The Inhibition of Wnt Restrain KRASG12V-Driven Metastasis in Non-Small-Cell Lung Cancer


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Different KRAS Mutation Subtypes Cause Distinct Cell Morphologies and Characteristics
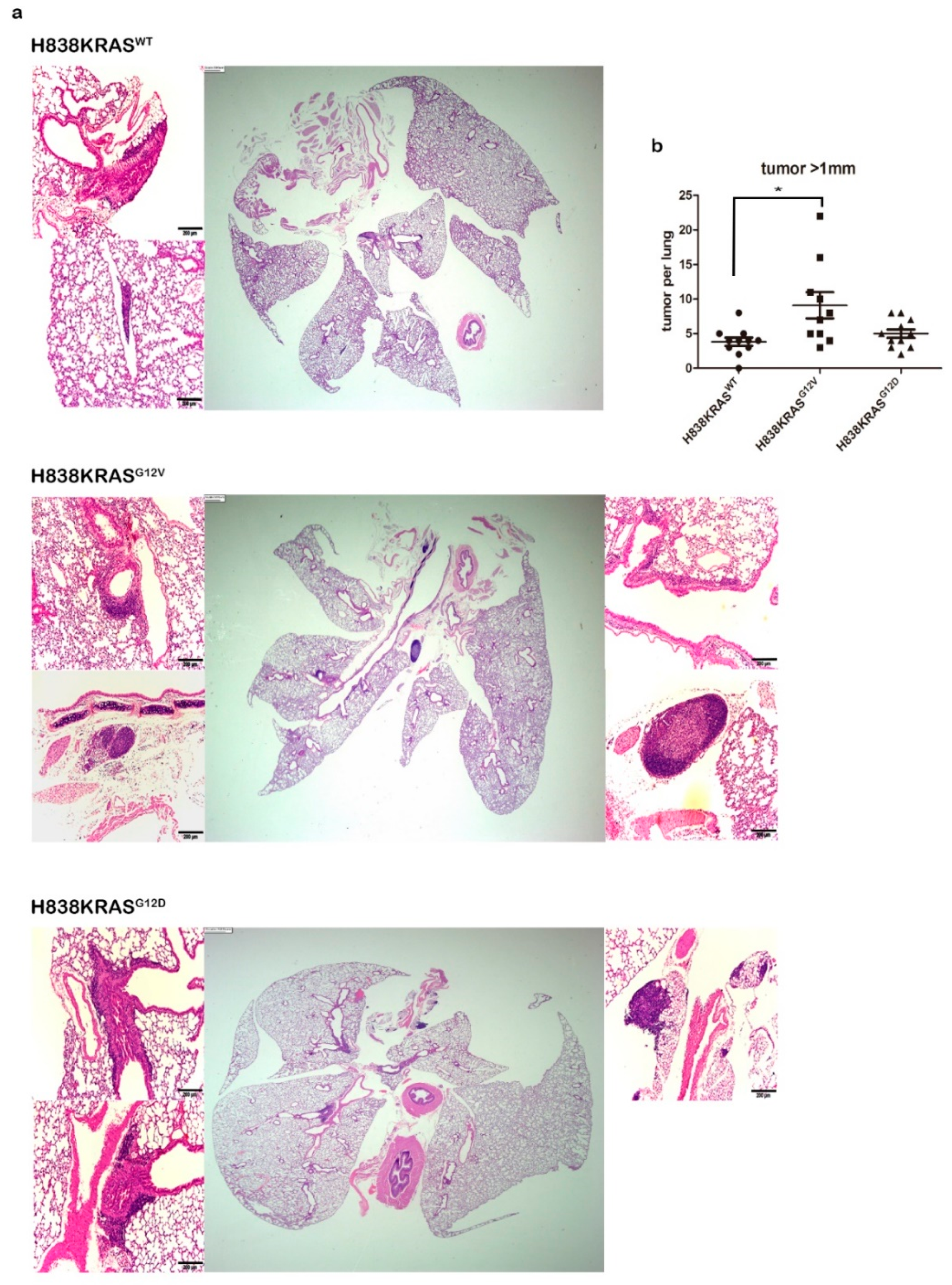
2.2. The Mutation of KRASG12V Confers a Greater Capacity in Metastasis
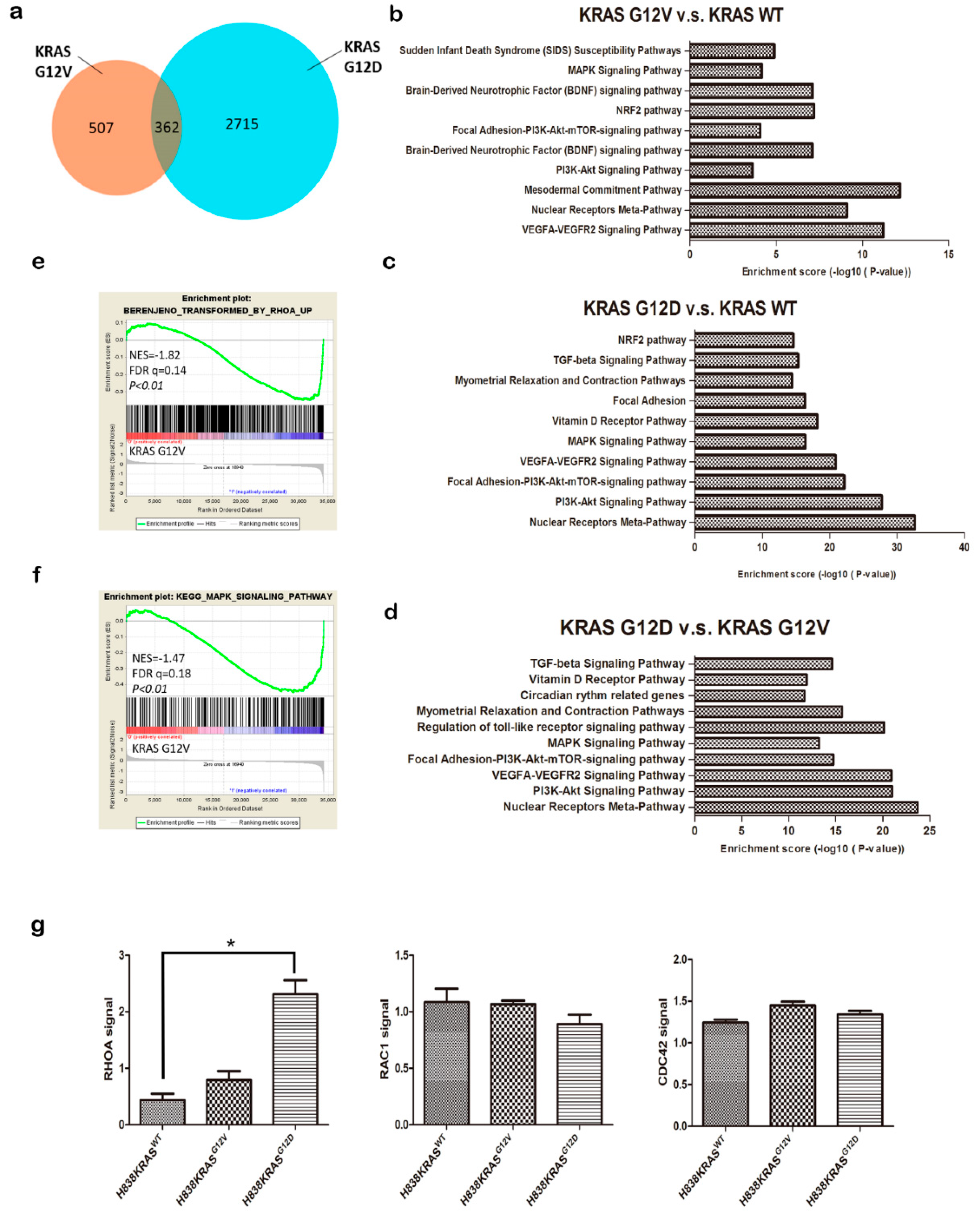
2.3. The Distinct Different Expression Profiling in KRASG12V and KRASG12D Mutation
2.4. The KRASG12D Induce RhoA Activation But Not KRASG12V
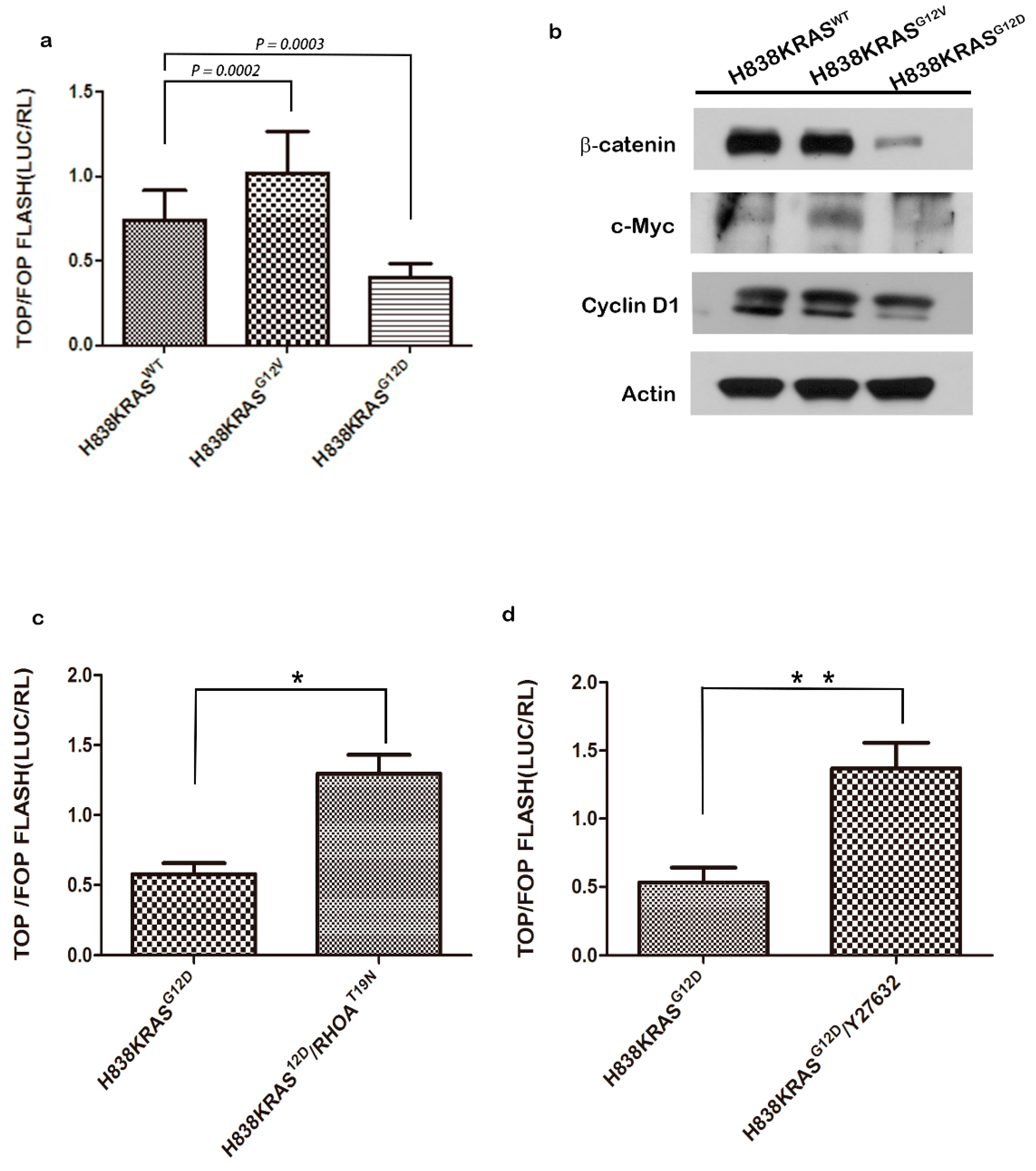
2.5. The H838 KRASG12D Cells Displayed Downregulated Levels of β-Catenin
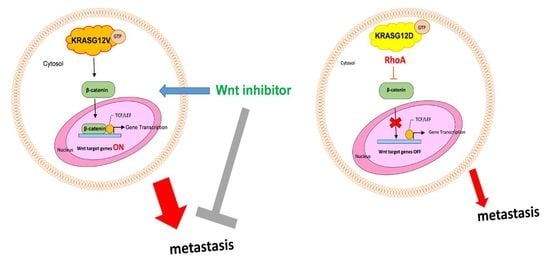
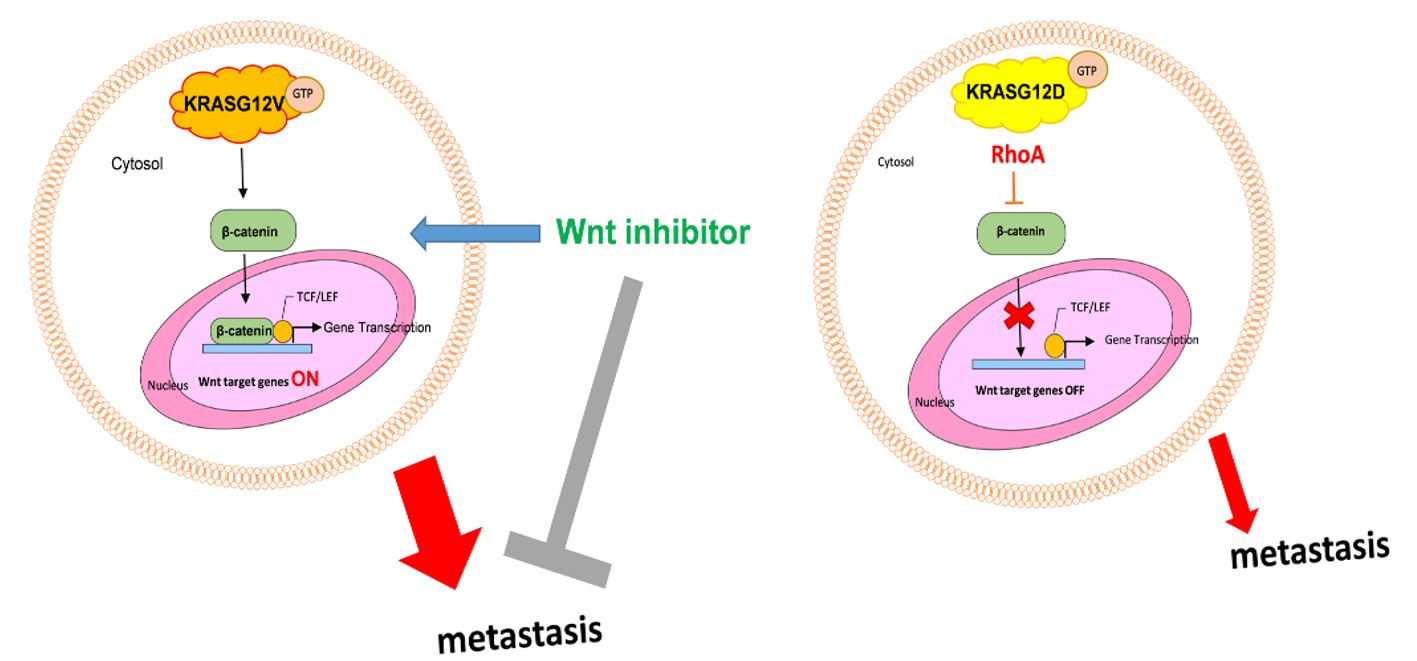
2.6. Activation of RhoA Suppresses the Activation of Wnt in H838 KRASG12D Cells
2.7. The Mutation of KRASG12V Promote Metastasis Through Wnt Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids
4.3. RT-qPCR
4.4. Microarray and Data Mining
4.5. Immunofluorescence Staining
4.6. Immunoblotting and Reagents
4.7. Cell Viability Assay
4.8. Migration, Invasion Assay and Colony Formation
4.9. Wound Healing Assay
4.10. GTPase Activation Assay
4.11. Luciferase Reporter Assay
4.12. Animal Experiments
4.13. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mullard, A. Cracking kras. Nat. Rev. Drug Discov. 2019, 18, 887–891. [Google Scholar] [CrossRef] [PubMed]
- McCormick, F. Sticking it to kras: Covalent inhibitors enter the clinic. Cancer Cell 2020, 37, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Ras oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The gtpase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. Kras: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef]
- Kranenburg, O. The kras oncogene: Past, present, and future. Biochim. Biophys. Acta 2005, 1756, 81–82. [Google Scholar] [CrossRef]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily gefs and gaps: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The cosmic (catalogue of somatic mutations in cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef]
- Riely, G.J.; Kris, M.G.; Rosenbaum, D.; Marks, J.; Li, A.; Chitale, D.A.; Nafa, K.; Riedel, E.R.; Hsu, M.; Pao, W.; et al. Frequency and distinctive spectrum of kras mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res. 2008, 14, 5731–5734. [Google Scholar] [CrossRef]
- Lindsay, C.R.; Jamal-Hanjani, M.; Forster, M.; Blackhall, F. Kras: Reasons for optimism in lung cancer. Eur. J. Cancer 2018, 99, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Sima, C.S.; Shen, R.; Kass, S.; Gainor, J.; Shaw, A.; Hames, M.; Iams, W.; Aston, J.; Lovly, C.M.; et al. Prognostic impact of kras mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J. Thorac. Oncol. 2015, 10, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Miller, L.D. Ras mutations and oncogenesis: Not all ras mutations are created equally. Front. Genet. 2011, 2, 100. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Jiang, T.; Li, X.; Zhao, C.; Zhang, L.; Zhao, S.; Liu, X.; Qiao, M.; Luo, J.; Shi, J.; et al. Characterization of distinct types of kras mutation and its impact on first-line platinum-based chemotherapy in chinese patients with advanced non-small cell lung cancer. Oncol. Lett. 2017, 14, 6525–6532. [Google Scholar] [CrossRef]
- Renaud, S.; Seitlinger, J.; Falcoz, P.E.; Schaeffer, M.; Voegeli, A.C.; Legrain, M.; Beau-Faller, M.; Massard, G. Specific kras amino acid substitutions and egfr mutations predict site-specific recurrence and metastasis following non-small-cell lung cancer surgery. Br. J. Cancer 2016, 115, 346–353. [Google Scholar] [CrossRef]
- Renaud, S.; Falcoz, P.E.; Schaeffer, M.; Guenot, D.; Romain, B.; Olland, A.; Reeb, J.; Santelmo, N.; Chenard, M.P.; Legrain, M.; et al. Prognostic value of the kras g12v mutation in 841 surgically resected caucasian lung adenocarcinoma cases. Br. J. Cancer 2015, 113, 1206–1215. [Google Scholar] [CrossRef]
- Cserepes, M.; Ostoros, G.; Lohinai, Z.; Raso, E.; Barbai, T.; Timar, J.; Rozsas, A.; Moldvay, J.; Kovalszky, I.; Fabian, K.; et al. Subtype-specific kras mutations in advanced lung adenocarcinoma: A retrospective study of patients treated with platinum-based chemotherapy. Eur. J. Cancer 2014, 50, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Garassino, M.C.; Marabese, M.; Rusconi, P.; Rulli, E.; Martelli, O.; Farina, G.; Scanni, A.; Broggini, M. Different types of k-ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann. Oncol. 2011, 22, 235–237. [Google Scholar] [CrossRef]
- Mellema, W.W.; Masen-Poos, L.; Smit, E.F.; Hendriks, L.E.; Aerts, J.G.; Termeer, A.; Goosens, M.J.; Smit, H.J.; van den Heuvel, M.M.; van der Wekken, A.J.; et al. Comparison of clinical outcome after first-line platinum-based chemotherapy in different types of kras mutated advanced non-small-cell lung cancer. Lung Cancer 2015, 90, 249–254. [Google Scholar] [CrossRef]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of kras oncogene substitutions on protein behavior: Implications for signaling and clinical outcome. J. Natl. Cancer Inst. 2012, 104, 228–239. [Google Scholar] [CrossRef]
- Ghimessy, A.K.; Gellert, A.; Schlegl, E.; Hegedus, B.; Raso, E.; Barbai, T.; Timar, J.; Ostoros, G.; Megyesfalvi, Z.; Gieszer, B.; et al. Kras mutations predict response and outcome in advanced lung adenocarcinoma patients receiving first-line bevacizumab and platinum-based chemotherapy. Cancers (Basel) 2019, 11, 1514. [Google Scholar] [CrossRef] [PubMed]
- Barbacid, M. Ras oncogenes: Their role in neoplasia. Eur. J. Clin. Investig. 1990, 20, 225–235. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and structural analysis of common cancer-associated kras mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S.; Hall, A. Rho gtpases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. Rho gtpases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho family proteins: Coordinating cell responses. Trends Cell Biol. 2001, 11, 471–477. [Google Scholar] [CrossRef]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small gtp-binding proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef]
- Haga, R.B.; Ridley, A.J. Rho gtpases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef]
- Vega, F.M.; Ridley, A.J. Rho gtpases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Hall, A. Ras and rho gtpases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef]
- Khosravi-Far, R.; Solski, P.A.; Clark, G.J.; Kinch, M.S.; Der, C.J. Activation of rac1, rhoa, and mitogen-activated protein kinases is required for ras transformation. Mol. Cell. Biol. 1995, 15, 6443–6453. [Google Scholar] [CrossRef]
- Qiu, R.G.; Chen, J.; Kirn, D.; McCormick, F.; Symons, M. An essential role for rac in ras transformation. Nature 1995, 374, 457–459. [Google Scholar] [CrossRef]
- Qiu, R.G.; Chen, J.; McCormick, F.; Symons, M. A role for rho in ras transformation. Proc. Natl. Acad. Sci. USA 1995, 92, 11781–11785. [Google Scholar] [CrossRef]
- Ren, Y.; Li, R.; Zheng, Y.; Busch, H. Cloning and characterization of gef-h1, a microtubule-associated guanine nucleotide exchange factor for rac and rho gtpases. J. Biol. Chem. 1998, 273, 34954–34960. [Google Scholar] [CrossRef] [PubMed]
- Cullis, J.; Meiri, D.; Sandi, M.J.; Radulovich, N.; Kent, O.A.; Medrano, M.; Mokady, D.; Normand, J.; Larose, J.; Marcotte, R.; et al. The rhogef gef-h1 is required for oncogenic ras signaling via ksr-1. Cancer Cell 2014, 25, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor gef-h1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002, 4, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Nalbant, P.; Chang, Y.C.; Birkenfeld, J.; Chang, Z.F.; Bokoch, G.M. Guanine nucleotide exchange factor-h1 regulates cell migration via localized activation of rhoa at the leading edge. Mol. Biol. Cell 2009, 20, 4070–4082. [Google Scholar] [CrossRef]
- Von Thun, A.; Preisinger, C.; Rath, O.; Schwarz, J.P.; Ward, C.; Monsefi, N.; Rodriguez, J.; Garcia-Munoz, A.; Birtwistle, M.; Bienvenut, W.; et al. Extracellular signal-regulated kinase regulates rhoa activation and tumor cell plasticity by inhibiting guanine exchange factor h1 activity. Mol. Cell. Biol. 2013, 33, 4526–4537. [Google Scholar] [CrossRef]
- Chen, J.; Xia, H.; Zhang, X.; Karthik, S.; Pratap, S.V.; Ooi, L.L.; Hong, W.; Hui, K.M. Ect2 regulates the rho/erk signalling axis to promote early recurrence in human hepatocellular carcinoma. J. Hepatol. 2015, 62, 1287–1295. [Google Scholar] [CrossRef]
- Matoba, K.; Kawanami, D.; Nagai, Y.; Takeda, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. Rho-kinase blockade attenuates podocyte apoptosis by inhibiting the notch signaling pathway in diabetic nephropathy. Int. J. Mol. Sci. 2017, 18, 1795. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, T.R.; Linseman, D.A. Rho family gtpases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.; Macaya, I.; Bazzocco, S.; Mazzolini, R.; Andretta, E.; Dopeso, H.; Mateo-Lozano, S.; Bilic, J.; Carton-Garcia, F.; Nieto, R.; et al. Rhoa inactivation enhances wnt signalling and promotes colorectal cancer. Nat. Commun. 2014, 5, 5458. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Pinedo, E.C.; Durham, A.C.; Stewart, K.M.; Goss, A.M.; Lu, M.M.; Demayo, F.J.; Morrisey, E.E. Wnt/beta-catenin signaling accelerates mouse lung tumorigenesis by imposing an embryonic distal progenitor phenotype on lung epithelium. J. Clin. Investig. 2011, 121, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Hall, A. The small gtp-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Forti, F.L.; Armelin, H.A. Vasopressin triggers senescence in k-ras transformed cells via rhoa-dependent downregulation of cyclin d1. Endocr. Relat. Cancer 2007, 14, 1117–1125. [Google Scholar] [CrossRef]
- Ming, X.F.; Viswambharan, H.; Barandier, C.; Ruffieux, J.; Kaibuchi, K.; Rusconi, S.; Yang, Z. Rho gtpase/rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase b/akt in human endothelial cells. Mol. Cell. Biol. 2002, 22, 8467–8477. [Google Scholar] [CrossRef]
- Chew, T.W.; Liu, X.J.; Liu, L.; Spitsbergen, J.M.; Gong, Z.; Low, B.C. Crosstalk of ras and rho: Activation of rhoa abates kras-induced liver tumorigenesis in transgenic zebrafish models. Oncogene 2014, 33, 2717–2727. [Google Scholar] [CrossRef]
- Arango, D.; Laiho, P.; Kokko, A.; Alhopuro, P.; Sammalkorpi, H.; Salovaara, R.; Nicorici, D.; Hautaniemi, S.; Alazzouzi, H.; Mecklin, J.P.; et al. Gene-expression profiling predicts recurrence in dukes’ c colorectal cancer. Gastroenterology 2005, 129, 874–884. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Nelson, W.J.; Nusse, R. Convergence of wnt, beta-catenin, and cadherin pathways. Science 2004, 303, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-ras promotes tumorigenicity through suppression of non-canonical wnt signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical kras(g12c) inhibitor amg 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, P.-S.; Huang, M.-H.; Kuo, Y.-Y.; Yang, J.C.-H. The Inhibition of Wnt Restrain KRASG12V-Driven Metastasis in Non-Small-Cell Lung Cancer. Cancers 2020, 12, 837. https://doi.org/10.3390/cancers12040837
Hung P-S, Huang M-H, Kuo Y-Y, Yang JC-H. The Inhibition of Wnt Restrain KRASG12V-Driven Metastasis in Non-Small-Cell Lung Cancer. Cancers. 2020; 12(4):837. https://doi.org/10.3390/cancers12040837
Chicago/Turabian StyleHung, Pei-Shan, Ming-Hung Huang, Yuan-Yeh Kuo, and James Chih-Hsin Yang. 2020. "The Inhibition of Wnt Restrain KRASG12V-Driven Metastasis in Non-Small-Cell Lung Cancer" Cancers 12, no. 4: 837. https://doi.org/10.3390/cancers12040837
APA StyleHung, P.-S., Huang, M.-H., Kuo, Y.-Y., & Yang, J. C.-H. (2020). The Inhibition of Wnt Restrain KRASG12V-Driven Metastasis in Non-Small-Cell Lung Cancer. Cancers, 12(4), 837. https://doi.org/10.3390/cancers12040837