Triapine Derivatives Act as Copper Delivery Vehicles to Induce Deadly Metal Overload in Cancer Cells

 , , ,
, , ,  and
and 
Abstract
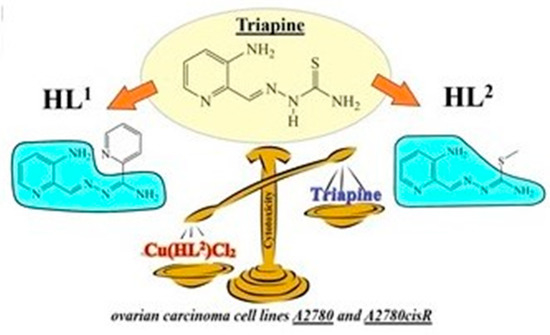
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.1.1. 3-Amino-2-pyridinecarboxaldehyde 2-pyridinamidrazone (HL1)
2.1.2. [Cu(HL1)Cl2]·H2O∙(1·H2O)
2.1.3. 3-Amino-2-pyridinecarboxaldehyde S-methylisothiosemicarbazone (HL2)
2.1.4. [Cu(HL2)Cl2] (2)
2.1.5. [Fe(L2)2NO3]∙0.75H2O (3∙0.75H2O)
2.2. Physical Measurements
2.3. Crystallographic Structure Determination
2.4. Electrochemistry and Spectroelectrochemistry
2.5. Cell Lines and Culture Conditions
2.6. Inhibition of Cell Viability Assay
2.7. Intracellular Accumulation
2.8. Spectrophotometric and pH-Potentiometric Measurements
2.9. Tyrosyl Radical Reduction in Human R2 RNR Protein
2.10. EPR Spin Trapping Experiments
2.11. Western Blotting Experiments
3. Results and Discussion
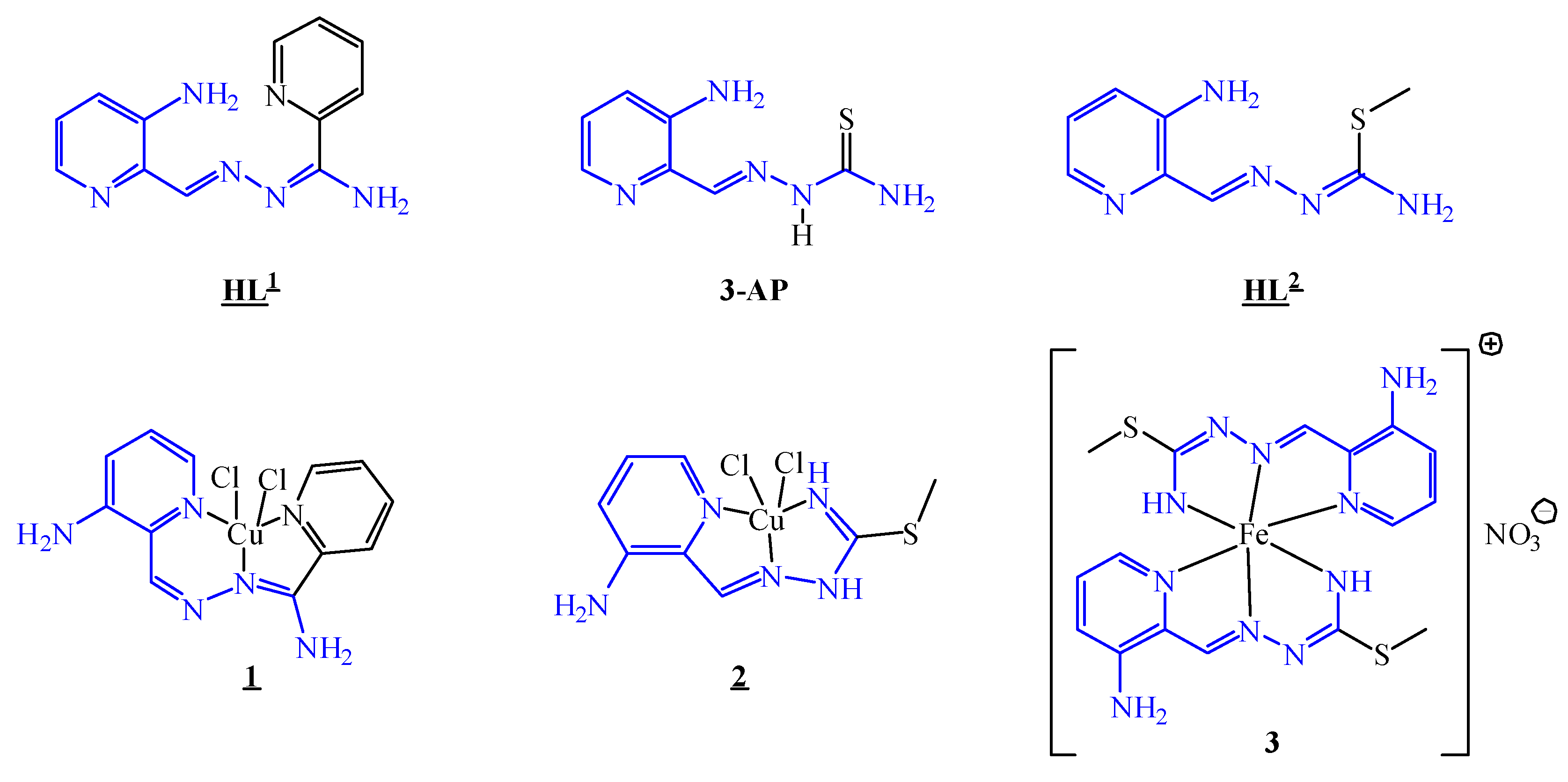
3.1. Synthesis and Characterisation of the HL1, HL2, Cu(II) Complexes 1 and 2 and Fe(III) Complex 3
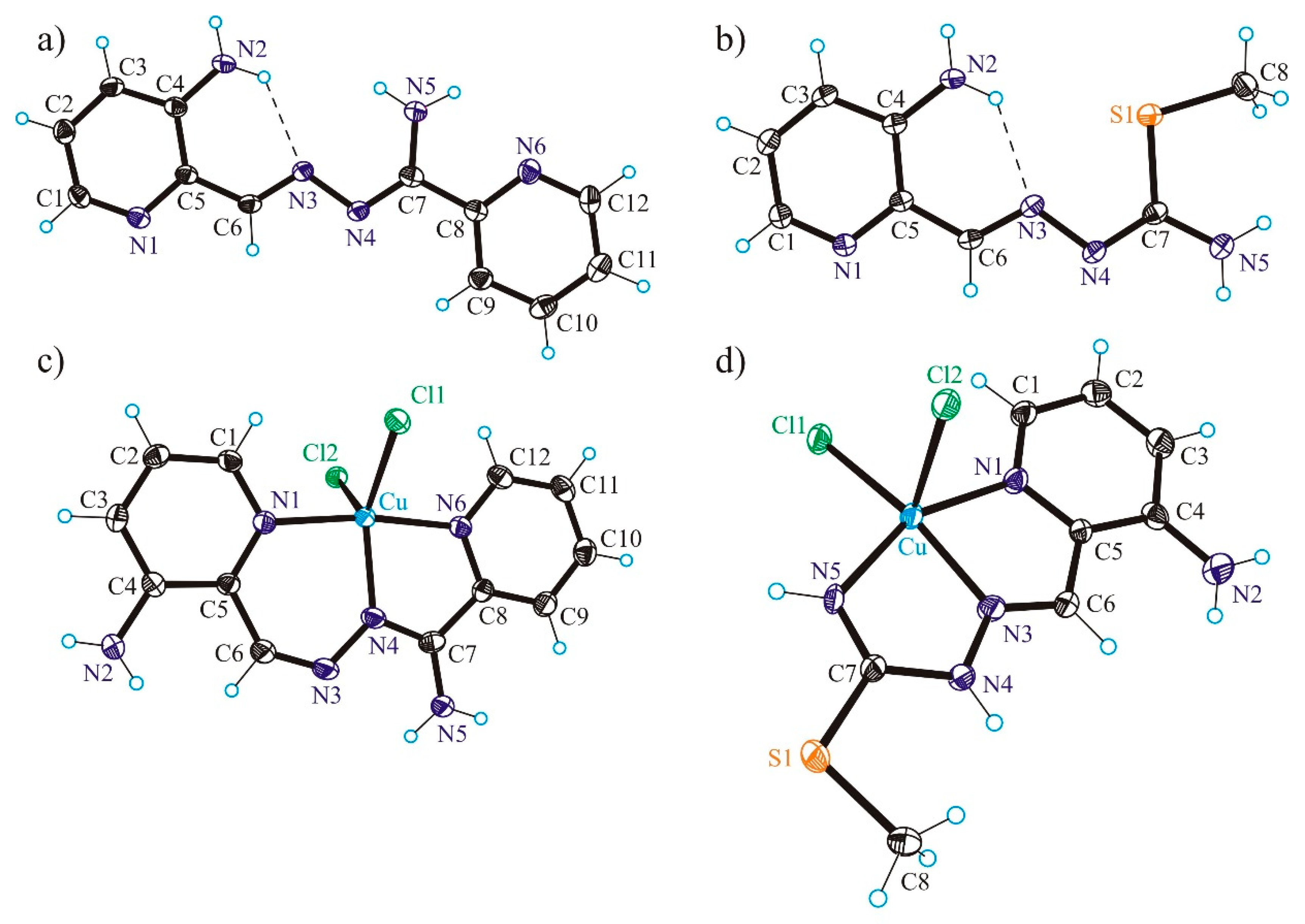
3.2. X-ray Crystallography
3.3. Solution Chemistry of HL1, HL2 and Their Cu(II) Complexes (1 and 2)
3.4. Cytotoxicity
4. Investigation of Mechanism of Action
4.1. Intracellular Accumulation
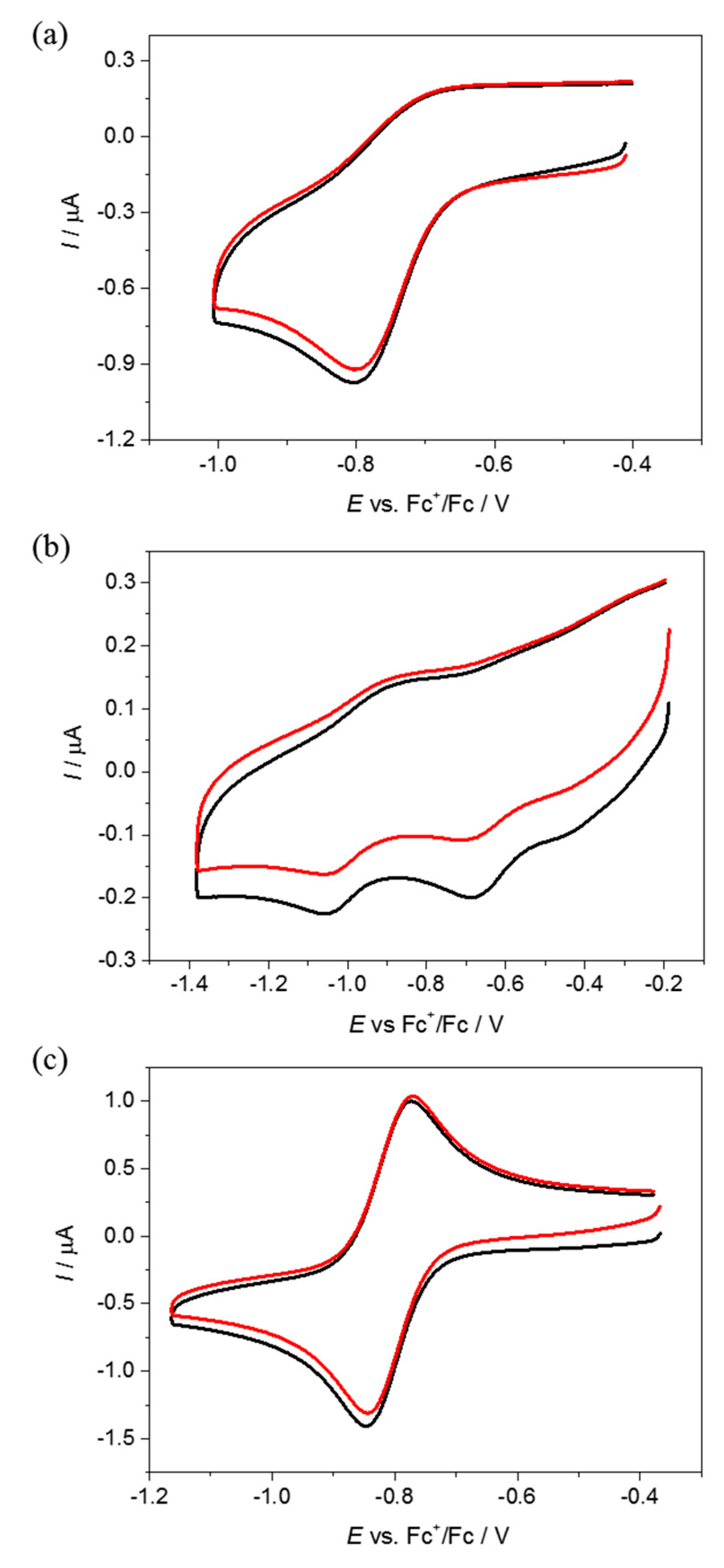
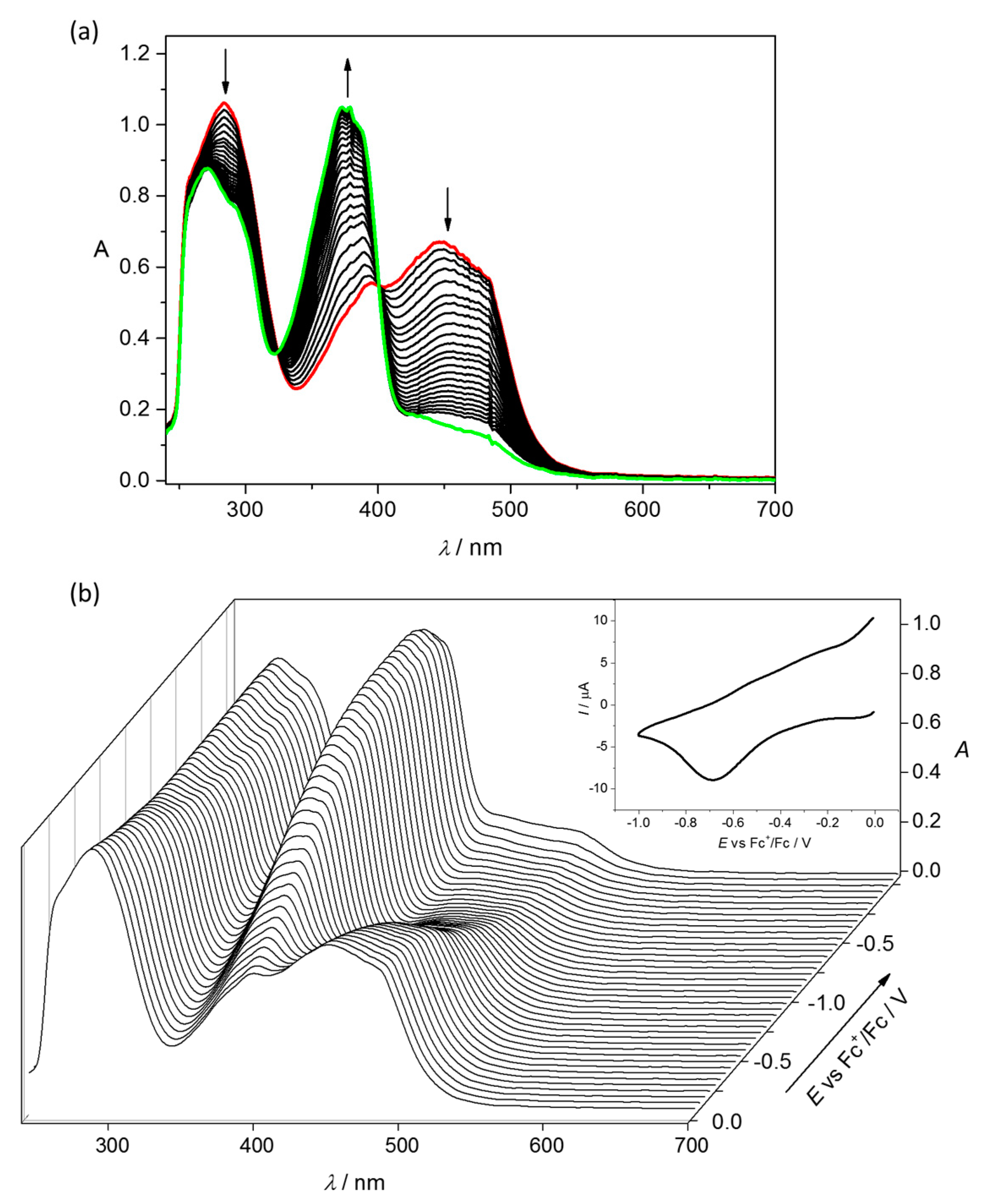
4.2. Electrochemistry
4.3. Intracellular Release of Metal Ions
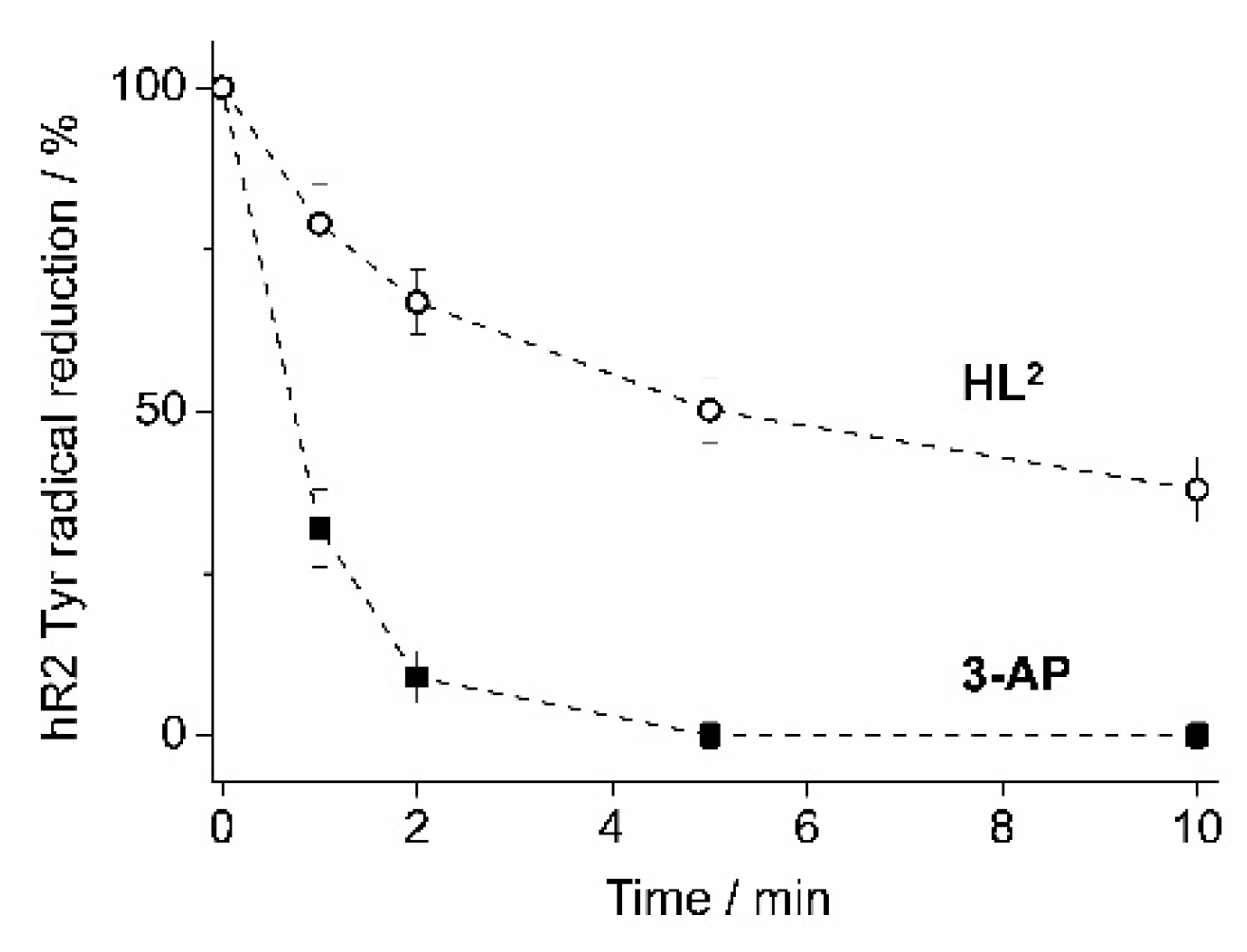
4.4. hR2 RNR Inhibition by HL2
4.5. The Effects of Cu Overload on Cancer Cell Signalling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brockman, R.W.; Thomson, J.R.; Bell, M.J.; Skipper, H.E. Observations on the Antileukemic Activity of Pyridine-2-Carboxaldehyde Thiosemicarbazone and Thiocarbohydrazone. Cancer Res. 1956, 16, 167–170. [Google Scholar]
- Kunos, C.A.; Chu, E.; Beumer, J.H.; Sznol, M.; Ivy, S.P. Phase I Trial of Daily Triapine in Combination with Cisplatin Chemotherapy for Advanced-Stage Malignancies. Cancer Chemother. Pharm. 2017, 79, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Nutting, C.M.; van Herpen, C.M.L.; Miah, A.B.; Bhide, S.A.; Machiels, J.-P.; Buter, J.; Kelly, C.; de Raucourt, D.; Harrington, K.J. Phase II Study of 3-AP Triapine in Patients with Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Ann. Oncol. 2009, 20, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.S.; Alberti, D.B.; Schelman, W.R.; Kolesar, J.M.; Thomas, J.P.; Marnocha, R.; Eickhoff, J.C.; Ivy, S.P.; Wilding, G.; Holen, K.D. The Maximum Tolerated Dose and Biologic Effects of 3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone (3-AP) in Combination with Irinotecan for Patients with Refractory Solid Tumors. Cancer Chemother. Pharm. 2010, 66, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Kunos, C.A.; Chu, E.; Makower, D.; Kaubisch, A.; Sznol, M.; Ivy, S.P. Phase I Trial of Triapine–Cisplatin–Paclitaxel Chemotherapy for Advanced Stage or Metastatic Solid Tumor Cancers. Front. Oncol. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schelman, W.R.; Morgan-Meadows, S.; Marnocha, R.; Lee, F.; Eickhoff, J.; Huang, W.; Pomplun, M.; Jiang, Z.; Alberti, D.; Kolesar, J.M.; et al. A Phase I Study of Triapine® in Combination with Doxorubicin in Patients with Advanced Solid Tumors. Cancer Chemother. Pharm. 2009, 63, 1147–1156. [Google Scholar] [CrossRef]
- Mortazavi, A.; Ling, Y.; Martin, L.K.; Wei, L.; Phelps, M.A.; Liu, Z.; Harper, E.J.; Ivy, S.P.; Wu, X.; Zhou, B.-S.; et al. A Phase I Study of Prolonged Infusion of Triapine in Combination with Fixed Dose Rate Gemcitabine in Patients with Advanced Solid Tumors. Investig. New Drugs 2013, 31, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Finch, R.A.; Liu, M.-C.; Cory, A.H.; Cory, J.G.; Sartorelli, A.C. Triapine (3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone; 3-AP): An Inhibitor of Ribonucleotide Reductase with Antineoplastic Activity. Adv. Enz. Regul. 1999, 39, 3–12. [Google Scholar] [CrossRef]
- Aye, Y.; Long, M.J.C.; Stubbe, J. Mechanistic Studies of Semicarbazone Triapine Targeting Human Ribonucleotide Reductase in Vitro and in Mammalian Cells: Tyrosyl Radical Quenching not Involving Reactive Oxygen Species. J. Biol. Chem. 2012, 287, 35768–35778. [Google Scholar] [CrossRef]
- Popović-Bijelić, A.; Kowol, C.R.; Lind, M.E.S.; Luo, J.; Himo, F.; Enyedy, É.A.; Arion, V.B.; Gräslund, A. Ribonucleotide Reductase Inhibition by Metal Complexes of Triapine (3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone): A Combined Experimental and Theoretical Study. J. Inorg. Biochem. 2011, 105, 1422–1431. [Google Scholar] [CrossRef]
- Shao, J.; Zhou, B.; Di Bilio, A.J.; Zhu, L.; Wang, T.; Qi, C.; Shih, J.; Yen, Y. A Ferrous-Triapine Complex Mediates Formation of Reactive Oxygen Species That Inactivate Human Ribonucleotide Reductase. Mol. Cancer 2006, 5, 586–592. [Google Scholar] [CrossRef]
- Knox, J.J.; Hotte, S.J.; Kollmannsberger, C.; Winquist, E.; Fisher, B.; Eisenhauer, E.A. Phase II Study of Triapine® in Patients with Metastatic Renal Cell Carcinoma: A Trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC IND.161). Investig. New Drugs 2007, 25, 471–477. [Google Scholar] [CrossRef]
- Yen, Y.; Margolin, K.; Doroshow, J.; Fishman, M.; Johnson, B.; Clairmont, C.; Sullivan, D.; Sznol, M. A Phase I Trial of 3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone in Combination with Gemcitabine for Patients with Advanced Cancer. Cancer Chemother. Pharm. 2004, 54, 331–342. [Google Scholar] [CrossRef]
- Traynor, A.M.; Lee, J.-W.; Bayer, G.K.; Tate, J.M.; Thomas, S.P.; Mazurczak, M.; Graham, D.L.; Kolesar, J.M.; Schiller, J.H. A Phase II Trial of Triapine® (NSC# 663249) and Gemcitabine as Second Line Treatment of Advanced Non-Small Cell Lung Cancer: Eastern Cooperative Oncology Group Study 1503. Investig. New Drugs 2010, 28, 91–97. [Google Scholar]
- Mackenzie, M.J.; Saltman, D.; Hirte, H.; Low, J.; Johnson, C.; Pond, G.; Moore, M.J. A Phase II Study of 3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone (3-AP) and Gemcitabine in Advanced Pancreatic Carcinoma. A Trial of the Princess Margaret Hospital Phase II Consortium. Investig. New Drugs 2007, 25, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Attia, S.; Kolesar, J.; Mahoney, M.R.; Pitot, H.C.; Laheru, D.; Heun, J.; Huang, W.; Eickhoff, J.; Erlichman, C.; Holen, K.D. A Phase 2 Consortium (P2C) Trial of 3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone (3-AP) for Advanced Adenocarcinoma of the Pancreas. Investig. New Drugs 2008, 26, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Kowol, C.R.; Trondl, R.; Heffeter, P.; Arion, V.B.; Jakupec, M.A.; Roller, A.; Galanski, M.; Berger, W.; Keppler, B.K. Impact of Metal Coordination on Cytotoxicity of 3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone (Triapine) and Novel Insights into Terminal Dimethylation. J. Med. Chem. 2009, 52, 5032–5043. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P.J.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Novel Thiosemicarbazones of the ApT and DpT Series and Their Copper Complexes: Identification of Pronounced Redox Activity and Characterization of Their Antitumor Activity. J. Med. Chem. 2010, 53, 5759–5769. [Google Scholar] [CrossRef]
- Bacher, F.; Dömötör, O.; Chugunova, A.; Nagy, N.V.; Filipović, L.; Radulović, S.; Enyedy, É.A.; Arion, V.B. Strong Effect of Copper(ii) Coordination on Antiproliferative Activity of Thiosemicarbazone–Piperazine and Thiosemicarbazone–Morpholine Hybrids. Dalton Trans. 2015, 44, 9071–9090. [Google Scholar] [CrossRef]
- Dobrova, A.; Platzer, S.; Bacher, F.; Milunovic, M.N.M.; Dobrov, A.; Spengler, G.; Enyedy, É.A.; Novitchi, G.; Arion, V.B. Structure–Antiproliferative Activity Studies on l -Proline- and Homoproline-4-N-Pyrrolidine-3-Thiosemicarbazone Hybrids and Their Nickel(II), Palladium(II) and Copper(II) Complexes. Dalton Trans. 2016, 45, 13427–13439. [Google Scholar] [CrossRef]
- Ohui, K.; Afanasenko, E.; Bacher, F.; Ting, R.L.X.; Zafar, A.; Blanco-Cabra, N.; Torrents, E.; Dömötör, O.; May, N.V.; Darvasiova, D.; et al. New Water-Soluble Copper(II) Complexes with Morpholine–Thiosemicarbazone Hybrids: Insights into the Anticancer and Antibacterial Mode of Action. J. Med. Chem. 2019, 62, 512–530. [Google Scholar] [CrossRef] [PubMed]
- Adsule, S.; Barve, V.; Chen, D.; Ahmed, F.; Dou, Q.P.; Padhye, S.; Sarkar, F.H. Novel Schiff Base Copper Complexes of Quinoline-2 Carboxaldehyde as Proteasome Inhibitors in Human Prostate Cancer Cells. J. Med. Chem. 2006, 49, 7242–7246. [Google Scholar] [CrossRef] [PubMed]
- Kalaiarasi, G.; Rex Jeya Rajkumar, S.; Dharani, S.; Rath, N.P.; Prabhakaran, R. In Vitro Cytotoxicity of New Water Soluble Copper(II) Metallates Containing 7-Hydroxy-4-Oxo-4H-Chromene Thiosemicarbazones. Polyhedron 2019, 173, 114120. [Google Scholar] [CrossRef]
- Bisceglie, F.; Musiari, A.; Pinelli, S.; Alinovi, R.; Menozzi, I.; Polverini, E.; Tarasconi, P.; Tavone, M.; Pelosi, G. Quinoline-2-Carboxaldehyde Thiosemicarbazones and Their Cu(II) and Ni(II) Complexes as Topoisomerase IIa Inhibitors. J. Inorg. Biochem. 2015, 152, 10–19. [Google Scholar] [CrossRef]
- Da Silva, J.G.; Recio Despaigne, A.A.; Louro, S.R.W.; Bandeira, C.C.; Souza-Fagundes, E.M.; Beraldo, H. Cytotoxic Activity, Albumin and DNA Binding of New Copper(II) Complexes with Chalcone-Derived Thiosemicarbazones. Eur. J. Med. Chem. 2013, 65, 415–426. [Google Scholar] [CrossRef]
- Palanimuthu, D.; Shinde, S.V.; Somasundaram, K.; Samuelson, A.G. In Vitro and in Vivo Anticancer Activity of Copper Bis(Thiosemicarbazone) Complexes. J. Med. Chem. 2013, 56, 722–734. [Google Scholar] [CrossRef]
- Milunović, M.N.M.; Dobrova, A.; Novitchi, G.; Gligorijević, N.; Radulović, S.; Kožišek, J.; Rapta, P.; Enyedy, E.A.; Arion, V.B. Effects of Terminal Substitution and Iron Coordination on Antiproliferative Activity of l-Proline-Salicylaldehyde-Thiosemicarbazone Hybrids: Effects of Terminal Substitution and Iron Coordination on Antiproliferative Activity of L-Proline-Salicylaldehyde-Thiosemicarbazone Hybrids. Eur. J. Inorg. Chem. 2017, 4773–4783. [Google Scholar]
- Enyedy, É.A.; Primik, M.F.; Kowol, C.R.; Arion, V.B.; Kiss, T.; Keppler, B.K. Interaction of Triapine and Related Thiosemicarbazones with Iron(III)/(II) and Gallium(III): A Comparative Solution Equilibrium Study. Dalton Trans. 2011, 40, 5895–5905. [Google Scholar] [CrossRef]
- Pogorzelska, A.; Sławiński, J.; Kawiak, A.; Żołnowska, B.; Chojnacki, J.; Stasiłojć, G.; Ulenberg, S.; Szafrański, K.; Bączek, T. Synthesis, Molecular Structure, and Metabolic Stability of New Series of N’-(2-Alkylthio-4-Chloro-5-Methylbenzenesulfonyl)-1-(5-Phenyl-1H-Pyrazol-1-Yl)Amidine as Potential Anti-Cancer Agents. Eur. J. Med. Chem. 2018, 155, 670–680. [Google Scholar] [CrossRef]
- Al-Qtaitat, M.A.; El-Abadelah, M.M.; Sabbah, D.A.; Bardaweel, S.; Sweidan, K.; Sabri, S.S.; Mubarak, M.S. Synthesis, Characterization, and Bioactivity of New Bisamidrazone Derivatives as Possible Anticancer Agents. Med. Chem. Res. 2018, 27, 1419–1431. [Google Scholar] [CrossRef]
- Gokhale, N.; Padhye, S.; Rathbone, D.; Billington, D.; Lowe, P.; Schwalbe, C.; Newton, C. The Crystal Structure of First Copper(II) Complex of a Pyridine-2-Carboxamidrazone–A Potential Antitumor Agent. Inorg. Chem. Commun. 2001, 4, 26–29. [Google Scholar] [CrossRef]
- Mazur, L.; Modzelewska-Banachiewicz, B.; Paprocka, R.; Zimecki, M.; Wawrzyniak, U.E.; Kutkowska, J.; Ziółkowska, G. Synthesis, Crystal Structure and Biological Activities of a Novel Amidrazone Derivative and Its Copper(II) Complex–A Potential Antitumor Drug. J. Inorg. Biochem. 2012, 114, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Ohui, K.; Babak, M.V.; Darvasiova, D.; Roller, A.; Vegh, D.; Rapta, P.; Guan, G.R.S.; Ou, Y.H.; Pastorin, G.; Arion, V.B. Redox-Active Organoruthenium(II)– and Organoosmium(II)–Copper(II) Complexes, with an Amidrazone–Morpholine Hybrid and [CuICl2]− as Counteranion and Their Antiproliferative Activity. Organometallics 2019, 38, 2307–2318. [Google Scholar] [CrossRef]
- Arion, V.B. Coordination Chemistry of S-Substituted Isothiosemicarbazides and Isothiosemicarbazones. Coord. Chem. Rev. 2019, 387, 348–397. [Google Scholar] [CrossRef]
- Guo, T.; Guan, R.; Zou, J.; Liu, J.; Ying, L.; Yang, W.; Wu, H.; Cao, Y. Red Light-Emitting Hyperbranched Fluorene-Alt-Carbazole Copolymers with an Iridium Complex as the Core. Polym. Chem. 2011, 2, 2193–2203. [Google Scholar] [CrossRef]
- Venuti, M.C.; Stephenson, R.A.; Alvarez, R.; Bruno, J.J.; Strosberg, A.M. Inhibitors of Cyclic AMP Phosphodiesterase. 3. Synthesis and Biological Evaluation of Pyrido and Imidazolyl Analogs of 1,2,3,5-Tetrahydro-2-Oxoimidazo[2,1-b]Quinazoline. J. Med. Chem. 1988, 31, 2136–2145. [Google Scholar] [CrossRef]
- Freund, M.; Paradies, T. Zur Kenntnis Des Tetrazols. Ber. Dtsch. Chem. Ges. 1901, 34, 3110–3122. [Google Scholar] [CrossRef]
- SAINT-Plus and APEX2; Bruker-Nonius AXS Inc.: Madison, WI, USA, 2016.
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. A Found. Cryst. 2008, 64, 112–122. [Google Scholar]
- Burnett, M.N.; Johnson, C.K. ORTEP III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations; Report ORNL-6895; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1996. [Google Scholar]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A Study of Some Problems in Determining the Stoichiometric Proton Dissociation Constants of Complexes by Potentiometric Titrations Using a Glass Electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of Equilibria in Solution. Determination of Equilibrium Constants with the HYPERQUAD Suite of Programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Voevodskaya, N.; Lendzian, F.; Gräslund, A. A Stable FeIII–FeIV Replacement of Tyrosyl Radical in a Class I Ribonucleotide Reductase. Biochem. Biophys. Res. Commun. 2005, 330, 1213–1216. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.J.; Gräslund, A.; Ochiai, E.I.; Ingemarson, R.; Thelander, L. Purification and Characterization of Recombinant Mouse and Herpes Simplex Virus Ribonucleotide Reductase R2 Subunit. Biochemistry 1991, 30, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.H.; Padhye, S.S.; Padhye, S.B.; Anson, C.E.; Powell, A.K. Copper Complexes of Carboxamidrazone Derivatives as Anticancer Agents. 3. Synthesis, Characterization and Crystal Structure of [Cu(Appc)Cl2], (Appc=N1-(2-Acetylpyridine)Pyridine-2-Carboxamidrazone). Inorg. Chim. Acta 2001, 319, 90–94. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen–Sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[1,7-Bis(N-Methylbenzimidazol-2′-Yl)-2,6-Dithiaheptane]Copper(II) Perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Nagy, N.V.; Zsigó, É.; Kowol, C.R.; Arion, V.B.; Keppler, B.K.; Kiss, T. Comparative Solution Equilibrium Study of the Interactions of Copper(II), Iron(II) and Zinc(II) with Triapine (3-Aminopyridine-2-Carbaldehyde Thiosemicarbazone) and Related Ligands. Eur. J. Inorg. Chem. 2010, 1717–1728. [Google Scholar] [CrossRef]
- Kallus, S.; Uhlik, L.; van Schoonhoven, S.; Pelivan, K.; Berger, W.; Enyedy, É.A.; Hofmann, T.; Heffeter, P.; Kowol, C.R.; Keppler, B.K. Synthesis and Biological Evaluation of Biotin-Conjugated Anticancer Thiosemicarbazones and Their Iron(III) and Copper(II) Complexes. J. Inorg. Biochem. 2019, 190, 85–97. [Google Scholar] [CrossRef]
- Saporito-Magriñá, C.M.; Musacco-Sebio, R.N.; Andrieux, G.; Kook, L.; Orrego, M.T.; Tuttolomondo, M.V.; Desimone, M.F.; Boerries, M.; Borner, C.; Repetto, M.G. Copper-Induced Cell Death and the Protective Role of Glutathione: The Implication of Impaired Protein Folding Rather than Oxidative Stress. Metallomics 2018, 10, 1743–1754. [Google Scholar] [CrossRef]
- Panjehpour, M.; Taher, M.-A.; Bayesteh, M. The Growth Inhibitory Effects of Cadmium and Copper on the MDA-MB468 Human Breast Cancer Cells. J. Res. Med. Sci. 2010, 15, 279–286. [Google Scholar]
- Richardson, D.R.; Sharpe, P.C.; Lovejoy, D.B.; Senaratne, D.; Kalinowski, D.S.; Islam, M.; Bernhardt, P.V. Dipyridyl Thiosemicarbazone Chelators with Potent and Selective Antitumor Activity Form Iron Complexes with Redox Activity. J. Med. Chem. 2006, 49, 6510–6521. [Google Scholar] [CrossRef]
- Yu, Y.; Wong, J.; Lovejoy, D.B.; Kalinowski, D.S.; Richardson, D.R. Chelators at the Cancer Coalface: Desferrioxamine to Triapine and Beyond. Clin. Cancer Res. 2006, 12, 6876–6883. [Google Scholar] [CrossRef]
- Nurtjahja-Tjendraputra, E.; Fu, D.; Phang, J.M.; Richardson, D.R. Iron Chelation Regulates Cyclin D1 Expression via the Proteasome: A Link to Iron Deficiency–Mediated Growth Suppression. Blood 2007, 109, 4045–4054. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; White, A.R. Copper as a Key Regulator of Cell Signalling Pathways. Expert Rev. Mol. Med. 2014, 16, e11. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Fouani, L.; Jansson, P.J.; Wooi, D.; Sahni, S.; Lane, D.J.R.; Palanimuthu, D.; Lok, H.C.; Kovačević, Z.; Huang, M.L.H.; et al. Copper and Conquer: Copper Complexes of Di-2-Pyridylketone Thiosemicarbazones as Novel Anti-Cancer Therapeutics. Metallomics 2016, 8, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Sîrbu, A.; Palamarciuc, O.; Babak, M.V.; Lim, J.M.; Ohui, K.; Enyedy, E.A.; Shova, S.; Darvasiová, D.; Rapta, P.; Ang, W.H.; et al. Copper(II) Thiosemicarbazone Complexes Induce Marked ROS Accumulation and Promote Nrf2-Mediated Antioxidant Response in Highly Resistant Breast Cancer Cells. Dalton Trans. 2017, 46, 3833–3847. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HL1 | [H3L1a]Cl2·2H2O | HL2 | 1 | 2 |
|---|---|---|---|---|---|
| empirical formula | C12H12N6 | C12H18Cl2N6O2 | C8H11N5S | C12H12Cl2CuN6 | C8H11Cl2CuN5S |
| fw | 240.28 | 349.22 | 209.28 | 374.72 | 343.72 |
| space group | Pna21 | P21/c | P21/c | P21/c | P |
| a, [Å] | 16.9760 (18) | 8.8057 (3) | 10.9320 (4) | 8.3933 (3) | 8.0926 (13) |
| b, [Å] | 6.1689 (7) | 28.3257 (9) | 7.0723 (3) | 11.5392 (2) | 8.9713 (15) |
| c, [Å] | 10.9516 (12) | 6.8074 (3) | 12.6838 (5) | 15.1025 (5) | 9.1504 (15) |
| α, [°] | 78.368 (7) | ||||
| β, [°] | 109.7135 (14) | 93.627 (2) | 104.188 (2) | 78.276 (6) | |
| γ, [°] | 74.508 (6) | ||||
| V [Å3] | 1146.9(2) | 1598.4 (1) | 978.29 (7) | 1418.09 (7) | 619.15 (18) |
| Z | 4 | 4 | 4 | 4 | 2 |
| λ, [Å] | 0.71073 | 0.71073 | 0.71073 | 1.54186 | 0.71073 |
| ρcalcd, [g cm−3] | 1.392 | 1.451 | 1.421 | 1.755 | 1.844 |
| cryst size, [mm3] | 0.12 × 0.10 × 0.06 | 0.35 × 0.10 × 0.10 | 0.20 × 0.18 × 0.12 | 0.22 × 0.04 × 0.04 | 0.20 × 0.18 × 0.07 |
| T [K] | 120 (2) | 120(2) | 130 (2) | 100 (2) | 200 (2) |
| μ, [mm−1] | 0.092 | 0.422 | 0.298 | 5.662 | 2.347 |
| R1a | 0.0329 | 0.0607 | 0.0309 | 0.0358 | 0.0555 |
| wR2b | 0.0807 | 0.2349 | 0.0873 | 0.0950 | 0.2121 |
| GOF c | 1.099 | 1.180 | 1.048 | 0.958 | 1.071 |
| Compound | IC50 [μM] a | Cellular Accumulation d, nmol Cu/mg Protein | ||||
|---|---|---|---|---|---|---|
| A2780 | A2780cis | RF b | HEK293 | SF c | A2780 | |
| HL1 | 242 ± 84 | 221 ± 79 | 0.9 | 253 ± 39 | 1.0 | - |
| HL2 | 202 ± 49 | 211 ± 8 | 1.0 | 187 ± 24 | 0.9 | - |
| 1 | 17 ± 2 | 15 ± 3 | 0.9 | 28 ± 2 | 1.6 | 0.76 ± 0.10 |
| 2 | 1.4 ± 0.2 | 0.92 ± 0.11 | 0.7 | 1.5 ± 0.2 | 1.1 | 0.64 ± 0.18 |
| 3 | 41 ± 12 | 36 ± 8 | 0.9 | 48 ± 9 | 1.2 | - |
| Triapine e | 0.67 ± 0.22 | 1.1 ± 0.1 | 1.6 | 0.39 ± 0.05 | 0.6 | - |
| Cisplatin e | 0.44 ± 0.13 | 4.6 ± 0.3 | 10.5 | n.d. f | - | - |
| CuCl2 g | 83 ± 12 | 82 ± 3 | 1.0 | 187 ± 37 | 2.3 | 0.64 ± 0.19 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohui, K.; Stepanenko, I.; Besleaga, I.; Babak, M.V.; Stafi, R.; Darvasiova, D.; Giester, G.; Pósa, V.; Enyedy, E.A.; Vegh, D.; et al. Triapine Derivatives Act as Copper Delivery Vehicles to Induce Deadly Metal Overload in Cancer Cells. Biomolecules 2020, 10, 1336. https://doi.org/10.3390/biom10091336
Ohui K, Stepanenko I, Besleaga I, Babak MV, Stafi R, Darvasiova D, Giester G, Pósa V, Enyedy EA, Vegh D, et al. Triapine Derivatives Act as Copper Delivery Vehicles to Induce Deadly Metal Overload in Cancer Cells. Biomolecules. 2020; 10(9):1336. https://doi.org/10.3390/biom10091336
Chicago/Turabian StyleOhui, Kateryna, Iryna Stepanenko, Iuliana Besleaga, Maria V. Babak, Radu Stafi, Denisa Darvasiova, Gerald Giester, Vivien Pósa, Eva A. Enyedy, Daniel Vegh, and et al. 2020. "Triapine Derivatives Act as Copper Delivery Vehicles to Induce Deadly Metal Overload in Cancer Cells" Biomolecules 10, no. 9: 1336. https://doi.org/10.3390/biom10091336
APA StyleOhui, K., Stepanenko, I., Besleaga, I., Babak, M. V., Stafi, R., Darvasiova, D., Giester, G., Pósa, V., Enyedy, E. A., Vegh, D., Rapta, P., Ang, W. H., Popović-Bijelić, A., & Arion, V. B. (2020). Triapine Derivatives Act as Copper Delivery Vehicles to Induce Deadly Metal Overload in Cancer Cells. Biomolecules, 10(9), 1336. https://doi.org/10.3390/biom10091336