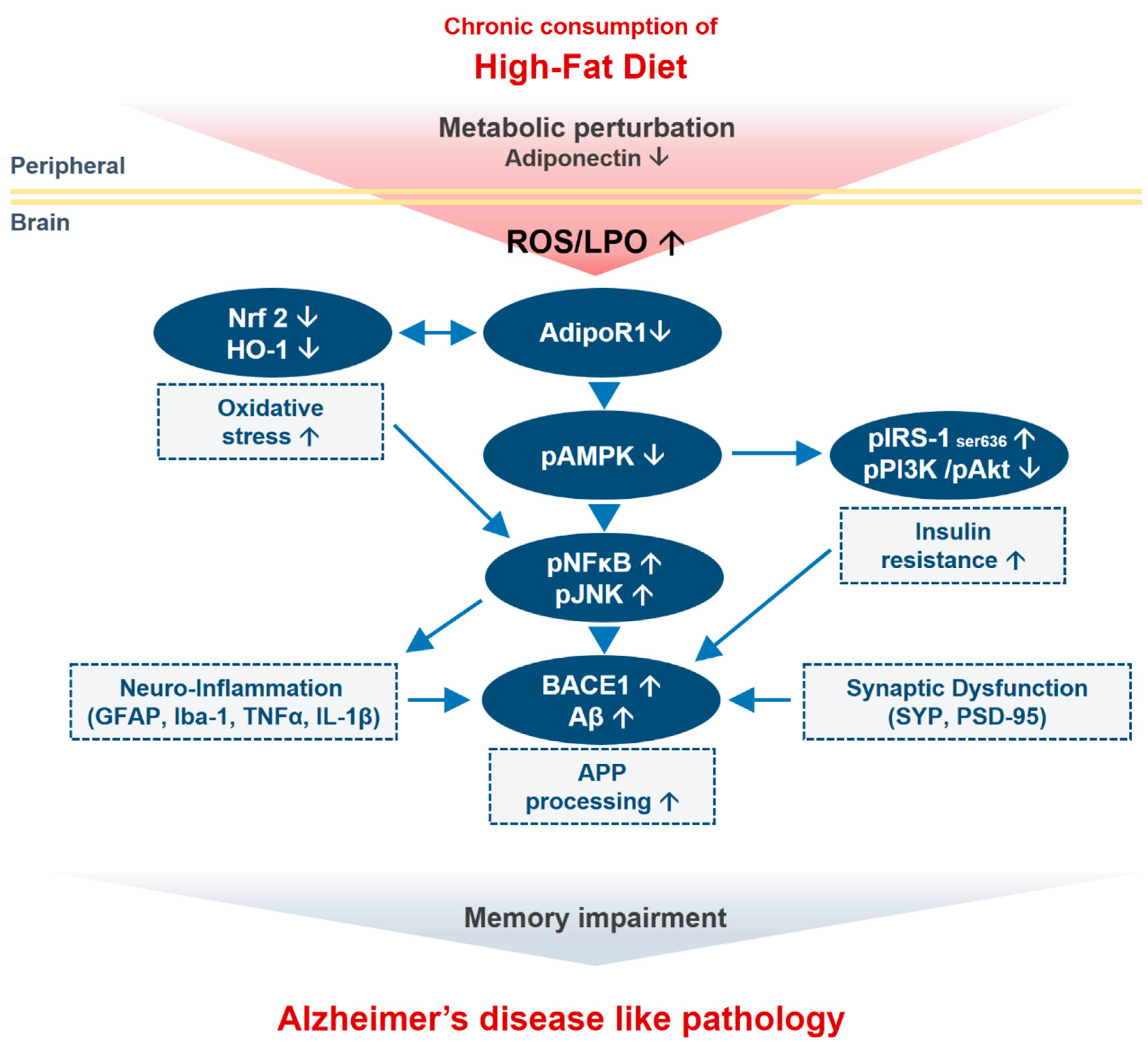
Metabolic Stress Alters Antioxidant Systems, Suppresses the Adiponectin Receptor 1 and Induces Alzheimer’s Like Pathology in Mice Brain

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Diets
2.3. Behavioral Analysis: Morris Water Maze (MWM) and Y-Maze Test
2.4. Insulin Sensitivity and Glucose Tolerance Tests
2.5. Extraction of Murine Brain Proteins
2.6. Western Blotting Analysis
2.7. Cell Culturing
2.8. Cell Transfection
2.9. Detection of Oxidative Stress (In Vivo and In Vitro)
2.10. Assessment of Lipid Peroxidation (In Vivo and In Vitro)
2.11. Glutathione Assays
2.12. Immunofluorescence
2.13. Antibodies
2.14. Thioflavin S Staining of Aβ Plaques
2.15. Data and Statistical Analysis
3. Results
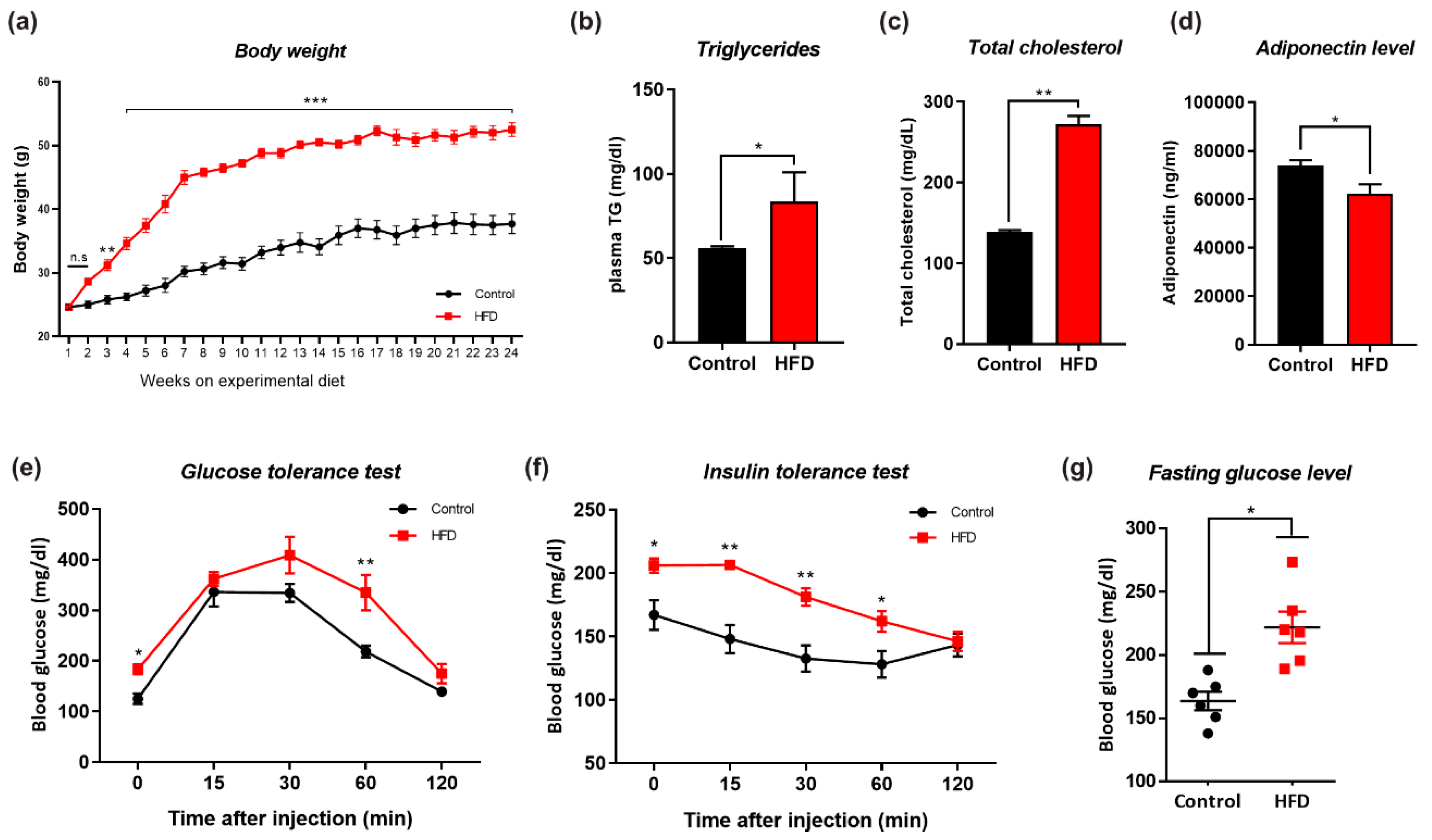
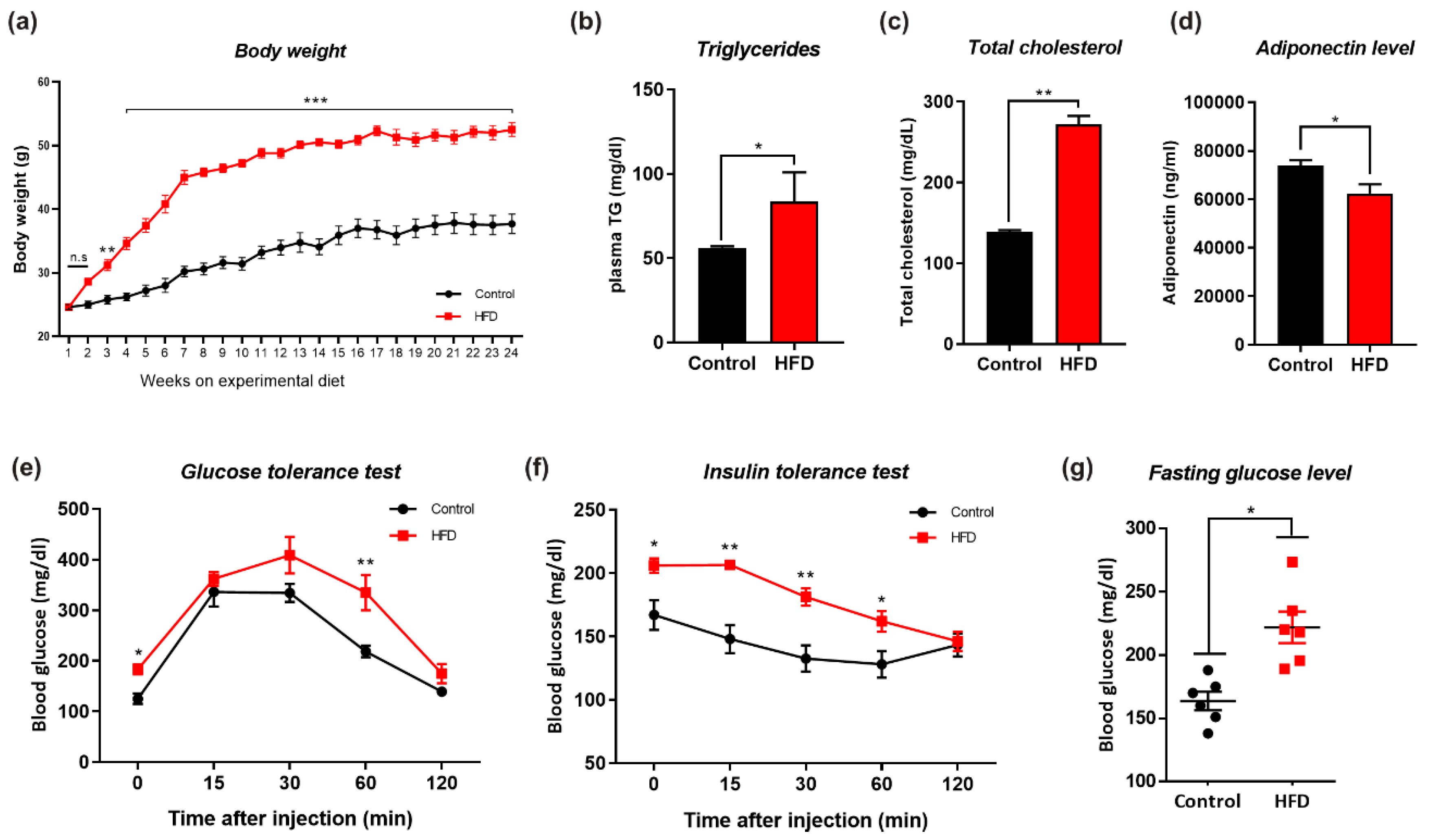
3.1. HFD-Induced Obesity Deregulates Metabolic Parameters in Mice
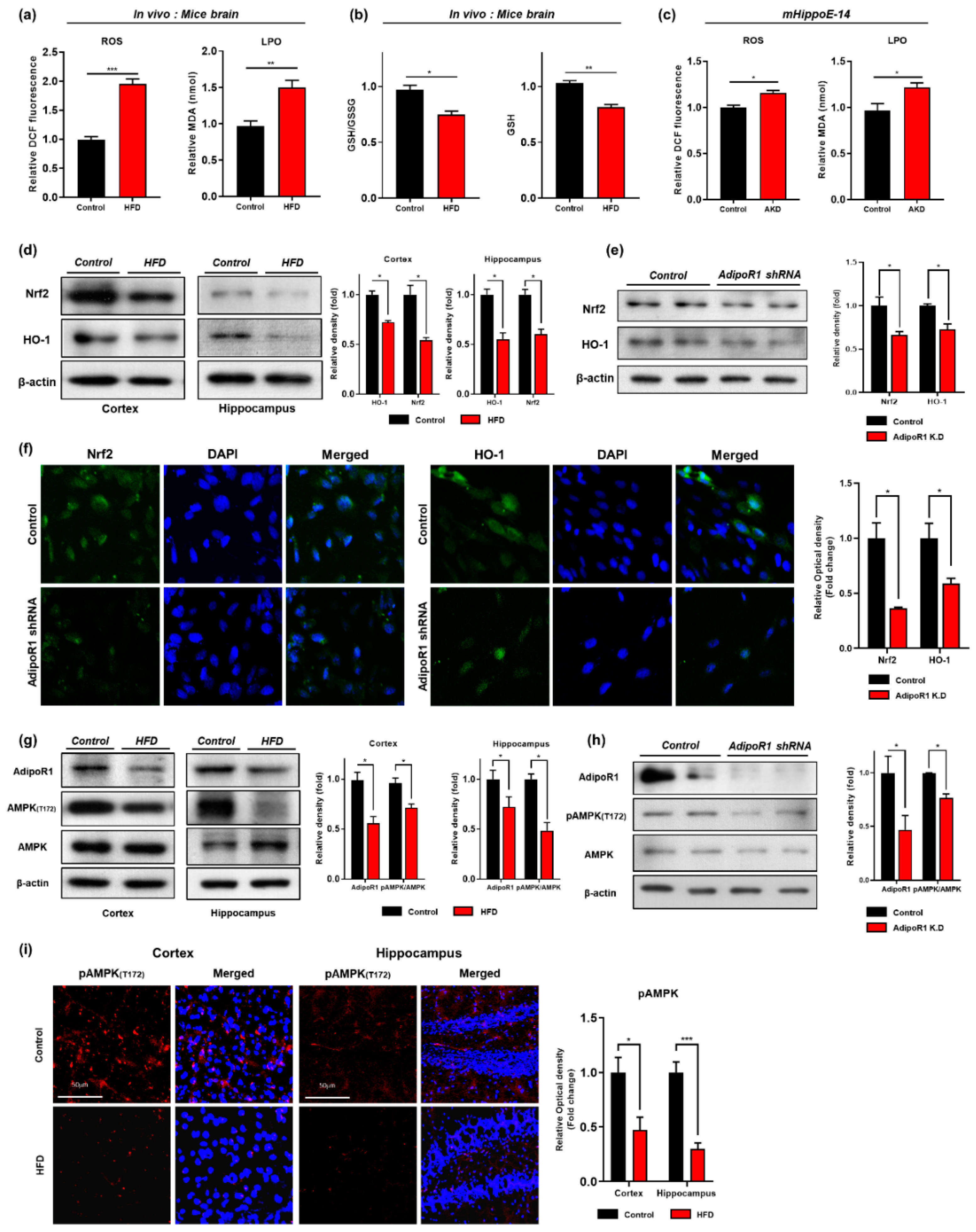
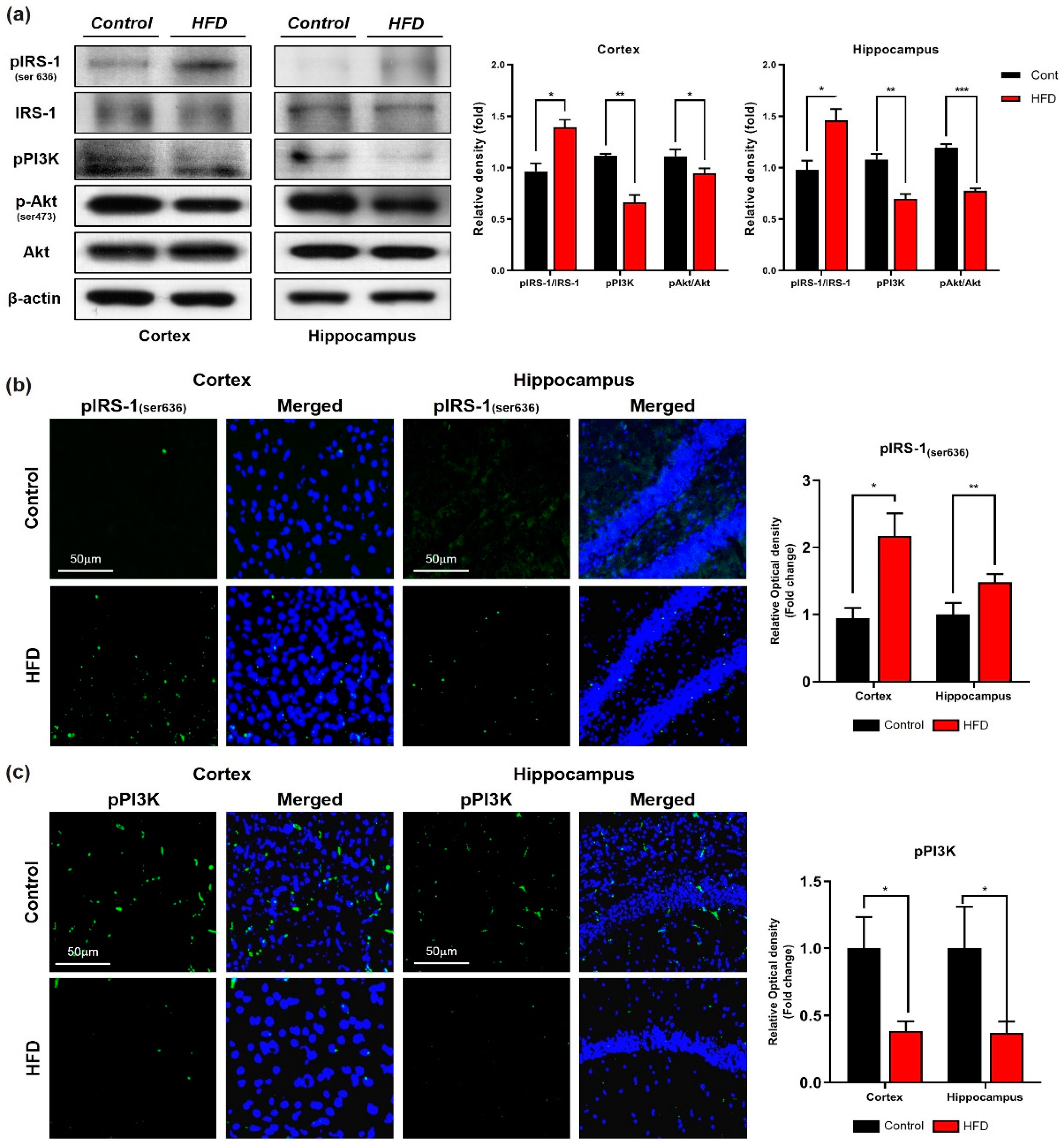
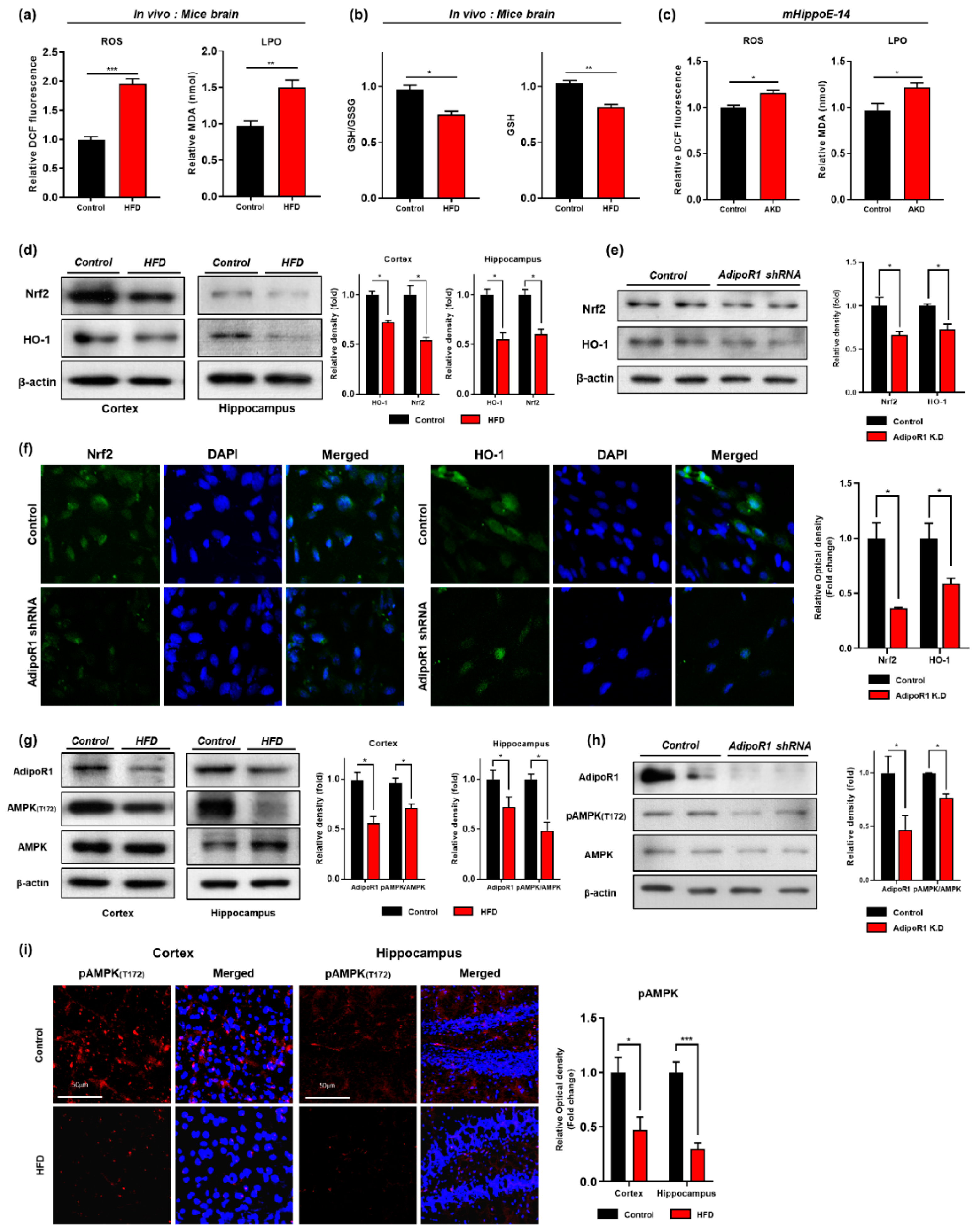
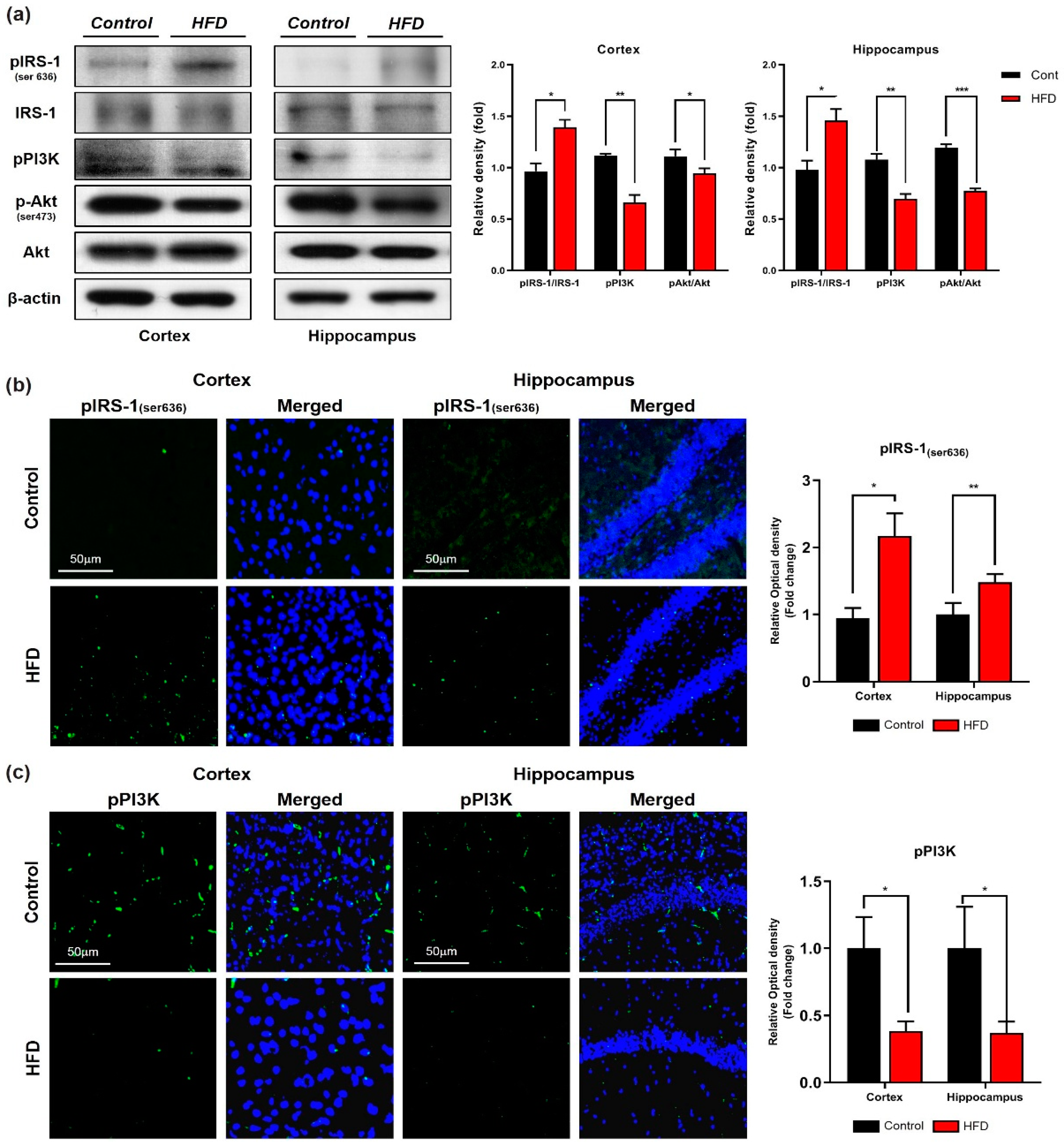
3.2. HFD Induces Oxidative-Stress-Mediated Brain Insulin Resistance by Impairing AdipoR1/P-AMPK Signaling Both In Vivo and In Vitro
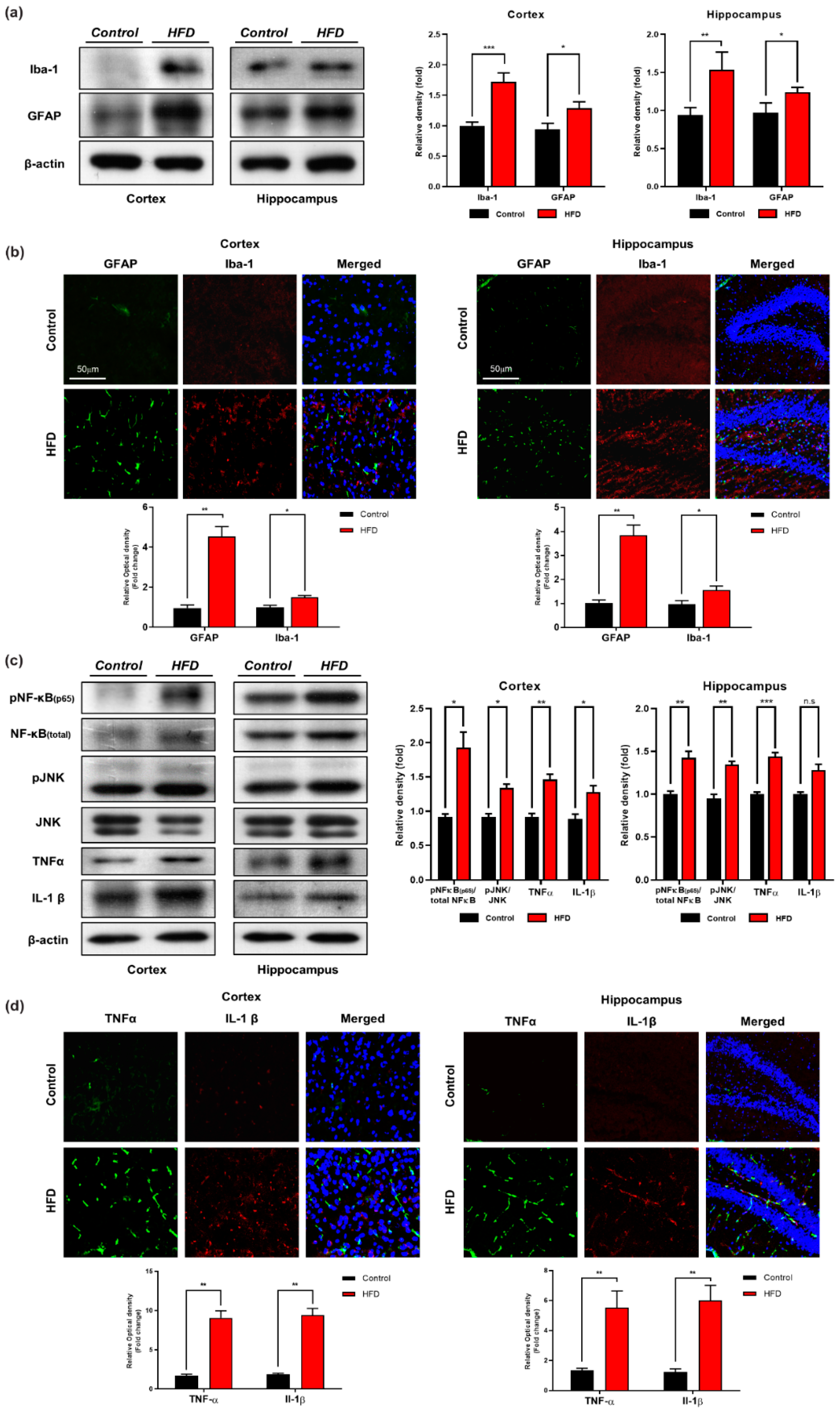
3.3. Chronic HFD Consumption Activates Astrocytes and Microglia and Exacerbates Neuroinflammation in Mouse Brains
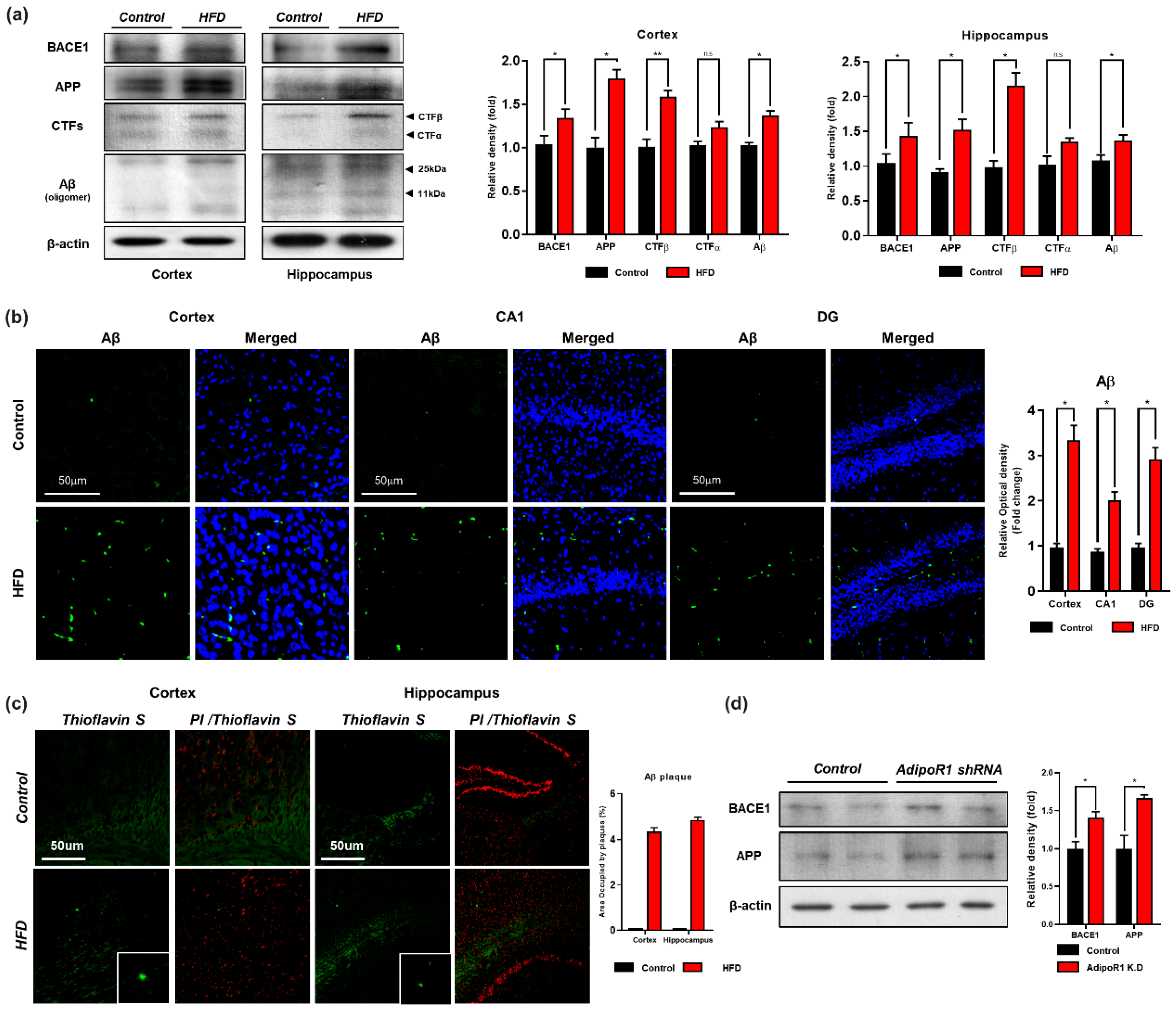
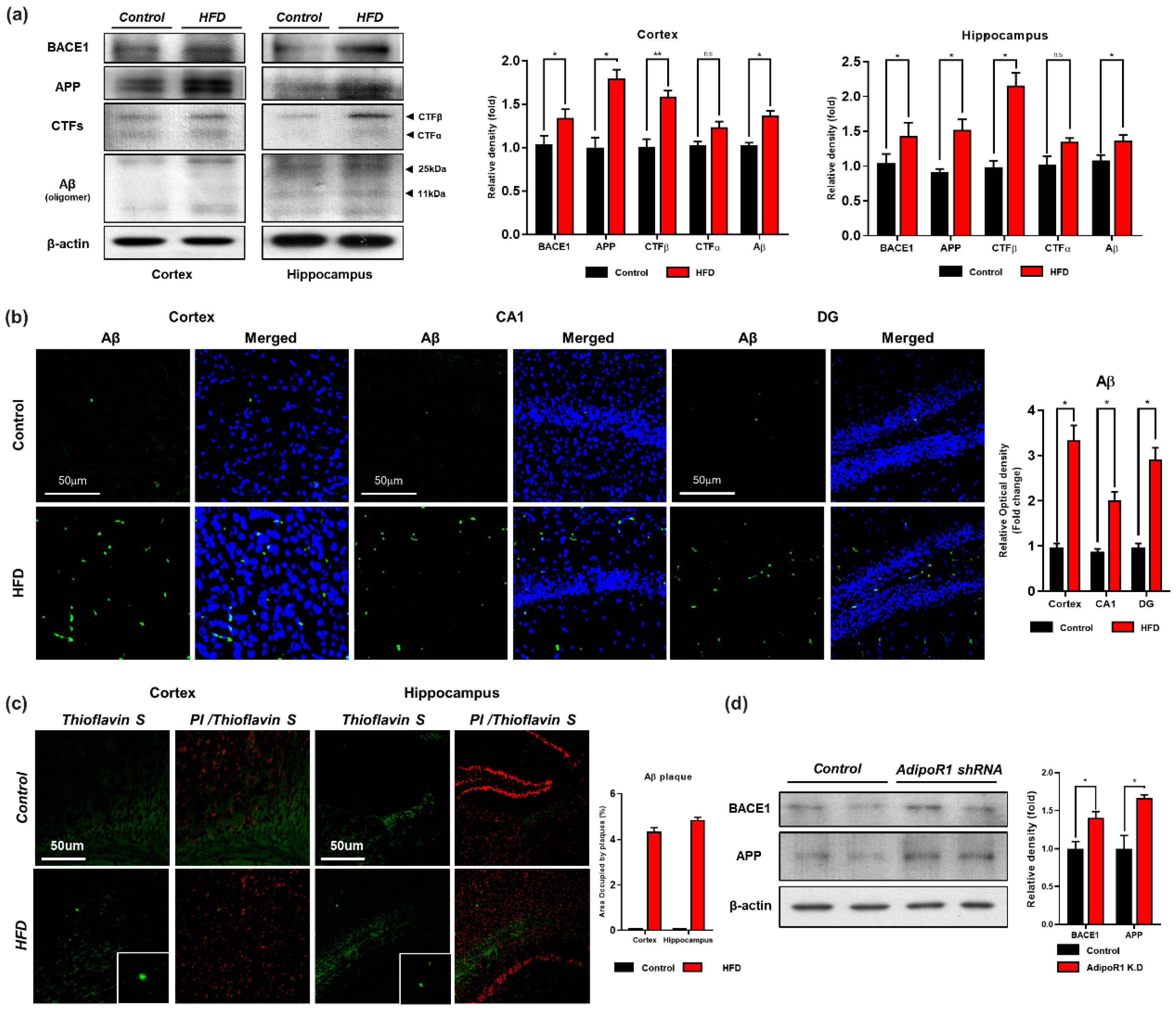
3.4. HFD Induces AD-Like Pathology in Mouse Brains
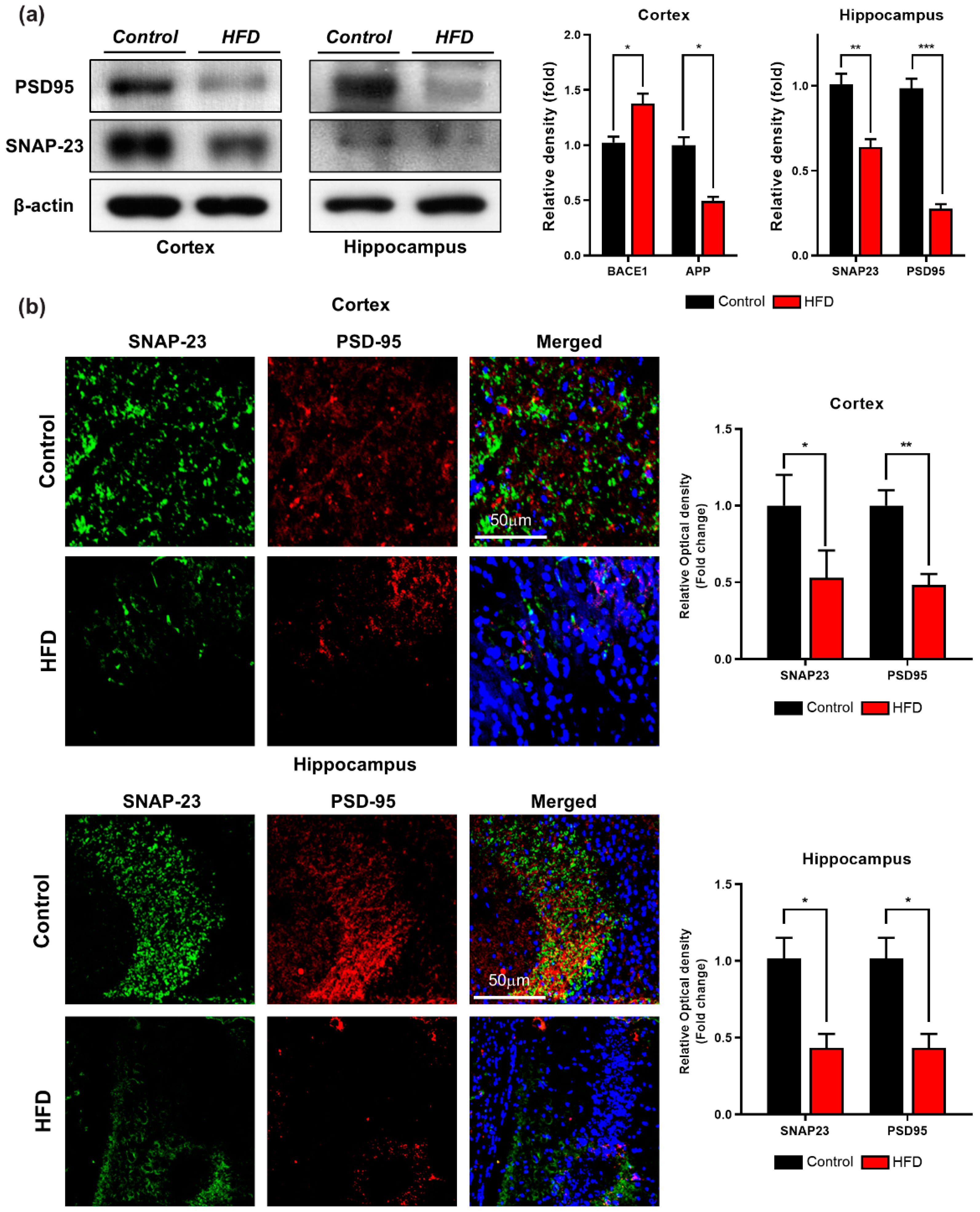
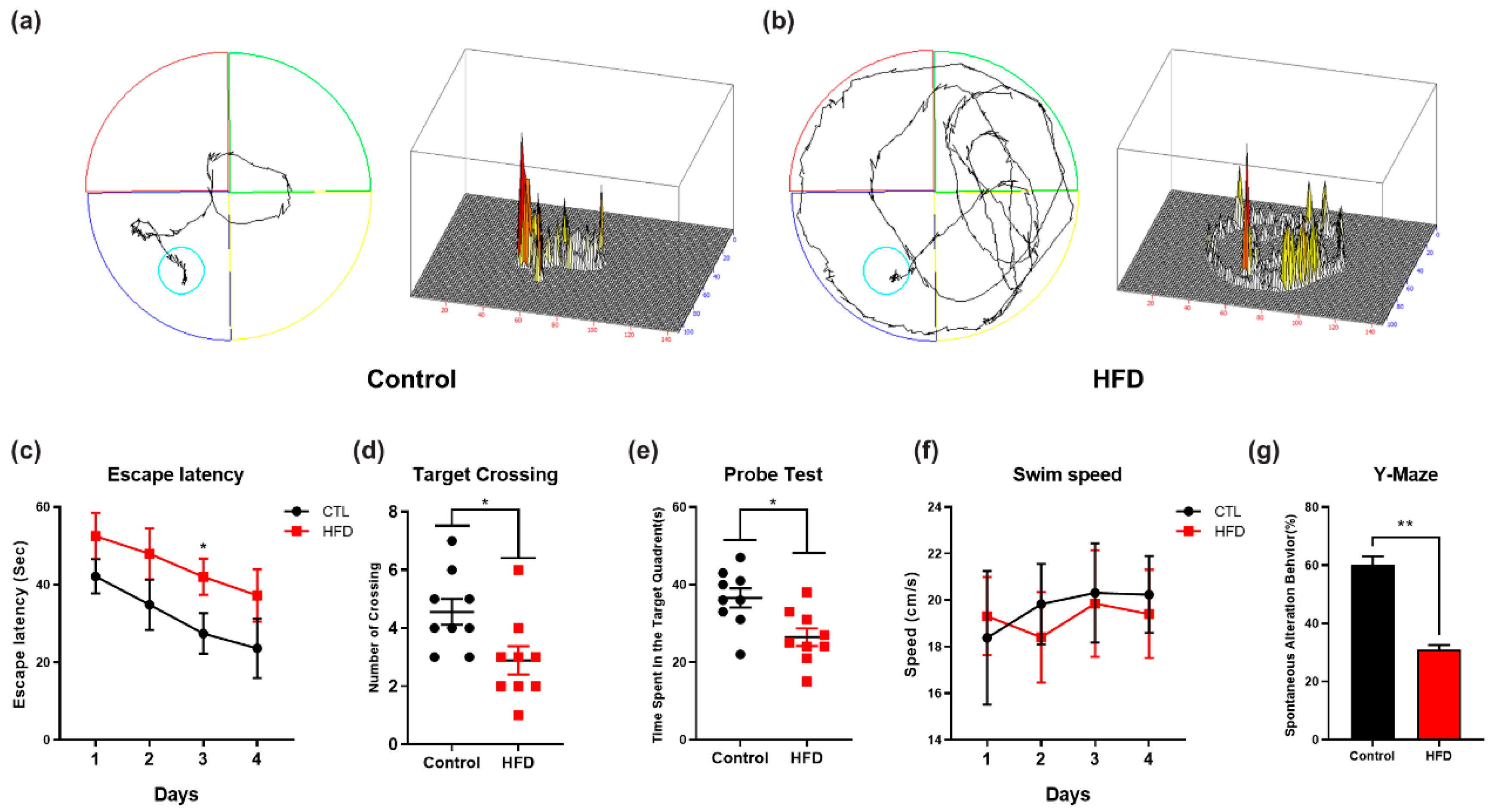
3.5. HFD Induces Synaptic Dysfunction and Memory Impairment in Mouse Brains
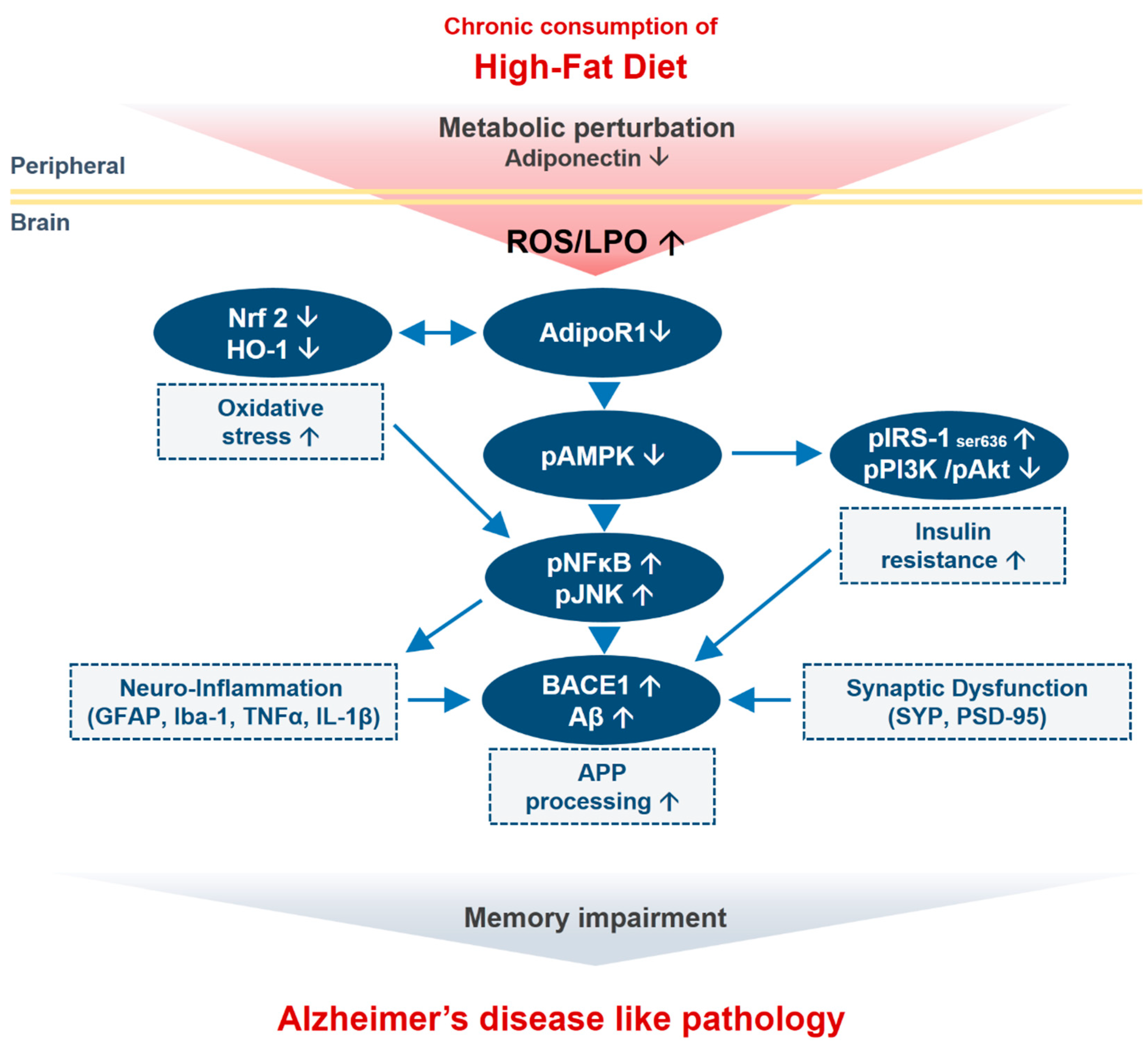
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Park, S.B.; Kang, J.Y.; Kim, J.M.; Park, S.K.; Yoo, S.K.; Lee, U.; Kim, D.O.; Heo, H.J. Effect of Aruncus dioicus var. kamtschaticus Extract on Neurodegeneration Improvement: Ameliorating Role in Cognitive Disorder Caused by High-Fat Diet Induced Obesity. Nutrients 2019, 11, 1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.W.; Lee, M.; Oh, K.J. Adipose Tissue-Derived Signatures for Obesity and Type 2 Diabetes: Adipokines, Batokines and MicroRNAs. J. Clin. Med. 2019, 8, 854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, K.B.; Lemberg, L. Obesity and the metabolic syndrome. Am. J. Crit. Care 2003, 12, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metabolism 2016, 65, 1376–1390. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Caloric intake and the risk of Alzheimer disease. Arch. Neurol. 2002, 59, 1258–1263. [Google Scholar] [CrossRef]
- Ruan, Y.; Tang, J.; Guo, X.; Li, K.; Li, D. Dietary Fat Intake and Risk of Alzheimer’s Disease and Dementia: A Meta-Analysis of Cohort Studies. Curr. Alzheimer Res. 2018, 15, 869–876. [Google Scholar] [CrossRef]
- Pannacciulli, N.; Del Parigi, A.; Chen, K.; Le, D.S.; Reiman, E.M.; Tataranni, P.A. Brain abnormalities in human obesity: A voxel-based morphometric study. Neuroimage 2006, 31, 1419–1425. [Google Scholar] [CrossRef]
- Forny-Germano, L.; De Felice, F.G.; Vieira, M. The Role of Leptin and Adiponectin in Obesity-Associated Cognitive Decline and Alzheimer’s Disease. Front. Neurosci. 2018, 12, 1027. [Google Scholar] [CrossRef] [Green Version]
- Bloemer, J.; Pinky, P.D.; Govindarajulu, M.; Hong, H.; Judd, R.; Amin, R.H.; Moore, T.; Dhanasekaran, M.; Reed, M.N.; Suppiramaniam, V. Role of Adiponectin in Central Nervous System Disorders. Neural Plast. 2018, 2018, 4593530. [Google Scholar] [CrossRef] [Green Version]
- Kubota, N.; Yano, W.; Kubota, T.; Yamauchi, T.; Itoh, S.; Kumagai, H.; Kozono, H.; Takamoto, I.; Okamoto, S.; Shiuchi, T.; et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007, 6, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Kang, S.M.; Kim, E.; Kim, C.H.; Song, H.T.; Lee, J.E. Adiponectin receptor-mediated signaling ameliorates cerebral cell damage and regulates the neurogenesis of neural stem cells at high glucose concentrations: An in vivo and in vitro study. Cell Death Dis. 2015, 6, 1844. [Google Scholar] [CrossRef]
- Shah, S.A.; Yoon, G.H.; Chung, S.S.; Abid, M.N.; Kim, T.H.; Lee, H.Y.; Kim, M.O. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol. Psychiatry 2017, 22, 407–416. [Google Scholar] [CrossRef]
- Kim, M.W.; Abid, N.B.; Jo, M.H.; Jo, M.G.; Yoon, G.H.; Kim, M.O. Suppression of adiponectin receptor 1 promotes memory dysfunction and Alzheimer’s disease-like pathologies. Sci. Rep. 2017, 7, 12435. [Google Scholar] [CrossRef]
- Ng, R.C.; Cheng, O.Y.; Jian, M.; Kwan, J.S.; Ho, P.W.; Cheng, K.K.; Yeung, P.K.; Zhou, L.L.; Hoo, R.L.; Chung, S.K.; et al. Chronic adiponectin deficiency leads to Alzheimer’s disease-like cognitive impairments and pathologies through AMPK inactivation and cerebral insulin resistance in aged mice. Mol. Neurodegener. 2016, 11, 71. [Google Scholar] [CrossRef] [Green Version]
- van der Heide, L.P.; Ramakers, G.M.; Smidt, M.P. Insulin signaling in the central nervous system: Learning to survive. Prog. Neurobiol. 2006, 79, 205–221. [Google Scholar] [CrossRef]
- de la Monte, S.M. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009, 42, 475–481. [Google Scholar] [CrossRef]
- Vandal, M.; White, P.J.; Tremblay, C.; St-Amour, I.; Chevrier, G.; Emond, V.; Lefrancois, D.; Virgili, J.; Planel, E.; Giguere, Y.; et al. Insulin reverses the high-fat diet-induced increase in brain Abeta and improves memory in an animal model of Alzheimer disease. Diabetes 2014, 63, 4291–4301. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Frisardi, V.; Solfrizzi, V.; Capurso, C.; Imbimbo, B.P.; Vendemiale, G.; Seripa, D.; Pilotto, A.; Panza, F. Is insulin resistant brain state a central feature of the metabolic-cognitive syndrome? J. Alzheimers Dis. 2010, 21, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Lester-Coll, N.; Rivera, E.J.; Soscia, S.J.; Doiron, K.; Wands, J.R.; de la Monte, S.M. Intracerebral streptozotocin model of type 3 diabetes: Relevance to sporadic Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 13–33. [Google Scholar] [CrossRef]
- De Felice, F.G. Alzheimer’s disease and insulin resistance: Translating basic science into clinical applications. J. Clin. Investig. 2013, 123, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Bailey-Downs, L.C.; Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Sonntag, W.E.; Csiszar, A.; Ungvari, Z. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: A paracrine mechanism contributing to vascular redox dysregulation and inflammation. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 780–792. [Google Scholar] [CrossRef] [Green Version]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1212–1226. [Google Scholar] [CrossRef]
- Duffy, C.M.; Hofmeister, J.J.; Nixon, J.P.; Butterick, T.A. High fat diet increases cognitive decline and neuroinflammation in a model of orexin loss. Neurobiol. Learn. Mem. 2019, 157, 41–47. [Google Scholar] [CrossRef]
- Bluher, M. Adipose tissue dysfunction in obesity. Exp. Clin. Endocrinol. Diabetes 2009, 117, 241–250. [Google Scholar] [CrossRef]
- Fujita, K.; Hayashi, T.; Matsushita, M.; Uemura, M.; Nonomura, N. Obesity, Inflammation, and Prostate Cancer. J. Clin. Med. 2019, 8, 201. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, S.T.; Clarke, J.R.; Bomfim, T.R.; De Felice, F.G. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimers Dement. 2014, 10, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Kim, B.; Feldman, E.L. Insulin resistance in the nervous system. Trends Endocrinol. Metab. 2012, 23, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Mediat. Inflamm. 2015, 2015, 105828. [Google Scholar] [CrossRef] [Green Version]
- Refolo, L.M.; Malester, B.; LaFrancois, J.; Bryant-Thomas, T.; Wang, R.; Tint, G.S.; Sambamurti, K.; Duff, K.; Pappolla, M.A. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 2000, 7, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Fitz, N.F.; Cronican, A.; Pham, T.; Fogg, A.; Fauq, A.H.; Chapman, R.; Lefterov, I.; Koldamova, R. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J. Neurosci. 2010, 30, 6862–6872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, C.; Tremblay, C.; Phivilay, A.; Berthiaume, L.; Emond, V.; Julien, P.; Calon, F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol. Aging 2010, 31, 1516–1531. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Rutten, B.P.F.; Kim, M.O. MST1 Regulates Neuronal Cell Death via JNK/Casp3 Signaling Pathway in HFD Mouse Brain and HT22 Cells. Int. J. Mol. Sci. 2019, 20, 2504. [Google Scholar] [CrossRef] [Green Version]
- Abbott, R.D.; Ross, G.W.; White, L.R.; Nelson, J.S.; Masaki, K.H.; Tanner, C.M.; Curb, J.D.; Blanchette, P.L.; Popper, J.S.; Petrovitch, H. Midlife adiposity and the future risk of Parkinson’s disease. Neurology 2002, 59, 1051–1057. [Google Scholar] [CrossRef]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kareholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef] [Green Version]
- Jurdak, N.; Lichtenstein, A.H.; Kanarek, R.B. Diet-induced obesity and spatial cognition in young male rats. Nutr. Neurosci. 2008, 11, 48–54. [Google Scholar] [CrossRef]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Ullah, R.; Khan, M.; Shah, S.A.; Saeed, K.; Kim, M.O. Natural Antioxidant Anthocyanins-A Hidden Therapeutic Candidate in Metabolic Disorders with Major Focus in Neurodegeneration. Nutrients 2019, 11, 1195. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Ali, T.; Rehman, S.U.; Khan, M.S.; Alam, S.I.; Ikram, M.; Muhammad, T.; Saeed, K.; Badshah, H.; Kim, M.O. Neuroprotective Effect of Quercetin Against the Detrimental Effects of LPS in the Adult Mouse Brain. Front. Pharmacol. 2018, 9, 1383. [Google Scholar] [CrossRef]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin Confers Neuroprotection by Regulating Nrf2/TLR4/NF-kappaB Signaling in an Abeta Mouse Model. Mol. Neurobiol. 2019, 10, 1007. [Google Scholar] [CrossRef]
- Jeon, B.T.; Jeong, E.A.; Shin, H.J.; Lee, Y.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes 2012, 61, 1444–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.A.; Khan, M.; Jo, M.H.; Jo, M.G.; Amin, F.U.; Kim, M.O. Melatonin Stimulates the SIRT1/Nrf2 Signaling Pathway Counteracting Lipopolysaccharide (LPS)-Induced Oxidative Stress to Rescue Postnatal Rat Brain. CNS Neurosci. Ther. 2017, 23, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.A.; Zeb, A.; Ali, T.; Muhammad, T.; Faheem, M.; Alam, S.I.; Saeed, K.; Koh, P.O.; Lee, K.W.; Kim, M.O. Identification of Proteins Differentially Expressed in the Striatum by Melatonin in a Middle Cerebral Artery Occlusion Rat Model-a Proteomic and in silico Approach. Front. Neurosci. 2018, 12, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine Modulates Cadmium-Induced Oxidative Stress, Neuroinflammation, and Cognitive Impairments by Regulating Nrf-2/HO-1 In Vivo and In Vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-kappaB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikram, M.; Saeed, K.; Khan, A.; Muhammad, T.; Khan, M.S.; Jo, M.G.; Rehman, S.U.; Kim, M.O. Natural Dietary Supplementation of Curcumin Protects Mice Brains against Ethanol-Induced Oxidative Stress-Mediated Neurodegeneration and Memory Impairment via Nrf2/TLR4/RAGE Signaling. Nutrients 2019, 11, 1082. [Google Scholar] [CrossRef] [Green Version]
- Ojala, J.O.; Sutinen, E.M. The Role of Interleukin-18, Oxidative Stress and Metabolic Syndrome in Alzheimer’s Disease. J. Clin. Med. 2017, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. (Lausanne) 2018, 9, 496. [Google Scholar] [CrossRef]
- Ng, R.C.; Chan, K.H. Potential Neuroprotective Effects of Adiponectin in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 592. [Google Scholar] [CrossRef] [Green Version]
- Lindholm, C.R.; Ertel, R.L.; Bauwens, J.D.; Schmuck, E.G.; Mulligan, J.D.; Saupe, K.W. A high-fat diet decreases AMPK activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats. J. Physiol. Biochem. 2013, 69, 165–175. [Google Scholar] [CrossRef]
- Yokoyama, H.; Takeda, R.; Kawai, E.; Ota, A.; Morita, E.; Imai, D.; Suzuki, Y.; Morioka, T.; Emoto, M.; Inaba, M.; et al. Inhibitory Effects of Intranasal Administration of Insulin on Fat Oxidation during Exercise Are Diminished in Young Overweight Individuals. J. Clin. Med. 2018, 7, 308. [Google Scholar] [CrossRef] [PubMed]
- Nerurkar, P.V.; Johns, L.M.; Buesa, L.M.; Kipyakwai, G.; Volper, E.; Sato, R.; Shah, P.; Feher, D.; Williams, P.G.; Nerurkar, V.R. Momordica charantia (bitter melon) attenuates high-fat diet-associated oxidative stress and neuroinflammation. J. Neuroinflamm. 2011, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Kappelle, L.J.; Utrecht Diabetic Encephalopathy Study, G. Increased risk of Alzheimer’s disease in Type II diabetes: Insulin resistance of the brain or insulin-induced amyloid pathology? Biochem. Soc. Trans. 2005, 33, 1041–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brands, A.M.; Biessels, G.J.; de Haan, E.H.; Kappelle, L.J.; Kessels, R.P. The effects of type 1 diabetes on cognitive performance: A meta-analysis. Diabetes Care 2005, 28, 726–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef]
- Vyas, Y.; Montgomery, J.M. The role of postsynaptic density proteins in neural degeneration and regeneration. Neural Regen. Res. 2016, 11, 906–907. [Google Scholar] [CrossRef]
- Arnold, S.E.; Lucki, I.; Brookshire, B.R.; Carlson, G.C.; Browne, C.A.; Kazi, H.; Bang, S.; Choi, B.R.; Chen, Y.; McMullen, M.F.; et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis. 2014, 67, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Ali, T.; Park, H.Y.; Badshah, H.; Rehman, S.U.; Kim, M.O. Neuroprotective Effect of Fisetin Against Amyloid-Beta-Induced Cognitive/Synaptic Dysfunction, Neuroinflammation, and Neurodegeneration in Adult Mice. Mol. Neurobiol. 2017, 54, 2269–2285. [Google Scholar] [CrossRef]
- Gallagher, J.J.; Minogue, A.M.; Lynch, M.A. Impaired performance of female APP/PS1 mice in the Morris water maze is coupled with increased Abeta accumulation and microglial activation. Neurodegener. Dis. 2013, 11, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Ahren, B. The high-fat diet-fed mouse: A model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004, 53, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Skinner, D.D.; Lane, T.E. Innate Immune Responses and Viral-Induced Neurologic Disease. J. Clin. Med. 2018, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Buckman, L.B.; Thompson, M.M.; Lippert, R.N.; Blackwell, T.S.; Yull, F.E.; Ellacott, K.L. Evidence for a novel functional role of astrocytes in the acute homeostatic response to high-fat diet intake in mice. Mol. Metab. 2015, 4, 58–63. [Google Scholar] [CrossRef]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef] [Green Version]
- de Kloet, A.D.; Pioquinto, D.J.; Nguyen, D.; Wang, L.; Smith, J.A.; Hiller, H.; Sumners, C. Obesity induces neuroinflammation mediated by altered expression of the renin-angiotensin system in mouse forebrain nuclei. Physiol. Behav. 2014, 136, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Plum, L.; Schubert, M.; Bruning, J.C. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar] [CrossRef]
- de la Monte, S.M. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef]
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Marino Gammazza, A.; Cappello, F.; Mule, F.; Di Carlo, M. Insulin resistance as common molecular denominator linking obesity to Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 723–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thundyil, J.; Pavlovski, D.; Sobey, C.G.; Arumugam, T.V. Adiponectin receptor signalling in the brain. Br. J. Pharmacol. 2012, 165, 313–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.A.; Amin, F.U.; Khan, M.; Abid, M.N.; Rehman, S.U.; Kim, T.H.; Kim, M.W.; Kim, M.O. Anthocyanins abrogate glutamate-induced AMPK activation, oxidative stress, neuroinflammation, and neurodegeneration in postnatal rat brain. J. Neuroinflamm. 2016, 13, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viollet, B.; Lantier, L.; Devin-Leclerc, J.; Hebrard, S.; Amouyal, C.; Mounier, R.; Foretz, M.; Andreelli, F. Targeting the AMPK pathway for the treatment of Type 2 diabetes. Front. Biosci. (Landmark Ed) 2009, 14, 3380–3400. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Qatanani, M.; Lazar, M.A. Mechanisms of obesity-associated insulin resistance: Many choices on the menu. Genes Dev. 2007, 21, 1443–1455. [Google Scholar] [CrossRef] [Green Version]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Liu, Z.; Patil, I.Y.; Jiang, T.; Sancheti, H.; Walsh, J.P.; Stiles, B.L.; Yin, F.; Cadenas, E. High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS ONE 2015, 10, e0128274. [Google Scholar] [CrossRef]
- Maesako, M.; Uemura, M.; Tashiro, Y.; Sasaki, K.; Watanabe, K.; Noda, Y.; Ueda, K.; Asada-Utsugi, M.; Kubota, M.; Okawa, K.; et al. High Fat Diet Enhances beta-Site Cleavage of Amyloid Precursor Protein (APP) via Promoting beta-Site APP Cleaving Enzyme 1/Adaptor Protein 2/Clathrin Complex Formation. PLoS ONE 2015, 10, e0131199. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s disease: Synaptic dysfunction and Abeta. Mol. Neurodegener. 2009, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busquets, O.; Ettcheto, M.; Pallàs, M.; Beas-Zarate, C.; Verdaguer, E.; Auladell, C.; Folch, J.; Camins, A. Long-term exposition to a high fat diet favors the appearance of β-amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer’s disease. Mech. Ageing Dev. 2017, 162, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Nadeem, M.; Lad, S.P.; Wuu, J.; Mufson, E.J. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J. Neuropathol. Exp. Neurol. 2006, 65, 592–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stranahan, A.M.; Mattson, M.P. Bidirectional metabolic regulation of neurocognitive function. Neurobiol. Learn. Mem. 2011, 96, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, C.E.; Winocur, G. Learning and memory impairment in rats fed a high saturated fat diet. Behav. Neural. Biol. 1990, 53, 74–87. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hahm, J.R.; Jo, M.H.; Ullah, R.; Kim, M.W.; Kim, M.O. Metabolic Stress Alters Antioxidant Systems, Suppresses the Adiponectin Receptor 1 and Induces Alzheimer’s Like Pathology in Mice Brain. Cells 2020, 9, 249. https://doi.org/10.3390/cells9010249
Hahm JR, Jo MH, Ullah R, Kim MW, Kim MO. Metabolic Stress Alters Antioxidant Systems, Suppresses the Adiponectin Receptor 1 and Induces Alzheimer’s Like Pathology in Mice Brain. Cells. 2020; 9(1):249. https://doi.org/10.3390/cells9010249
Chicago/Turabian StyleHahm, Jong Ryeal, Myeung Hoon Jo, Rahat Ullah, Min Woo Kim, and Myeong Ok Kim. 2020. "Metabolic Stress Alters Antioxidant Systems, Suppresses the Adiponectin Receptor 1 and Induces Alzheimer’s Like Pathology in Mice Brain" Cells 9, no. 1: 249. https://doi.org/10.3390/cells9010249