SPON1 Can Reduce Amyloid Beta and Reverse Cognitive Impairment and Memory Dysfunction in Alzheimer’s Disease Mouse Model

 , and
, and
Abstract

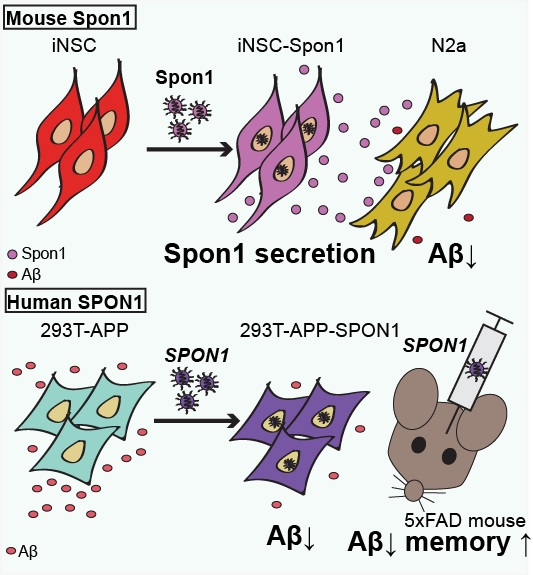{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Vector Construction
2.3. Therapeutic Engineering
2.4. Conventional and Quantitative Real-Time PCR
2.5. Immunocytochemistry
2.6. ELISA
2.7. In Vivo Experiments
2.8. Morris Water Maze Assay
2.9. Western Blot
2.10. Immunohistochemistry
2.11. Statistical Analysis
3. Results
3.1. Mouse Induced Neural Stem Cells Expressing Spon1 Maintain Neural Stem Cell Characteristics
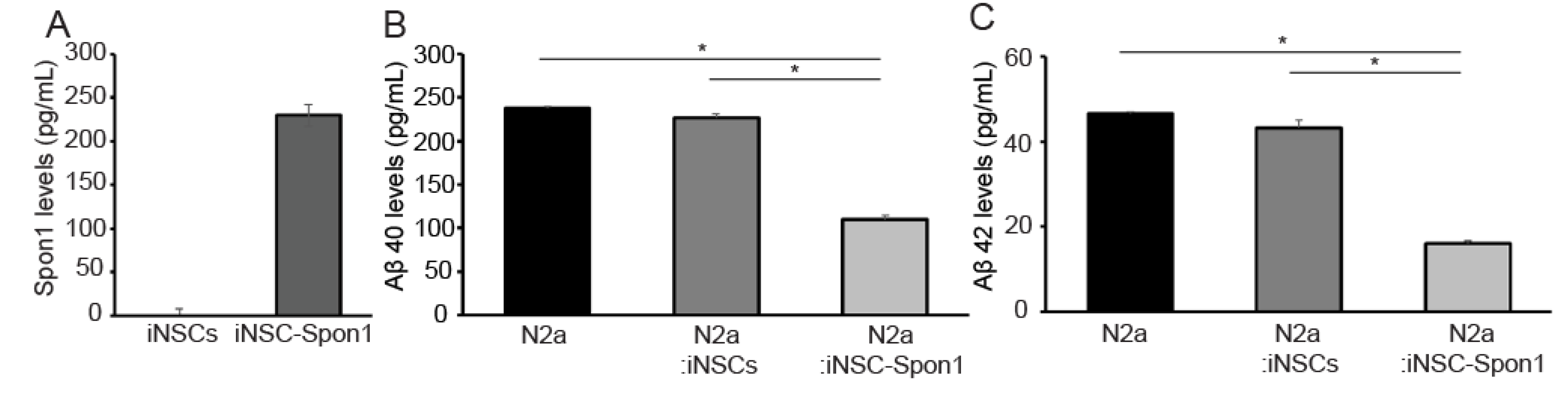
3.2. Mouse iNSCs Expressing Mouse Spon1 Reduce Amyloid Beta through the Bystander Effect
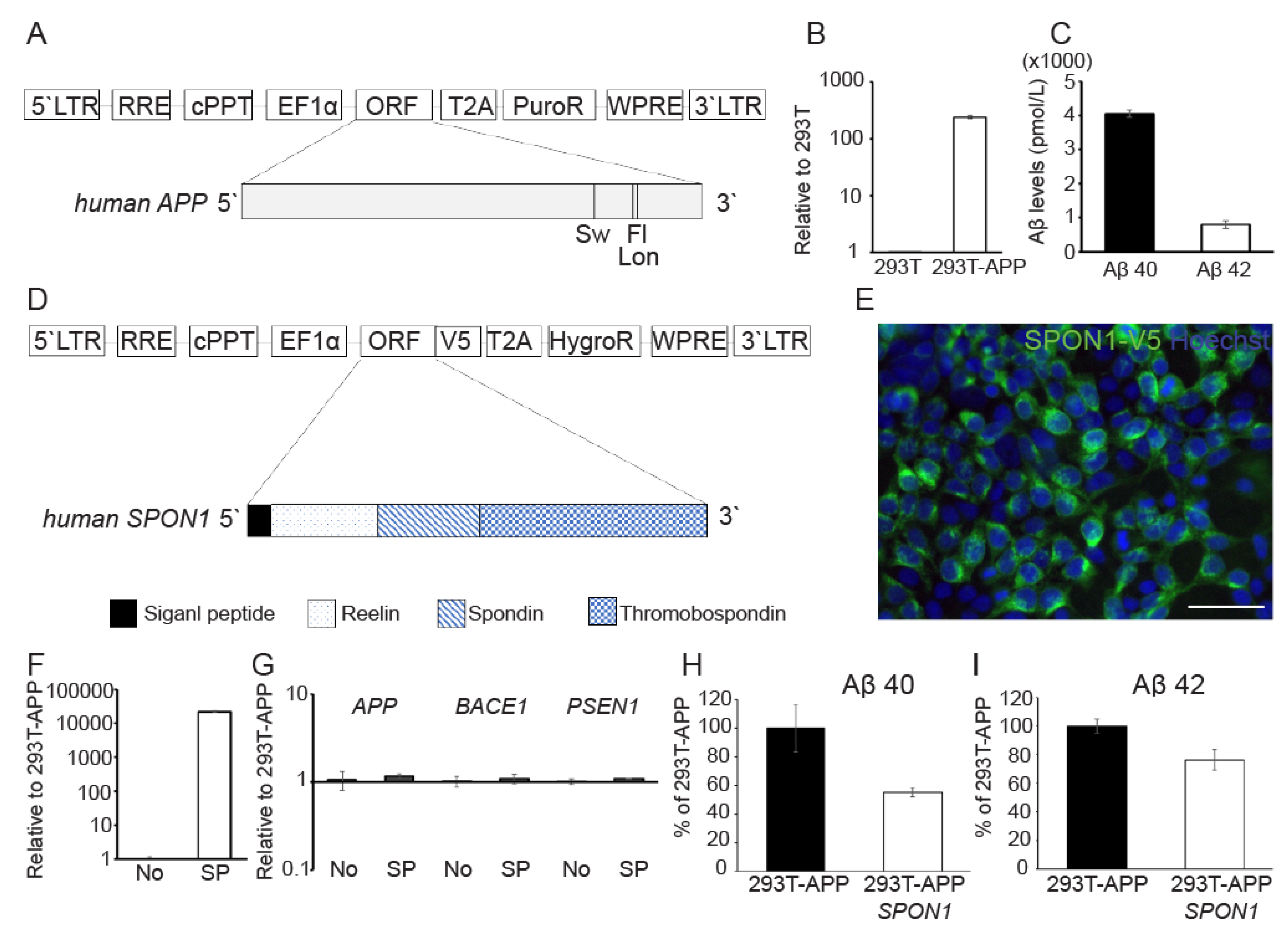
3.3. Human SPON1 Directly Reduces Amyloid Beta
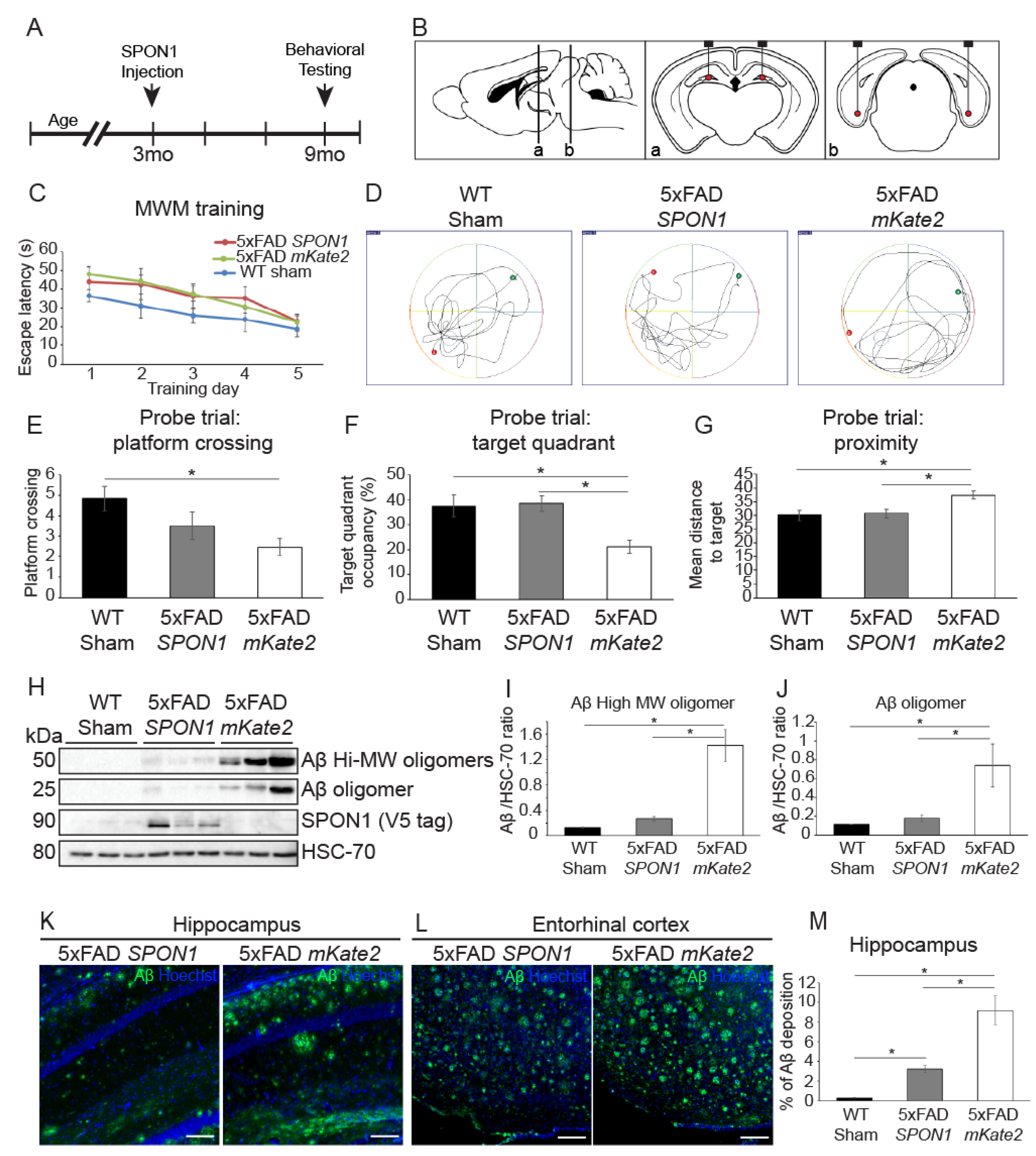
3.4. Human SPON1 Induces Improvement of Learning in the Alzheimer’s Disease Model
4. Discussion
4.1. The Effect of SPON1 on Decreasing Aβ Secretion In Vitro
4.2. Aβ Reduction and Reversal of Cognitive Impairment and Memory Dysfunction by Administration of SPON1 into 5xFAD Mice
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5xFAD | Mouse expressing human APP and PSEN1 transgenes with five AD-linked mutations |
| Aβ | Amyloid beta |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| BACE1 | Beta-site amyloid precursor protein cleaving enzyme 1 |
| EC | Entorhinal cortex |
| FBS | Fetal bovine serum |
| iNSCs | Induced neural stem cells |
| iNSC-Spon1 | Induced neural stem cells genetically engineered to express mouse Spon1 |
| N2a | Neuro 2a cells |
| OCT | Optimal cutting temperature compound |
| Oct4 | Octamer-binding transcription factor 4 |
| ROI | Region of interest |
| SPON1 | SPONDIN1, F-Spondin |
References
- Bachman, D.L.; Wolf, P.A.; Linn, R.; Knoefel, J.E.; Cobb, J.; Belanger, A.; D’Agostino, R.B.; White, L.R. Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham Study. Neurology 1992, 42, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Inv. Drug 2017, 26, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A Mouse Model of Amyloid beta Oligomers: Their Contribution to Synaptic Alteration, Abnormal Tau Phosphorylation, Glial Activation, and Neuronal Loss in Vivo. J. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef]
- Hu, J.G.; Akama, K.T.; Krafft, G.A.; Chromy, B.A.; Van Eldik, L.J. Amyloid-beta peptide activates cultured astrocytes: Morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998, 785, 195–206. [Google Scholar] [CrossRef]
- Ferretti, M.T.; Bruno, M.A.; Ducatenzeiler, A.; Klein, W.L.; Cuello, A.C. Intracellular Abeta-oligomers and early inflammation in a model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1329–1342. [Google Scholar] [CrossRef]
- Golde, T.E.; Petrucelli, L.; Lewis, J. Targeting Abeta and tau in Alzheimer’s disease, an early interim report. Exp. Neurol. 2010, 223, 252–266. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Weidemann, A.; Paliga, K.; Durrwang, U.; Reinhard, F.B.M.; Schuckert, O.; Evin, G.; Masters, C.L. Proteolytic processing of the Alzheimer’s disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J. Biol. Chem. 1999, 274, 5823–5829. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Sametsky, E.A.; Younkin, L.H.; Oakley, H.; Younkin, S.G.; Citron, M.; Vassar, R.; Disterhoft, J.F. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 2004, 41, 27–33. [Google Scholar] [CrossRef]
- Sastre, M.; Dewachter, I.; Rossner, S.; Bogdanovic, N.; Rosen, E.; Borghgraef, P.; Evert, B.O.; Dumitrescu-Ozimek, L.; Thal, D.R.; Landreth, G.; et al. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Nat. Acad. Sci. USA 2006, 103, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Hawkins, J.; Harrison, D.; Hille, C.; Wayne, G.; Cutler, L.; Buck, T.; Walter, D.; Demont, E.; Howes, C.; et al. Oral administration of a potent and selective non-peptidic BACE-1 inhibitor decreases beta-cleavage of amyloid precursor protein and amyloid-beta production in vivo. J. Neurochem. 2007, 100, 802–809. [Google Scholar] [CrossRef]
- Wong, H.K.; Sakurai, T.; Oyama, F.; Kaneko, K.; Wada, K.; Miyazaki, H.; Kurosawa, M.; De Strooper, B.; Saftig, P.; Nukina, N. Beta subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J. Biol. Chem. 2005, 280, 23009–23017. [Google Scholar] [CrossRef]
- Wang, H.; Song, L.; Laird, F.; Wong, P.C.; Lee, H.K. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J. Neurosci. 2008, 28, 8677–8681. [Google Scholar] [CrossRef]
- Willem, M.; Garratt, A.N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science 2006, 314, 664–666. [Google Scholar] [CrossRef]
- Vassar, R.; Kuhn, P.H.; Haass, C.; Kennedy, M.E.; Rajendran, L.; Wong, P.C.; Lichtenthaler, S.F. Function, therapeutic potential and cell biology of BACE proteases: Current status and future prospects. J. Neurochem. 2014, 130, 4–28. [Google Scholar] [CrossRef]
- Cheret, C.; Willem, M.; Fricker, F.R.; Wende, H.; Wulf-Goldenberg, A.; Tahirovic, S.; Nave, K.A.; Saftig, P.; Haass, C.; Garratt, A.N.; et al. Bace1 and Neuregulin-1 cooperate to control formation and maintenance of muscle spindles. Embo J. 2013, 32, 2015–2028. [Google Scholar] [CrossRef]
- Hu, X.Y.; Zhou, X.D.; He, W.X.; Yang, J.; Xiong, W.C.; Wong, P.; Wilson, C.G.; Yan, R.Q. BACE1 Deficiency Causes Altered Neuronal Activity and Neurodegeneration. J. Neurosci. 2010, 30, 8819–8829. [Google Scholar] [CrossRef]
- Peterziel, H.; Sackmann, T.; Strelau, J.; Kuhn, P.H.; Lichtenthaler, S.F.; Marom, K.; Klar, A.; Unsicker, K. F-spondin regulates neuronal survival through activation of disabled-1 in the chicken ciliary ganglion. Mol. Cell Neurosci. 2011, 46, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Burstyn-Cohen, T.; Tzarfaty, V.; Frumkin, A.; Feinstein, Y.; Stoeckli, E.; Klar, A. F-spondin is required for accurate pathfinding of commissural axons at the floor plate. Neuron 1999, 23, 233–246. [Google Scholar] [CrossRef]
- Ho, A.; Sudhof, T.C. Binding of F-spondin to amyloid-beta precursor protein: A candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc. Nat. Acad. Sci. USA 2004, 101, 2548–2553. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.S.; Wessner, D.; Beffert, U.; Becker, A.G.; Matsuoka, Y.; Rebeck, G.W. F-spondin interaction with the apolipoprotein E receptor ApoEr2 affects processing of amyloid precursor protein. Mol. Cell Biol. 2005, 25, 9259–9268. [Google Scholar] [CrossRef]
- Hafez, D.M.; Huang, J.Y.; Richardson, J.C.; Masliah, E.; Peterson, D.A.; Marr, R.A. F-spondin gene transfer improves memory performance and reduces amyloid-beta levels in mice. Neuroscience 2012, 223, 465–472. [Google Scholar] [CrossRef][Green Version]
- Pan, C.P.; Kumar, C.; Bohl, S.; Klingmueller, U.; Mann, M. Comparative Proteomic Phenotyping of Cell Lines and Primary Cells to Assess Preservation of Cell Type-specific Functions. Mol. Cell Proteom. 2009, 8, 443–450. [Google Scholar] [CrossRef]
- Heravi, M.; Dargahi, L.; Parsafar, S.; Marvian, A.T.; Aliakbari, F.; Morshedi, D. The primary neuronal cells are more resistant than PC12 cells to alpha-synuclein toxic aggregates. Neurosci. Lett. 2019, 701, 38–47. [Google Scholar] [CrossRef]
- McLaren, D.; Gorba, T.; Marguerie de Rotrou, A.; Pillai, G.; Chappell, C.; Stacey, A.; Lingard, S.; Falk, A.; Smith, A.; Koch, P.; et al. Automated large-scale culture and medium-throughput chemical screen for modulators of proliferation and viability of human induced pluripotent stem cell-derived neuroepithelial-like stem cells. J. Biomol. Screen 2013, 18, 258–268. [Google Scholar] [CrossRef]
- Lujan, E.; Chanda, S.; Ahlenius, H.; Sudhof, T.C.; Wernig, M. Direct conversion of mouse fibroblasts to self-renewing, tripotent neural precursor cells. Proc. Nat. Acad. Sci. USA 2012, 109, 2527–2532. [Google Scholar] [CrossRef]
- Mitchell, R.R. Activation of Neural Cell Fate Programs Toward Direct Conversion of Adult Human Fibroblasts into Tri-Potent Neural Progenitors Using OCT-4. Stem Cells Dev. 2016, 25, 1033–1033. [Google Scholar] [CrossRef]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, A.; Zhao, C.; Gage, F.H. Retrovirus-mediated single-cell gene knockout technique in adult newborn neurons in vivo. Nat. Protoc. 2006, 1, 3049–3055. [Google Scholar] [CrossRef] [PubMed]
- Blurton-Jones, M.; Kitazawa, M.; Martinez-Coria, H.; Castello, N.A.; Muller, F.J.; Loring, J.F.; Yamasaki, T.R.; Poon, W.W.; Green, K.N.; LaFerla, F.M. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Nat. Acad. Sci. USA 2009, 106, 13594–13599. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF Treatment Ameliorates Cell Loss in the Entorhinal Cortex of APP Transgenic Mice. J. Neurosci. 2013, 33, 15596–15602. [Google Scholar] [CrossRef] [PubMed]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef]
- Casali, B.T.; Landreth, G.E. Abeta Extraction from Murine Brain Homogenates. Bio Protoc. 2016, 6. [Google Scholar] [CrossRef]
- Phansalkar, N.; More, S.; Sabale, A.; Joshi, M. Adaptive local thresholding for detection of nuclei in diversity stained cytology images. In Proceedings of the International Conference on Communications and Signal. Processing (ICCSP), Calicut, India, 10–12 February 2011; pp. 218–220. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Nat. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef]
- Das, P.; Verbeeck, C.; Minter, L.; Chakrabarty, P.; Felsenstein, K.; Kukar, T.; Maharvi, G.; Fauq, A.; Osborne, B.A.; Golde, T.E. Transient pharmacologic lowering of Abeta production prior to deposition results in sustained reduction of amyloid plaque pathology. Mol. Neurodegener. 2012, 7, 39. [Google Scholar] [CrossRef]
- Devi, L.; Tang, J.; Ohno, M. Beneficial effects of the beta-secretase inhibitor GRL-8234 in 5XFAD Alzheimer’s transgenic mice lessen during disease progression. Curr. Alzheimer Res. 2015, 12, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Baldauf, K.; Wetzel, W.; Reymann, K.G. Behavioral and EEG changes in male 5xFAD mice. Physiol. Behav. 2014, 135, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Tohda, C. Icariin Improves Memory Impairment in Alzheimer’s Disease Model Mice (5xFAD) and Attenuates Amyloid beta-induced Neurite Atrophy. Phytother. Res. 2010, 24, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.; Schmoll, H.; Riederer, P.; Bauer, J.; Platt, D.; Popa-Wagner, A. Complement C1q and C3 mRNA expression in the frontal cortex of Alzheimer’s patients. J. Mol. Med. (Berl.) 1995, 73, 465–471. [Google Scholar] [CrossRef]
- Eichenbaum, H. Time cells in the hippocampus: A new dimension for mapping memories. Nat. Rev. Neurosci. 2014, 15, 732–744. [Google Scholar] [CrossRef]
- Van Giau, V.; Bagyinszky, E.; Yang, Y.S.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Yan, Y.; Somer, E.; Grau, V. Classification of amyloid PET images using novel features for early diagnosis of Alzheimer’s disease and mild cognitive impairment conversion. Nucl. Med. Commun. 2019, 40, 242–248. [Google Scholar] [CrossRef]
- Brazaca, L.C.; Moreto, J.R.; Martin, A.; Tehrani, F.; Wang, J.; Zucolotto, V. Colorimetric Paper-Based Immunosensor for Simultaneous Determination of Fetuin B and Clusterin toward Early Alzheimer’s Diagnosis. ACS Nano 2019, 13, 13325–13332. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.Y.; Kang, J.Y.; Lee, T.; Nam, D.; Jeon, C.-J.; Kim, J.B. SPON1 Can Reduce Amyloid Beta and Reverse Cognitive Impairment and Memory Dysfunction in Alzheimer’s Disease Mouse Model. Cells 2020, 9, 1275. https://doi.org/10.3390/cells9051275
Park SY, Kang JY, Lee T, Nam D, Jeon C-J, Kim JB. SPON1 Can Reduce Amyloid Beta and Reverse Cognitive Impairment and Memory Dysfunction in Alzheimer’s Disease Mouse Model. Cells. 2020; 9(5):1275. https://doi.org/10.3390/cells9051275
Chicago/Turabian StylePark, Soo Yong, Joo Yeong Kang, Taehee Lee, Donggyu Nam, Chang-Jin Jeon, and Jeong Beom Kim. 2020. "SPON1 Can Reduce Amyloid Beta and Reverse Cognitive Impairment and Memory Dysfunction in Alzheimer’s Disease Mouse Model" Cells 9, no. 5: 1275. https://doi.org/10.3390/cells9051275
APA StylePark, S. Y., Kang, J. Y., Lee, T., Nam, D., Jeon, C.-J., & Kim, J. B. (2020). SPON1 Can Reduce Amyloid Beta and Reverse Cognitive Impairment and Memory Dysfunction in Alzheimer’s Disease Mouse Model. Cells, 9(5), 1275. https://doi.org/10.3390/cells9051275