Inducible Cardiac-Specific Deletion of Sirt1 in Male Mice Reveals Progressive Cardiac Dysfunction and Sensitization of the Heart to Pressure Overload

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Results
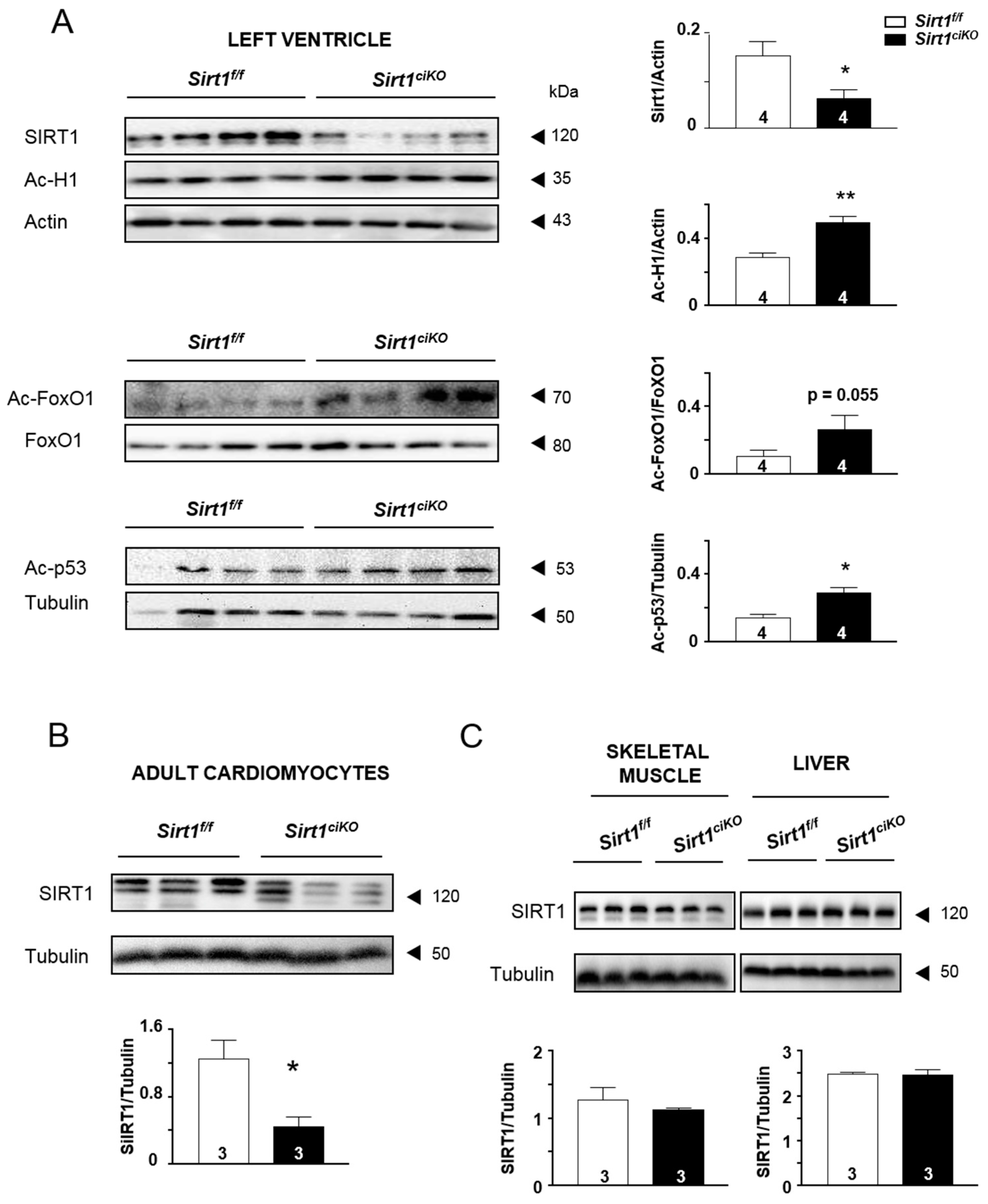
2.1. Cardiac Specific Tamoxifen-Induced Loss of Sirt1 in α-MHC-Cre/Flox Mice
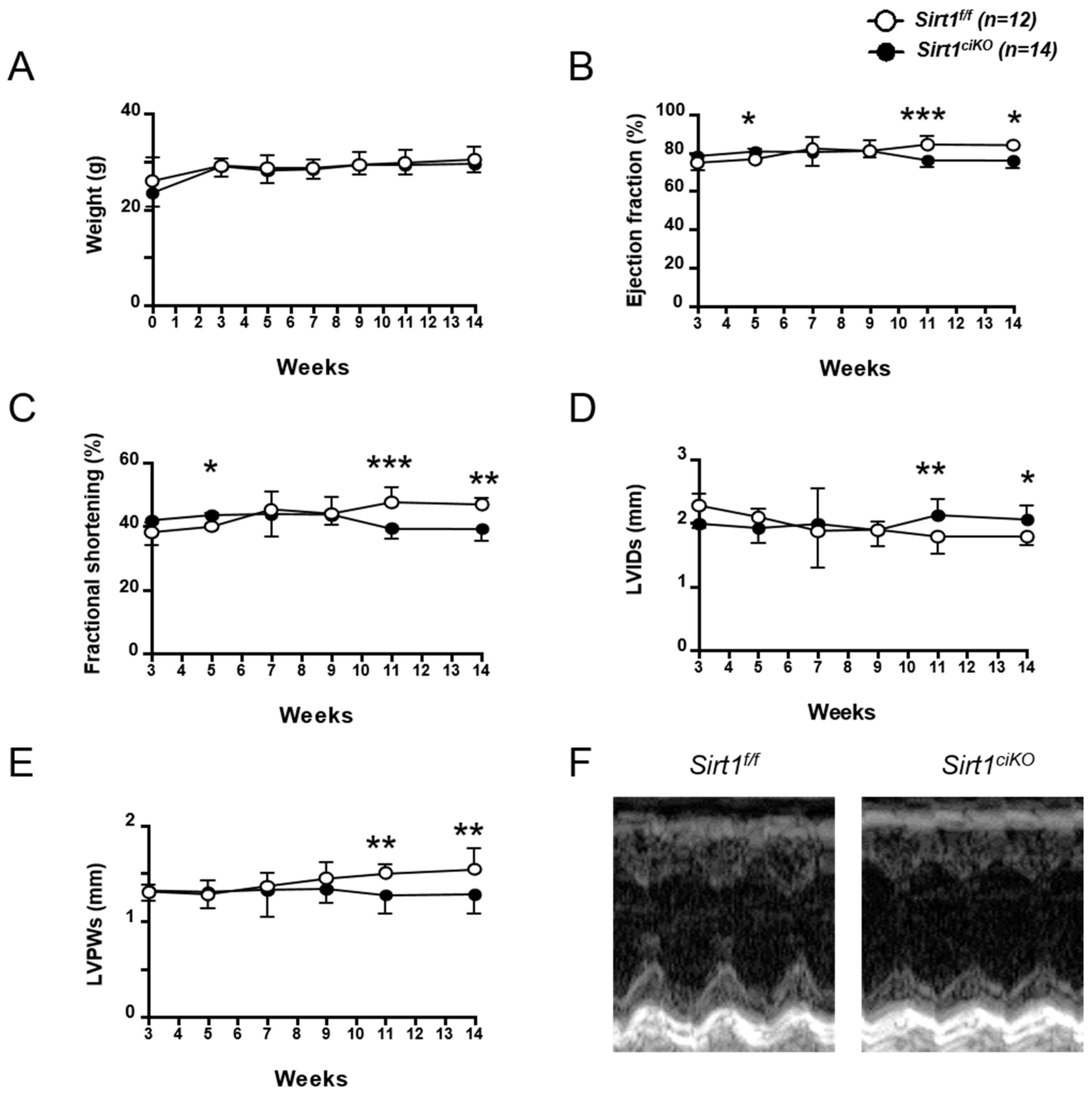
2.2. Sirt1ciKO Mutants Develop a Mild Left Ventricular Systolic Dysfunction
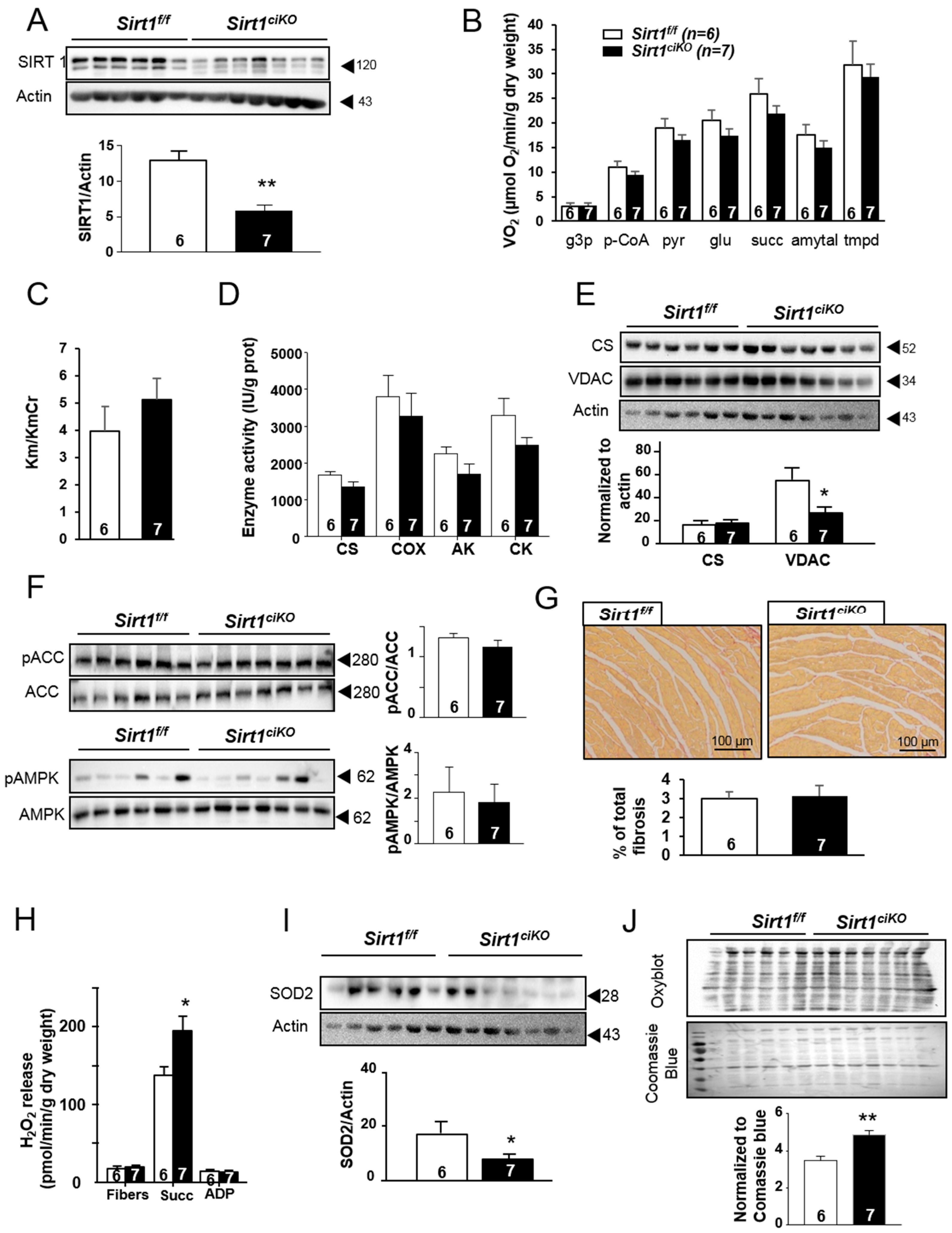
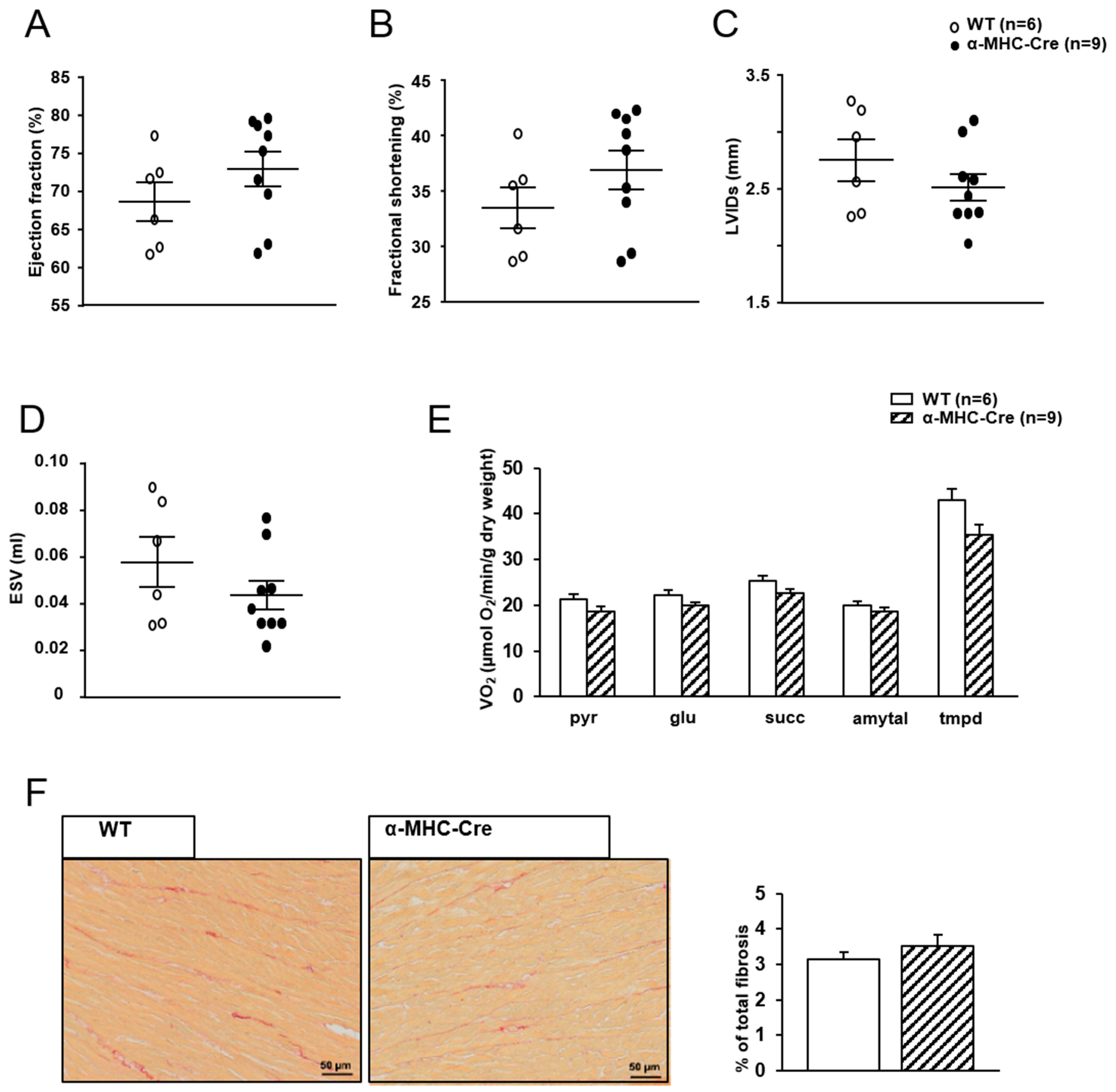
2.3. Mitochondrial Oxidative Capacities are Preserved in Cardiac-Specific Sirt1 Mutant Mice
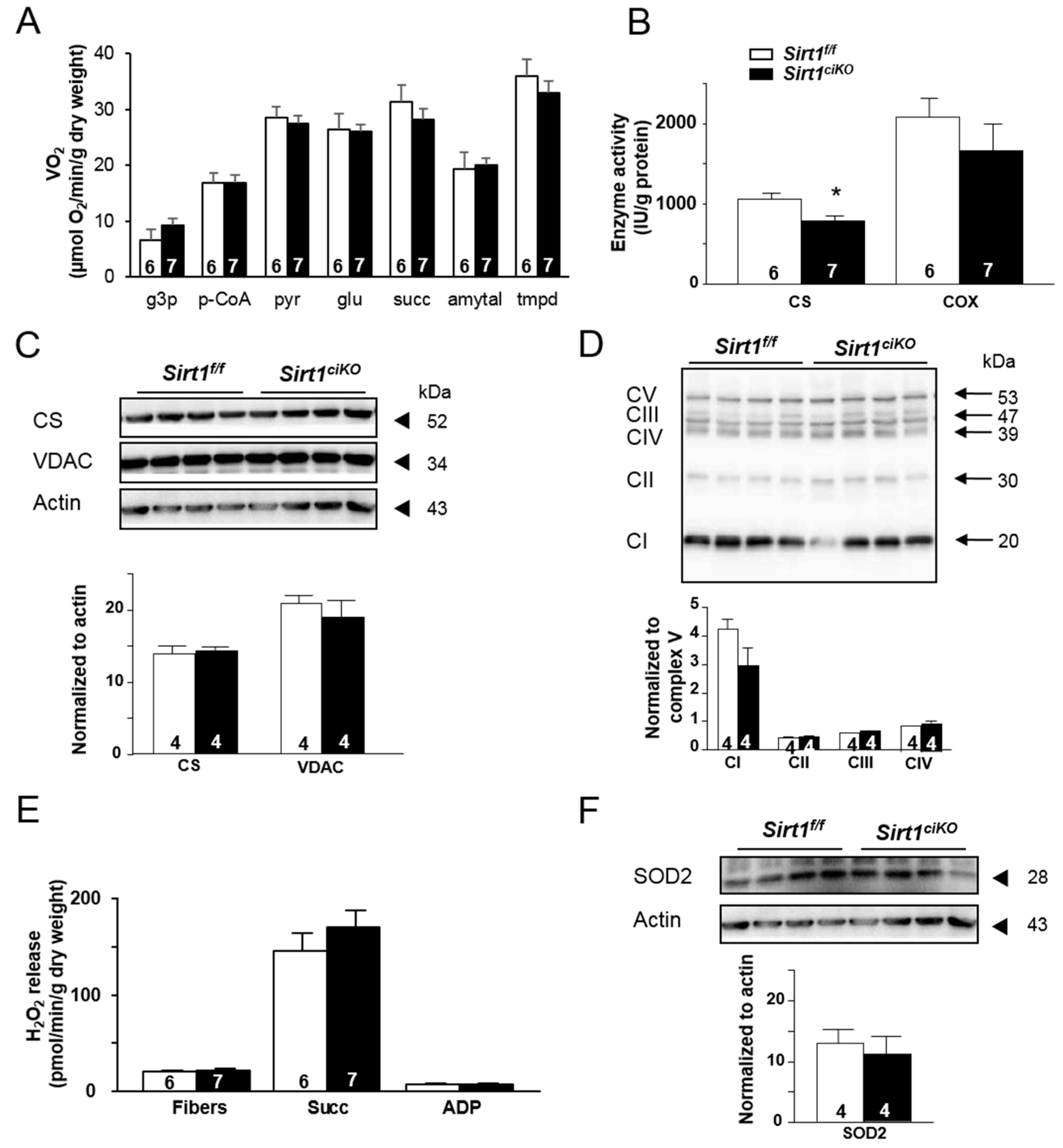
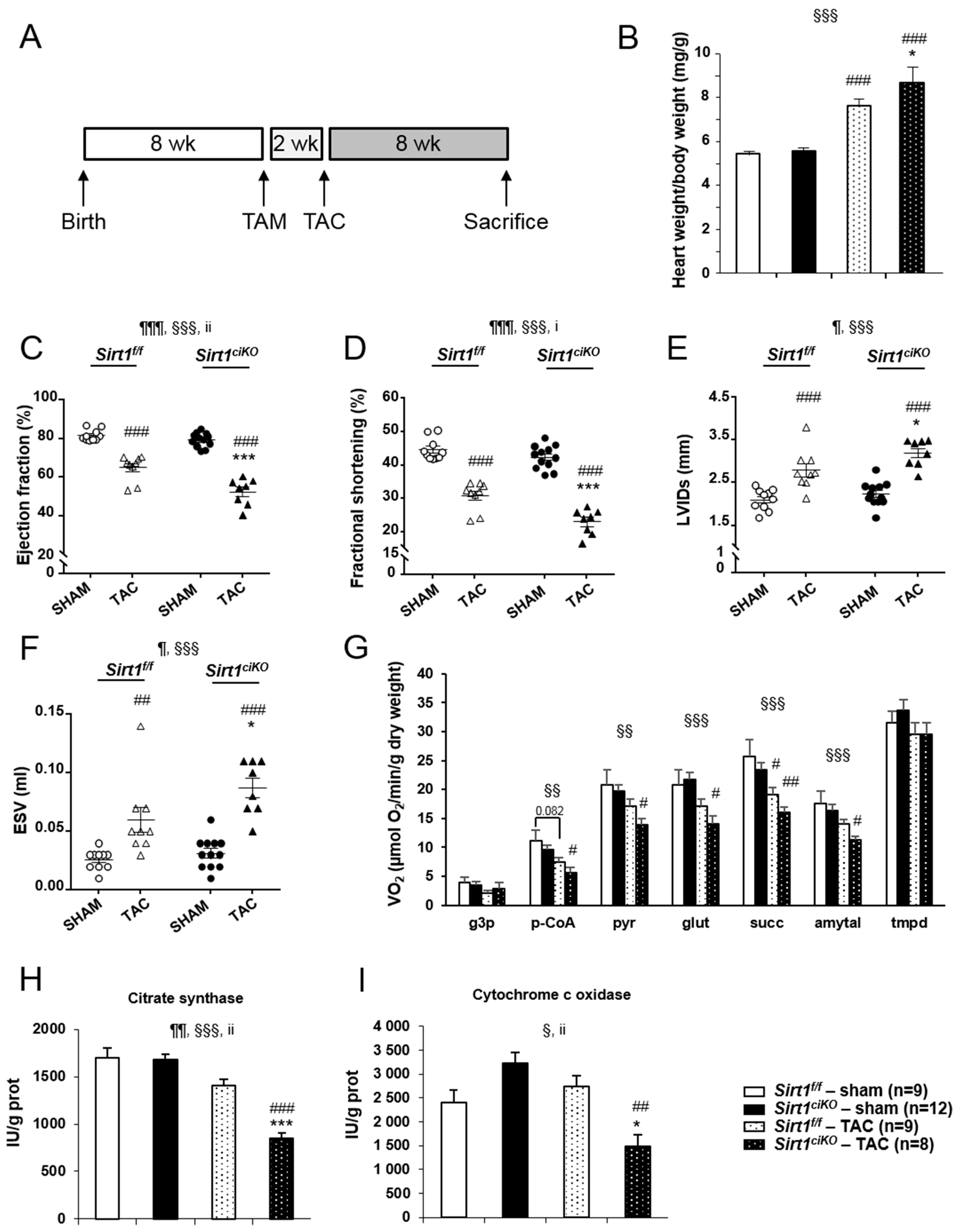
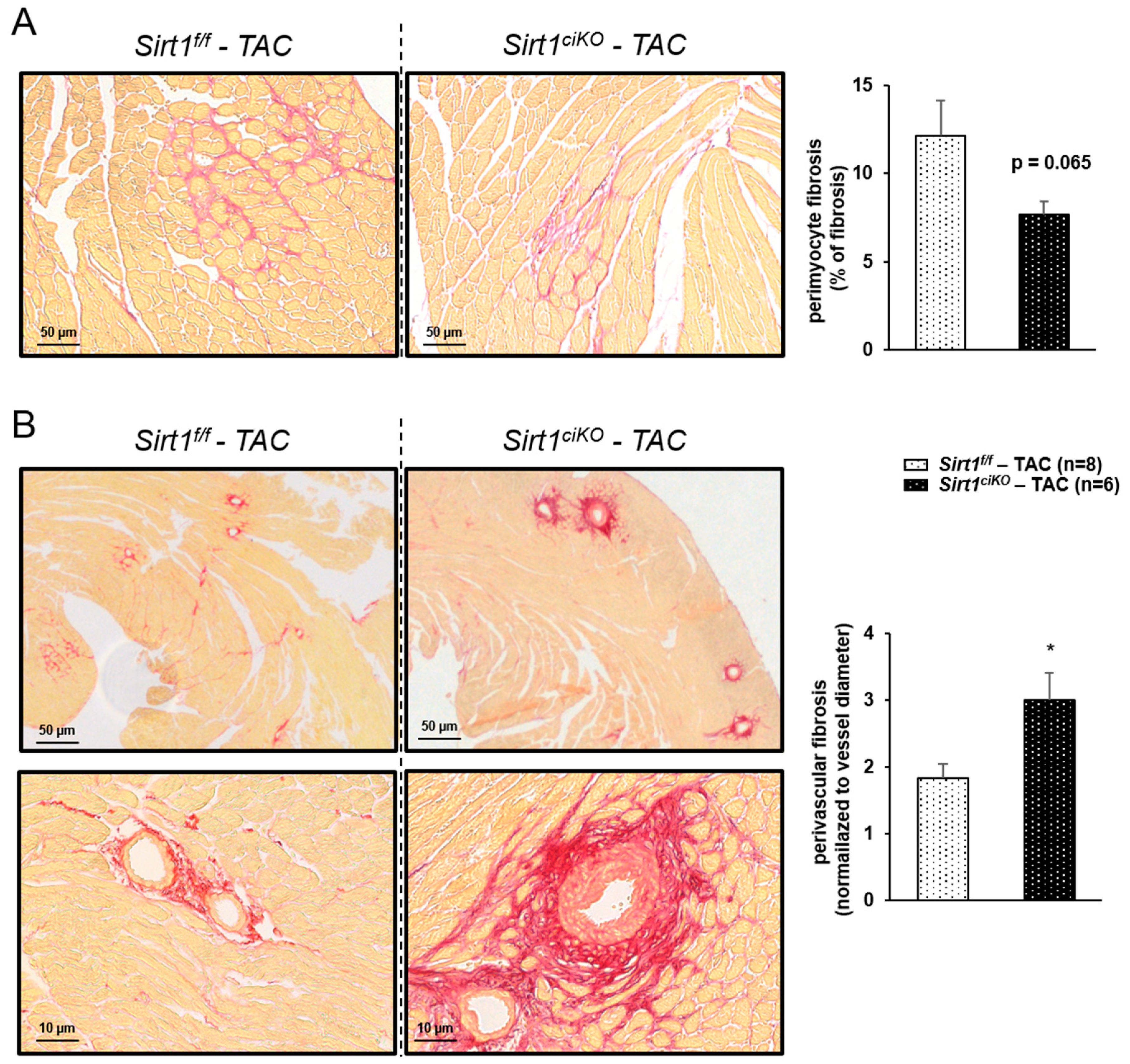
2.4. Sirt1ciKO Mice Are More Sensitive to Cardiac Pressure Overload
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Echocardiography
4.3. Ventricular Cardiomyocyte Isolation
4.4. Mitochondrial Functional Assays in Permeabilized Cardiac Fibers
4.5. Mitochondrial Respiration
4.6. Mitochondrial H2O2 Release
4.7. Enzyme Activity
4.8. Immunoblotting
4.9. Histological Analysis
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HF | Heart failure |
| CVD | Cardiovascular Diseases |
| SIRT | Sirtuin |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1α |
| MHC | Myosin heavy chain |
| TAC | Tranverse aortic constriction |
| LV | Left ventricle |
| CS | Citrate synthase |
| COX | Cytochrome c oxidase |
| p53 | Tumour suppressor p53 protein |
| H1 | Histone H1 |
| FoxO1 | Forkhead box protein O1 |
| LVEF | Left ventricular ejection fraction |
| LVFS | Left ventricular fractional shortening |
| LVPWs | End-systolic left ventricular posterior wall thickness |
| LVIDs | End-systolic left ventricular internal diameter |
| ESV | End-systolic volume |
| IVSs | End-systolic interventricular spetal thickness |
| HW | Heart weight |
| BW | Body weight |
| TL | Tibia length |
| AK | Adenylate kinase |
| CK | Creatine kinase |
| VDAC | Voltage-dependent anion channel |
| AMPK | AMP-activated protein kinase |
| ACC | Acetyl-Coa carboxylase |
| ROS | Reactive oxygen species |
| ER | Endoplasmic reticulum |
References
- Neubauer, S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Li, X. SIRT1 and energy metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Rogina, B.; Helfand, S.L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef]
- Tanno, M.; Kuno, A.; Horio, Y.; Miura, T. Emerging beneficial roles of sirtuins in heart failure. Basic Res. Cardiol. 2012, 107, 273. [Google Scholar] [CrossRef]
- Matsushima, S.; Sadoshima, J. The role of sirtuins in cardiac disease. Am. J. Physiol. 2015, 309, H1375–H1389. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Patten, I.S.; Arany, Z. PGC-1 coactivators in the cardiovascular system. Trends Endocrinol. Metab. 2012, 23, 90–97. [Google Scholar] [CrossRef]
- Garnier, A.; Fortin, D.; Delomenie, C.; Momken, I.; Veksler, V.; Ventura-Clapier, R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 2003, 551, 491–501. [Google Scholar] [CrossRef]
- Sihag, S.; Cresci, S.; Li, A.Y.; Sucharov, C.C.; Lehman, J.J. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J. Mol. Cell. Cardiol. 2009, 46, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease With Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1alpha. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox. Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef] [PubMed]
- Gorski, P.A.; Jang, S.P.; Jeong, D.; Lee, A.; Lee, P.; Oh, J.G.; Chepurko, V.; Yang, D.K.; Kwak, T.H.; Eom, S.H.; et al. Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca(2+)-ATPase in Heart Failure. Circ. Res. 2019, 124, e63–e80. [Google Scholar] [CrossRef] [PubMed]
- Bugyei-Twum, A.; Ford, C.; Civitarese, R.; Seegobin, J.; Advani, S.L.; Desjardins, J.F.; Kabir, G.; Zhang, Y.; Mitchell, M.; Switzer, J.; et al. Sirtuin 1 activation attenuates cardiac fibrosis in a rodent pressure overload model by modifying Smad2/3 transactivation. Cardiovasc. Res. 2018, 114, 1629–1641. [Google Scholar] [CrossRef]
- Alcendor, R.R.; Kirshenbaum, L.A.; Imai, S.; Vatner, S.F.; Sadoshima, J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res. 2004, 95, 971–980. [Google Scholar] [CrossRef]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Oka, S.; Alcendor, R.; Zhai, P.; Park, J.Y.; Shao, D.; Cho, J.; Yamamoto, T.; Tian, B.; Sadoshima, J. PPARalpha-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab. 2011, 14, 598–611. [Google Scholar] [CrossRef]
- Oka, S.; Zhai, P.; Alcendor, R.; Park, J.Y.; Tian, B.; Sadoshima, J. Suppression of ERR targets by a PPARalpha/Sirt1 complex in the failing heart. Cell Cycle 2012, 11, 856–864. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pillai, J.B.; Isbatan, A.; Imai, S.; Gupta, M.P. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J. Biol. Chem. 2005, 280, 43121–43130. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.M.; Tsai, J.Y.; Chen, Y.C.; Huang, C.Y.; Hsu, H.L.; Weng, C.F.; Shih, C.C.; Hsu, C.P. Downregulation of Sirt1 as aging change in advanced heart failure. J. Biomed. Sci. 2014, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Akkafa, F.; Halil Altiparmak, I.; Erkus, M.E.; Aksoy, N.; Kaya, C.; Ozer, A.; Sezen, H.; Oztuzcu, S.; Koyuncu, I.; Umurhan, B. Reduced SIRT1 expression correlates with enhanced oxidative stress in compensated and decompensated heart failure. Redox Biol. 2015, 6, 169–173. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kawashima, T.; Inuzuka, Y.; Okuda, J.; Kato, T.; Niizuma, S.; Tamaki, Y.; Iwanaga, Y.; Kawamoto, A.; Narazaki, M.; Matsuda, T.; et al. Constitutive SIRT1 overexpression impairs mitochondria and reduces cardiac function in mice. J. Mol. Cell. Cardiol. 2011, 51, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Dominguez, E.; Navarro, M.; Vinciguerra, M.; Iglesias, R.; Giralt, M.; Lope-Piedrafita, S.; Ruberte, J.; Villarroya, F. Dilated cardiomyopathy and mitochondrial dysfunction in Sirt1-deficient mice: A role for Sirt1-Mef2 in adult heart. J. Mol. Cell. Cardiol. 2012, 53, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.E.; Smith, G.; Hall, J.E.; Stec, D.E. Systolic dysfunction in cardiac-specific ligand-inducible MerCreMer transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H253–H260. [Google Scholar] [CrossRef]
- Buerger, A.; Rozhitskaya, O.; Sherwood, M.C.; Dorfman, A.L.; Bisping, E.; Abel, E.D.; Pu, W.T.; Izumo, S.; Jay, P.Y. Dilated cardiomyopathy resulting from high-level myocardial expression of Cre-recombinase. J. Card Fail. 2006, 12, 392–398. [Google Scholar] [CrossRef]
- Bersell, K.; Choudhury, S.; Mollova, M.; Polizzotti, B.D.; Ganapathy, B.; Walsh, S.; Wadugu, B.; Arab, S.; Kuhn, B. Moderate and high amounts of tamoxifen in alphaMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis. Model. Mech. 2013, 6, 1459–1469. [Google Scholar] [CrossRef]
- Lexow, J.; Poggioli, T.; Sarathchandra, P.; Santini, M.P.; Rosenthal, N. Cardiac fibrosis in mice expressing an inducible myocardial-specific Cre driver. Dis. Model. Mech. 2013, 6, 1470–1476. [Google Scholar] [CrossRef]
- Wang, L.; Quan, N.; Sun, W.; Chen, X.; Cates, C.; Rousselle, T.; Zhou, X.; Zhao, X.; Li, J. Cardiomyocyte-specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc. Res. 2018, 114, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef]
- Maizel, J.; Xavier, S.; Chen, J.; Lin, C.H.; Vasko, R.; Goligorsky, M.S. Sirtuin 1 ablation in endothelial cells is associated with impaired angiogenesis and diastolic dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1691–H1704. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.N.; Han, L.; Dasgupta, C.; Huang, L.; Yang, S.; Zhang, L. SIRT1 increases cardiomyocyte binucleation in the heart development. Oncotarget 2018, 9, 7996–8010. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Predominant expression of Sir2alpha, an NAD-dependent histone deacetylase, in the embryonic mouse heart and brain. FEBS Lett. 2004, 556, 281–286. [Google Scholar] [CrossRef]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef]
- Lompre, A.M.; Nadal-Ginard, B.; Mahdavi, V. Expression of the cardiac ventricular alpha- and beta-myosin heavy chain genes is developmentally and hormonally regulated. J. Biol. Chem. 1984, 259, 6437–6446. [Google Scholar]
- Piquereau, J.; Novotova, M.; Fortin, D.; Garnier, A.; Ventura-Clapier, R.; Veksler, V.; Joubert, F. Postnatal development of mouse heart: Formation of energetic microdomains. J. Physiol. 2010, 588, 2443–2454. [Google Scholar] [CrossRef]
- Hsu, Y.J.; Hsu, S.C.; Hsu, C.P.; Chen, Y.H.; Chang, Y.L.; Sadoshima, J.; Huang, S.M.; Tsai, C.S.; Lin, C.Y. Sirtuin 1 protects the aging heart from contractile dysfunction mediated through the inhibition of endoplasmic reticulum stress-mediated apoptosis in cardiac-specific Sirtuin 1 knockout mouse model. Int. J. Cardiol. 2017, 228, 543–552. [Google Scholar] [CrossRef]
- Dai, D.F.; Chen, T.; Johnson, S.C.; Szeto, H.; Rabinovitch, P.S. Cardiac aging: From molecular mechanisms to significance in human health and disease. Antioxid. Redox Signal. 2012, 16, 1492–1526. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Sadoshima, J. Protection of the heart against ischemia/reperfusion by silent information regulator 1. Trends Cardiovasc. Med. 2011, 21, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Jian, Z.; Zhu, Y.; Zhu, Y.; Chen, B.; Ma, R.; Tang, F.; Xiao, Y. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. Int. J. Mol. Med. 2019, 43, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- Prola, A.; Silva, J.P.; Guilbert, A.; Lecru, L.; Piquereau, J.; Ribeiro, M.; Mateo, P.; Gressette, M.; Fortin, D.; Boursier, C.; et al. SIRT1 protects the heart from ER stress-induced cell death through eIF2alpha deacetylation. Cell Death Differ. 2016, 24, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Moulin, M.; Zurlo, G.; Mateo, P.; Gressette, M.; Paul, J.L.; Lemaire, C.; Ventura-Clapier, R.; Veksler, V.; Garnier, A. Cobalamin and folate protect mitochondrial and contractile functions in a murine model of cardiac pressure overload. J. Mol. Cell. Cardiol. 2017, 102, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Heath, D.; Edwards, J.E. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation 1958, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.; Gupta, M.P. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 2011, 4, ra46. [Google Scholar] [CrossRef]
- Waldman, M.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Kornwoski, R.; Aravot, D.; Arad, M.; Hochhauser, E. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1alpha axis. Exp. Cell Res. 2018, 373, 112–118. [Google Scholar] [CrossRef]
- Geng, B.; Cai, Y.; Gao, S.; Lu, J.; Zhang, L.; Zou, J.; Liu, M.; Yu, S.; Ye, J.; Liu, P. PARP-2 knockdown protects cardiomyocytes from hypertrophy via activation of SIRT1. Biochem. Biophys. Res. Commun. 2013, 430, 944–950. [Google Scholar] [CrossRef]
- Sohal, D.S.; Nghiem, M.; Crackower, M.A.; Witt, S.A.; Kimball, T.R.; Tymitz, K.M.; Penninger, J.M.; Molkentin, J.D. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 2001, 89, 20–25. [Google Scholar] [CrossRef]
- Feil, S.; Valtcheva, N.; Feil, R. Inducible Cre mice. Methods Mol. Biol. 2009, 530, 343–363. [Google Scholar] [PubMed]
- Shioya, T. A simple technique for isolating healthy heart cells from mouse models. J. Physiol. Sci. 2007, 57, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Veksler, V.; Gellerich, F.N.; Saks, V.; Margreiter, R.; Kunz, W.S. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat. Protoc. 2008, 3, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Godin, R.; Deschenes, S.; Bessi, V.L.; Mofarrahi, M.; Hussain, S.N.; Burelle, Y. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy 2013, 9, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Wharton, D.C.; Tzagoloff, A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 1967, 10, 245–250. [Google Scholar]
- Veksler, V.I.; Kuznetsov, A.V.; Anflous, K.; Mateo, P.; van Deursen, J.; Wieringa, B.; Ventura-Clapier, R. Muscle creatine kinase-deficient mice. II. Cardiac and skeletal muscles exhibit tissue-specific adaptation of the mitochondrial function. J. Biol. Chem. 1995, 270, 19921–19929. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 14 Weeks | 11 Months | 2-Way ANOVA | |||
|---|---|---|---|---|---|
| Sirt1f/f | Sirt1ciKO | Sirt1f/f | Sirt1ciKO | ||
| BW (g) | 30.4 ± 0.4 | 29.9 ± 0.3 | 33.4 ± 1.5 $$ | 29.6 ± 0.6 ** | ¶¶, i |
| TL (mm) | 17.2 ±0.2 | 17.2 ± 0.1 | 18.8 ± 0.3 $$ | 18.6 ± 0.7 $ | §§§ |
| HW (mg) | 143.2 ± 4.4 | 144.8 ± 3.1 | 173.6 ± 3.9 $$ | 162.6 ± 10.4 $ | §§§ |
| HW/BW (mg/g) | 4.7 ± 0.1 | 4.8 ± 0.1 | 5.2 ± 0.2 | 5.5 ± 0.3 $ | §§ |
| HW/TL (mg/mm) | 8.3 ± 0.2 | 8.6 ± 0.1 | 9.2 ± 0.2 | 8.8 ± 0.2 | |
| LW/BW (mg/g) | 4.5 ± 0.1 | 4.9 ± 0.2 | 5.2 ± 0.2 $$ | 5.3 ± 0.1 | §§§ |
| LW/TL (mg/mm) | 8.1 ± 0.2 | 8.6 ± 0.3 | 9.3 ± 0.5 | 8.7 ± 0.4 | / |
| KW/BW (mg/g) | 11.8 ± 0.2 | 12.1 ± 0.3 | 12.6 ± 0.9 | 12.2 ± 0.6 | / |
| KW/TL (mg/mm) | 20.9 ± 0.4 | 21.5 ± 0.5 | 22.2 ± 0.9 | 18.7 ± 1.2 $,* | i |
| HR (bpm) | 506 ± 7 | 498 ± 11 | 542 ± 13 | 549 ± 16 | §§ |
| IVSd (mm) | 1.04 ± 0.04 | 0.93 ± 0.03 | 0.82 ± 0.12 | 0.82 ± 0.07 | §§ |
| IVSs (mm) | 1.64 ± 0.03 | 1.44 ± 0.03 ** | 1.44 ± 0.06 $$ | 1.28 ± 0.03 $,* | ¶¶¶, §§§ |
| LVIDd (mm) | 3.36 ± 0.06 | 3.39 ± 0.06 | 3.82 ± 0.15 $ | 3.81 ± 0.13 $$ | §§§ |
| LVIDs (mm) | 1.78 ± 0.04 | 2.06 ± 0.06 * | 2.09 ± 0.12$ | 2.58 ± 0.12 $$$,*** | ¶¶¶, §§§ |
| LVPWd (mm) | 0.95 ± 0.04 | 0.81 ± 0.05 | 0.81 ± 0.04 | 0.84 ± 0.11 | / |
| LVPWs (mm) | 1.55 ± 0.06 | 1.28 ± 0.06 | 1.53 ± 0.11 | 1.27 ± 0.02 | ¶¶ |
| EDV (mL) | 0.098 ± 0.005 | 0.102 ± 0.006 | 0.142 ± 0.016 $ | 0.14 ± 0.014 $$ | §§§ |
| ESV (mL) | 0.016 ± 0.001 | 0.024 ± 0.002 * | 0.025 ± 0.004 $ | 0.046 ± 0.006 $$$,*** | ¶¶¶, §§§, i |
| LVEF (%) | 84.1 ± 0.5 | 76.3 ± 1.1 * | 82.6 ± 1.4 | 66.7 ± 5 $$$,*** | ¶¶¶, §§§, i |
| LVFS (%) | 46.9 ± 0.6 | 39.4 ± 1 ** | 45.6 ± 1.5 | 32.2 ± 3.3 $$$,*** | ¶¶¶, §§§, ii |
| SV (mL) | 0.083 ± 0.005 | 0.77 ± 0.004 | 0.117 ± 0.012 $$ | 0.094 ± 0.013 $,* | §§ |
| LV (mg) | 109 ± 6 | 94 ± 7 | 98 ± 5 | 102 ± 14 | / |
| CO (mL/min) | 42.0 ± 2.4 | 38.0 ± 2.1 | 63.3 ± 6.9 $$ | 51.1 ± 5.8 $,* | ¶, §§§ |
| Sirt1f/f | Sirt1ciKO | 2 Way ANOVA | |||
|---|---|---|---|---|---|
| SHAM | TAC | SHAM | TAC | ||
| BW (g) | 28.3 ± 0.4 | 28.5 ± 0.7 | 28.7 ± 0.5 | 28.7 ± 0.8 | / |
| TL (mm) | 17.6 ±0.1 | 17.8 ± 0.1 | 17.6 ± 0.1 | 18.2 ± 0.1 | / |
| HW (mg) | 153.4 ± 4.4 | 217.7 ± 12.8 $ | 158.9 ± 4.0 | 249.6 ± 24.7 $$$ | §§§ |
| HW/BW (mg/g) | 5.4 ± 0.1 | 7.6 ± 0.3 $$$ | 5.5 ± 0.1 | 8.7 ± 0.7 $$$,* | §§§ |
| HW/TL (mg/mm) | 8.7 ± 0.2 | 12.2 ± 0.7 $$ | 9.0 ± 0.3 | 13.7 ± 1.3$$$ | §§§ |
| LW/BW (mg/g) | 5.2 ± 0.1 | 5.4 ± 0.2 | 5.1 ± 0.1 | 6.2 ± 0.9 | / |
| LW/TL (mg/mm) | 8.3 ± 0.2 | 8.7 ± 0.5 | 8.4 ± 0.2 | 9.9 ± 1.6 | / |
| KW/BW (mg/g) | 12..3 ± 0.3 | 11.2 ± 0.3 $ | 12.1 ± 0.2 | 11.5 ± 0.3 | § |
| KW/TL (mg/mm) | 19.8 ± 0.6 | 17.9 ± 0.6 | 19.7 ± 0.6 | 18.2 ± 0.7 | § |
| HR (bpm) | 542 ± 19 | 541 ± 13 | 514 ± 23 | 543 ± 14 | / |
| IVSd (mm) | 0.72 ± 0.05 | 0.93 ± 0.07 | 0.71 ± 0.04 | 0.83 ± 0.04 | §§ |
| IVSs (mm) | 1.36 ± 0.05 | 1.39 ± 0.07 | 1.34 ± 0.03 | 1.21 ± 0.04 * | / |
| LVIDd (mm) | 3.79 ± 0.09 | 4.03 ± 0.17 | 3.88 ± 0.1 | 4.14 ± 0.11 | § |
| LVIDs (mm) | 2.11 ± 0.08 | 2.79 ± 0.16 $$$ | 2.24 ± 0.08 | 3.19 ± 0.11 $$$,* | ¶, §§§ |
| LVPWd (mm) | 0.71 ± 0.03 | 0.81 ± 0.08 | 0.67 ± 0.03 | 0.87 ± 0.04$ | §§ |
| LVPWs (mm) | 1.25 ± 0.04 | 1.23 ± 0.06 | 1.21 ± 0.04 | 1.14 ± 0.02 | / |
| EDV (mL) | 0.140 ± 0.009 | 0.152 ± 0.014 | 0.149 ± 0.011 | 0.179 ± 0.012 | / |
| ESV (mL) | 0.026 ± 0.003 | 0.061 ± 0.011 $$ | 0.031 ± 0.003 | 0.086 ± 0.008 $$$,* | ¶, §§§ |
| LVEF (%) | 81.7 ± 0.9 | 65.0 ± 2.2 $$$ | 79.5 ± 1.1 | 52.5 ± 2.4 $$$,*** | ¶¶¶, §§§, ii |
| LVFS (%) | 44.5 ± 1.0 | 30.8 ± 1.4 $$$ | 42.4 ± 1.0 | 23.0 ± 1.3 $$$,*** | ¶¶¶, §§§, i |
| SV (mL) | 0.11 ± 0.1 | 0.11 ± 0.01 | 0.12 ±0.01 | 0.09 ± 0.01 | / |
| LV (mg) | 92 ± 4 | 136 ± 17 $$ | 91 ± 5 | 135 ± 10 $$$ | §§§ |
| CO (mL/min) | 61.3 ± 3.9 | 54.3 ± 5.3 | 60 ± 4.6 | 50.8 ± 4 | / |
| Antibody | Company | Catalog No | Dilution |
|---|---|---|---|
| SIRT1 | Abcam | ab110304 | 1000 |
| Ac-H1 | Sigma | H7789 | 1000 |
| FoxO1 | Cell signaling | 2880S | 250 |
| Ac-FoxO1 | Santa Cruz | sc49437 | 1000 |
| Ac-p53 | Cell signaling | 2570 | 250 |
| CS | Abcam | ab96600 | 1000 |
| VDAC1 | Cell signaling | 4866 | 1000 |
| ACC | Cell signaling | 3676 | 1000 |
| pACC | Cell signaling | 3661 | 1000 |
| AMPK | Cell signaling | 2532 | 500 |
| pAMPK | Cell signaling | 2531 | 500 |
| SOD2 | Abcam | ab16956 | 500 |
| Total OXPHOS | MitoSciences | MS604 | 250 |
| Actin | Santa Cruz | SC-8432 | 200 |
| Tubulin | Sigma | T6199 | 1000 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanz, M.-N.; Grimbert, L.; Moulin, M.; Gressette, M.; Rucker-Martin, C.; Lemaire, C.; Mericskay, M.; Veksler, V.; Ventura-Clapier, R.; Garnier, A.; et al. Inducible Cardiac-Specific Deletion of Sirt1 in Male Mice Reveals Progressive Cardiac Dysfunction and Sensitization of the Heart to Pressure Overload. Int. J. Mol. Sci. 2019, 20, 5005. https://doi.org/10.3390/ijms20205005
Sanz M-N, Grimbert L, Moulin M, Gressette M, Rucker-Martin C, Lemaire C, Mericskay M, Veksler V, Ventura-Clapier R, Garnier A, et al. Inducible Cardiac-Specific Deletion of Sirt1 in Male Mice Reveals Progressive Cardiac Dysfunction and Sensitization of the Heart to Pressure Overload. International Journal of Molecular Sciences. 2019; 20(20):5005. https://doi.org/10.3390/ijms20205005
Chicago/Turabian StyleSanz, Maria-Nieves, Lucile Grimbert, Maryline Moulin, Mélanie Gressette, Catherine Rucker-Martin, Christophe Lemaire, Mathias Mericskay, Vladimir Veksler, Renée Ventura-Clapier, Anne Garnier, and et al. 2019. "Inducible Cardiac-Specific Deletion of Sirt1 in Male Mice Reveals Progressive Cardiac Dysfunction and Sensitization of the Heart to Pressure Overload" International Journal of Molecular Sciences 20, no. 20: 5005. https://doi.org/10.3390/ijms20205005
APA StyleSanz, M.-N., Grimbert, L., Moulin, M., Gressette, M., Rucker-Martin, C., Lemaire, C., Mericskay, M., Veksler, V., Ventura-Clapier, R., Garnier, A., & Piquereau, J. (2019). Inducible Cardiac-Specific Deletion of Sirt1 in Male Mice Reveals Progressive Cardiac Dysfunction and Sensitization of the Heart to Pressure Overload. International Journal of Molecular Sciences, 20(20), 5005. https://doi.org/10.3390/ijms20205005