive redox homeostasis mechanisms caused by KRIT1 loss-of-function. Data Brief 2018, 16, 929–938. [Google Scholar] [CrossRef]
- Gambelunghe, A.; Giovagnoli, S.; Di Michele, A.; Boncompagni, S.; Dell’Omo, M.; Leopold, K.; Iavicoli, I.; Talesa, V.N.; Antognelli, C. Redox-Sensitive Glyoxalase 1 Up-Regulation Is Crucial for Protecting Human Lung Cells from Gold Nanoparticles Toxicity. Antioxidants 2020, 9, 697. [Google Scholar] [CrossRef]
- James, D.; Devaraj, S.; Bellur, P.; Lakkanna, S.; Vicini, J.; Boddupalli, S. Novel concepts of broccoli sulforaphanes and disease: Induction of phase II antioxidant and detoxification enzymes by enhanced-glucoraphanin broccoli. Nutr. Rev. 2012, 70, 654–665. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Rizzo, B.; Barbalace, M.C.; Fabbri, D.; Hrelia, S. Neuroprotective effect of sulforaphane against methylglyoxal cytotoxicity. Chem. Res. Toxicol. 2015, 28, 1234–1245. [Google Scholar] [CrossRef]
- Alfarano, M.; Pastore, D.; Fogliano, V.; Schalkwijk, C.G.; Oliviero, T. The Effect of Sulforaphane on Glyoxalase I Expression and Activity in Peripheral Blood Mononuclear Cells. Nutrients 2018, 10, 1773. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.; Fernandes, R.; Crisóstomo, J.; Seiça, R.M.; Sena, C.M. The Sulforaphane and pyridoxamine supplementation normalize endothelial dysfunction associated with type 2 diabetes. Sci. Rep. 2017, 7, 14357. [Google Scholar] [CrossRef]
- Nishimoto, S.; Koike, S.; Inoue, N.; Suzuki, T.; Ogasawara, Y. Activation of Nrf2 attenuates carbonyl stress induced by methylglyoxal in human neuroblastoma cells: Increase in GSH levels is a critical event for the detoxification mechanism. Biochem. Biophys. Res. Commun. 2017, 483, 874–879. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Gambelunghe, A.; Muzi, G.; Talesa, V.N. Peroxynitrite-mediated glyoxalase I epigenetic inhibition drives apoptosis in airway epithelial cells exposed to crystalline silica via a novel mechanism involving argpyrimidine-modified Hsp70, JNK, and NF-κB. Free Radic. Biol. Med. 2015, 84, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef]
- Rauh, D.; Fischer, F.; Gertz, M.; Lakshminarasimhan, M.; Bergbrede, T.; Aladini, F.; Kambach, C.; Becker, C.F.; Zerweck, J.; Schutkowski, M.; et al. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat. Commun. 2013, 4, 2327. [Google Scholar] [CrossRef]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef]
- Reiniger, N.; Lau, K.; McCalla, D.; Eby, B.; Cheng, B.; Lu, Y.; Qu, W.; Quadri, N.; Ananthakrishnan, R.; Furmansky, M.; et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes 2010, 59, 2043–2054. [Google Scholar] [CrossRef]
- Irshad, Z.; Xue, M.; Ashour, A.; Larkin, J.R.; Thornalley, P.J.; Rabbani, N. Activation of the unfolded protein response in high glucose treated endothelial cells is mediated by methylglyoxal. Sci. Rep. 2019, 9, 7889. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Jiang, S.; Chang, M.L.; Volkin, J.; Cassalman, C.; Smith, K.M.; Streeter, M.D.; Spiegel, D.A.; Moreira-Neto, C.; Rabbani, N.; et al. A low glycemic diet protects disease-prone Nrf2-deficient mice against age-related macular degeneration. Free Radic. Biol. Med. 2020, 150, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, B.; Goldstein, B.; Thornalley, P.J.; Rabbani, N.; Tschoepe, D. Intracellular Accumulation of Methylglyoxal by Glyoxalase 1 Knock Down Alters Collagen Homoeostasis in L6 Myoblasts. Int. J. Mol. Sci. 2017, 18, 480. [Google Scholar] [CrossRef]
- Nigro, C.; Leone, A.; Fiory, F.; Prevenzano, I.; Nicolò, A.; Mirra, P.; Beguinot, F.; Miele, C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Du, X.; D’Agati, V.D.; Milne, R.; Sui, G.; Geoffrion, M.; Brownlee, M. Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes 2014, 63, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Kwon, D.M.; Kwon, K.; Park, C. Generation and characterization of mouse knockout for glyoxalase 1. Biochem. Biophys. Res. Commun. 2017, 490, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Shafie, A.; Xue, M.; Barker, G.; Zehnder, D.; Thornalley, P.J.; Rabbani, N. Reappraisal of putative glyoxalase 1-deficient mouse and dicarbonyl stress on embryonic stem cells in vitro. Biochem. J. 2016, 473, 4255–4270. [Google Scholar] [CrossRef]
- Moraru, A.; Wiederstein, J.; Pfaff, D.; Fleming, T.; Miller, A.K.; Nawroth, P.; Teleman, A.A. Elevated Levels of the Reactive Metabolite Methylglyoxal Recapitulate Progression of Type 2 Diabetes. Cell Metab. 2018, 27, 926–934. [Google Scholar] [CrossRef]
- Morcos, M.; Du, X.; Pfisterer, F.; Hutter, H.; Sayed, A.A.; Thornalley, P.; Ahmed, N.; Baynes, J.; Thorpe, S.; Kukudov, G.; et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 2008, 7, 260–269. [Google Scholar] [CrossRef]
- Distler, M.G.; Plant, L.D.; Sokoloff, G.; Hawk, A.J.; Aneas, I.; Wuenschell, G.E.; Termini, J.; Meredith, S.C.; Nobrega, M.A.; Palmer, A.A. Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J. Clin. Investig. 2012, 122, 2306–2315. [Google Scholar] [CrossRef]
- Brouwers, O.; Niessen, P.M.; Ferreira, I.; Miyata, T.; Scheffer, P.G.; Teerlink, T.; Schrauwen, P.; Brownlee, M.; Stehouwer, C.D.; Schalkwijk, C.G. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J. Biol. Chem. 2011, 286, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Berner, A.K.; Brouwers, O.; Pringle, R.; Klaassen, I.; Colhoun, L.; McVicar, C.; Brockbank, S.; Curry, J.W.; Miyata, T.; Brownlee, M.; et al. Protection against methylglyoxal-derived AGEs by regulation of glyoxalase 1 prevents retinal neuroglial and vasodegenerative pathology. Diabetologia 2012, 55, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, O.; Niessen, P.M.; Miyata, T.; Østergaard, J.A.; Flyvbjerg, A.; Peutz-Kootstra, C.J.; Sieber, J.; Mundel, P.H.; Brownlee, M.; Janssen, B.J.; et al. Glyoxalase-1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia 2014, 57, 224–235. [Google Scholar] [CrossRef]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef]
- Dobler, D.; Ahmed, N.; Song, L.; Eboigbodin, K.E.; Thornalley, P.J. Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes 2006, 55, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.G.; Smith, D.G.; Bhat, M.; Nagaraj, R.H. Glyoxalase I is critical for human retinal capillary pericyte survival under hyperglycemic conditions. J. Biol. Chem. 2006, 281, 11864–11871. [Google Scholar] [CrossRef]
- Barati, M.T.; Merchant, M.L.; Kain, A.B.; Jevans, A.W.; McLeish, K.R.; Klein, J.B. Proteomic analysis defines altered cellular redox pathways and advanced glycation end-product metabolism in glomeruli of db/db diabetic mice. Am. J. Physiol. Ren. Physiol. 2007, 293, F1157–F1165. [Google Scholar] [CrossRef]
- Palsamy, P.; Subramanian, S. Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2-Keap1 signaling. Biochim. Et Biophys. Acta 2011, 1812, 719–731. [Google Scholar] [CrossRef]
- Miller, A.G.; Tan, G.; Binger, K.J.; Pickering, R.J.; Thomas, M.C.; Nagaraj, R.H.; Cooper, M.E.; Wilkinson-Berka, J.L. Candesartan attenuates diabetic retinal vascular pathology by restoring glyoxalase-I function. Diabetes 2010, 59, 3208–3215. [Google Scholar] [CrossRef]
- Phillips, S.A.; Mirrlees, D.; Thornalley, P.J. Modification of the glyoxalase system in streptozotocin-induced diabetic rats. Effect of the aldose reductase inhibitor Statil. Biochem. Pharmacol. 1993, 46, 805–811. [Google Scholar] [CrossRef]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnölzer, M.; Lasitschka, F.; et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef]
- Atkins, T.W.; Thornally, P.J. Erythrocyte glyoxalase activity in genetically obese (ob/ob) and streptozotocin diabetic mice. Diabetes Res. 1989, 11, 125–129. [Google Scholar] [PubMed]
- McLellan, A.C.; Thornalley, P.J. Glyoxalase activity in human red blood cells fractioned by age. Mech. Ageing Dev. 1989, 48, 63–71. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Rabbani, N.; Thornalley, P.J. Glyoxalase in ageing. Semin. Cell Dev. Biol. 2011, 22, 293–301. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Methylglyoxal-induced dicarbonyl stress in aging and disease: First steps towards glyoxalase 1-based treatments. Clin. Sci. 2016, 130, 1677–1696. [Google Scholar] [CrossRef]
- Sharma-Luthra, R.; Kale, R.K. Age related changes in the activity of the glyoxalase system. Mech. Ageing Dev. 1994, 73, 39–45. [Google Scholar] [CrossRef]
- Amicarelli, F.; Di Ilio, C.; Masciocco, L.; Bonfigli, A.; Zarivi, O.; D’Andrea, M.R.; Di Giulio, C.; Miranda, M. Aging and detoxifying enzymes responses to hypoxic or hyperoxic treatment. Mech. Ageing Dev. 1997, 97, 215–226. [Google Scholar] [CrossRef]
- Kirk, J.E. The glyoxalase I activity of arterial tissue in individuals of various ages. J. Gerontol. 1960, 15, 139–141. [Google Scholar] [CrossRef]
- Haik, G.M., Jr.; Lo, T.W.; Thornalley, P.J. Methylglyoxal concentration and glyoxalase activities in the human lens. Exp. Eye Res. 1994, 59, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Mailankot, M.; Padmanabha, S.; Pasupuleti, N.; Major, D.; Howell, S.; Nagaraj, R.H. Glyoxalase I activity and immunoreactivity in the aging human lens. Biogerontology 2009, 10, 711–720. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kuhla, B.; Boeck, K.; Lüth, H.J.; Schmidt, A.; Weigle, B.; Schmitz, M.; Ogunlade, V.; Münch, G.; Arendt, T. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol. Aging 2006, 27, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Li, X.H.; Peng, Y.D.; Wang, J.B.; Tang, J.F.; Wang, Y.F. Association of two glyoxalase I gene polymorphisms with nephropathy and retinopathy in Type 2 diabetes. J. Endocrinol. Investig. 2011, 34, e343–e348. [Google Scholar] [CrossRef]
- Rasul, A.; Rashid, A.; Waheed, P.; Khan, S.A. Expression analysis of glyoxalase I gene among patients of diabetic retinopathy. Pak. J. Med. Sci. 2018, 34, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.; Waheed, P.; Rashid, A.; Khan, S.A. Gene Expression of Glyoxalase II in Diabetic Retinopathy. J. Coll. Physicians Surg. Pak. 2018, 28, 523–526. [Google Scholar] [CrossRef]
- Sachdeva, R.; Schlotterer, A.; Schumacher, D.; Matka, C.; Mathar, I.; Dietrich, N.; Medert, R.; Kriebs, U.; Lin, J.; Nawroth, P.; et al. TRPC proteins contribute to development of diabetic retinopathy and regulate glyoxalase 1 activity and methylglyoxal accumulation. Mol. Metab. 2018, 9, 156–167. [Google Scholar] [CrossRef]
- Arai, M.; Nihonmatsu-Kikuchi, N.; Itokawa, M.; Rabbani, N.; Thornalley, P.J. Measurement of glyoxalase activities. Biochem. Soc. Trans. 2014, 42, 491–494. [Google Scholar] [CrossRef]
- Peters, A.S.; Wortmann, M.; Fleming, T.H.; Nawroth, P.P.; Bruckner, T.; Böckler, D.; Hakimi, M. Effect of metformin treatment in patients with type 2 diabetes with respect to glyoxalase 1 activity in atherosclerotic lesions. Vasa. Z. Fur Gefasskrankh. 2019, 48, 186–192. [Google Scholar] [CrossRef]
- Cheng, A.S.; Cheng, Y.H.; Chiou, C.H.; Chang, T.L. Resveratrol upregulates Nrf2 expression to attenuate methylglyoxal-induced insulin resistance in Hep G2 cells. J. Agric. Food Chem. 2012, 60, 9180–9187. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Dargusch, R.; Ehren, J.L.; Okada, S.; Sharma, K.; Schubert, D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS ONE 2011, 6, e21226. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Zhu, X.; Zhang, L.; Lu, Q.; Wang, J.Y.; Zhang, F.; Guo, H.; Yin, J.L.; Yin, X.X. Up-regulation of glyoxalase 1 by mangiferin prevents diabetic nephropathy progression in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2013, 721, 355–364. [Google Scholar] [CrossRef]
- Liu, Y.W.; Cheng, Y.Q.; Liu, X.L.; Hao, Y.C.; Li, Y.; Zhu, X.; Zhang, F.; Yin, X.X. Mangiferin Upregulates Glyoxalase 1 Through Activation of Nrf2/ARE Signaling in Central Neurons Cultured with High Glucose. Mol. Neurobiol. 2017, 54, 4060–4070. [Google Scholar] [CrossRef] [PubMed]
- Suantawee, T.; Thilavech, T.; Cheng, H.; Adisakwattana, S. Cyanidin Attenuates Methylglyoxal-Induced Oxidative Stress and Apoptosis in INS-1 Pancreatic β-Cells by Increasing Glyoxalase-1 Activity. Nutrients 2020, 12, 1319. [Google Scholar] [CrossRef]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef]
- Angeloni, C.; Turroni, S.; Bianchi, L.; Fabbri, D.; Motori, E.; Malaguti, M.; Leoncini, E.; Maraldi, T.; Bini, L.; Brigidi, P.; et al. Novel targets of sulforaphane in primary cardiomyocytes identified by proteomic analysis. PLoS ONE 2013, 8, e83283. [Google Scholar] [CrossRef]
- Maeda, S.; Matsui, T.; Ojima, A.; Takeuchi, M.; Yamagishi, S. Sulforaphane inhibits advanced glycation end product-induced pericyte damage by reducing expression of receptor for advanced glycation end products. Nutr. Res. 2014, 34, 807–813. [Google Scholar] [CrossRef]
- Lv, J.; Bao, S.; Liu, T.; Wei, L.; Wang, D.; Ye, W.; Wang, N.; Song, S.; Li, J.; Chudhary, M.; et al. Sulforaphane delays diabetes-induced retinal photoreceptor cell degeneration. Cell Tissue Res. 2020. [Google Scholar] [CrossRef]
- Oh, S.; Ahn, H.; Park, H.; Lee, J.I.; Park, K.Y.; Hwang, D.; Lee, S.; Son, K.H.; Byun, K. The attenuating effects of pyridoxamine on adipocyte hypertrophy and inflammation differ by adipocyte location. J. Nutr. Biochem. 2019, 72, 108173. [Google Scholar] [CrossRef]
- Stitt, A.; Gardiner, T.A.; Alderson, N.L.; Canning, P.; Frizzell, N.; Duffy, N.; Boyle, C.; Januszewski, A.S.; Chachich, M.; Baynes, J.W.; et al. The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes 2002, 51, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhou, C.; Huang, M.; Tang, C.; Liu, X.; Yue, Y.; Diao, Q.; Zheng, Z.; Liu, D. Glyoxalase system: A systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed Pharm. 2020, 131, 110663. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aragonès, G.; Rowan, S.; G Francisco, S.; Yang, W.; Weinberg, J.; Taylor, A.; Bejarano, E. Glyoxalase System as a Therapeutic Target against Diabetic Retinopathy. Antioxidants 2020, 9, 1062. https://doi.org/10.3390/antiox9111062
Aragonès G, Rowan S, G Francisco S, Yang W, Weinberg J, Taylor A, Bejarano E. Glyoxalase System as a Therapeutic Target against Diabetic Retinopathy. Antioxidants. 2020; 9(11):1062. https://doi.org/10.3390/antiox9111062
Chicago/Turabian StyleAragonès, Gemma, Sheldon Rowan, Sarah G Francisco, Wenxin Yang, Jasper Weinberg, Allen Taylor, and Eloy Bejarano. 2020. "Glyoxalase System as a Therapeutic Target against Diabetic Retinopathy" Antioxidants 9, no. 11: 1062. https://doi.org/10.3390/antiox9111062
APA StyleAragonès, G., Rowan, S., G Francisco, S., Yang, W., Weinberg, J., Taylor, A., & Bejarano, E. (2020). Glyoxalase System as a Therapeutic Target against Diabetic Retinopathy. Antioxidants, 9(11), 1062. https://doi.org/10.3390/antiox9111062