Thrombogenic Risk Induced by Intravascular Mesenchymal Stem Cell Therapy: Current Status and Future Perspectives


{kind=link}
Abstract
1. Introduction
2. Hemostasis
2.1. Platelet Activation
2.2. The Coagulation
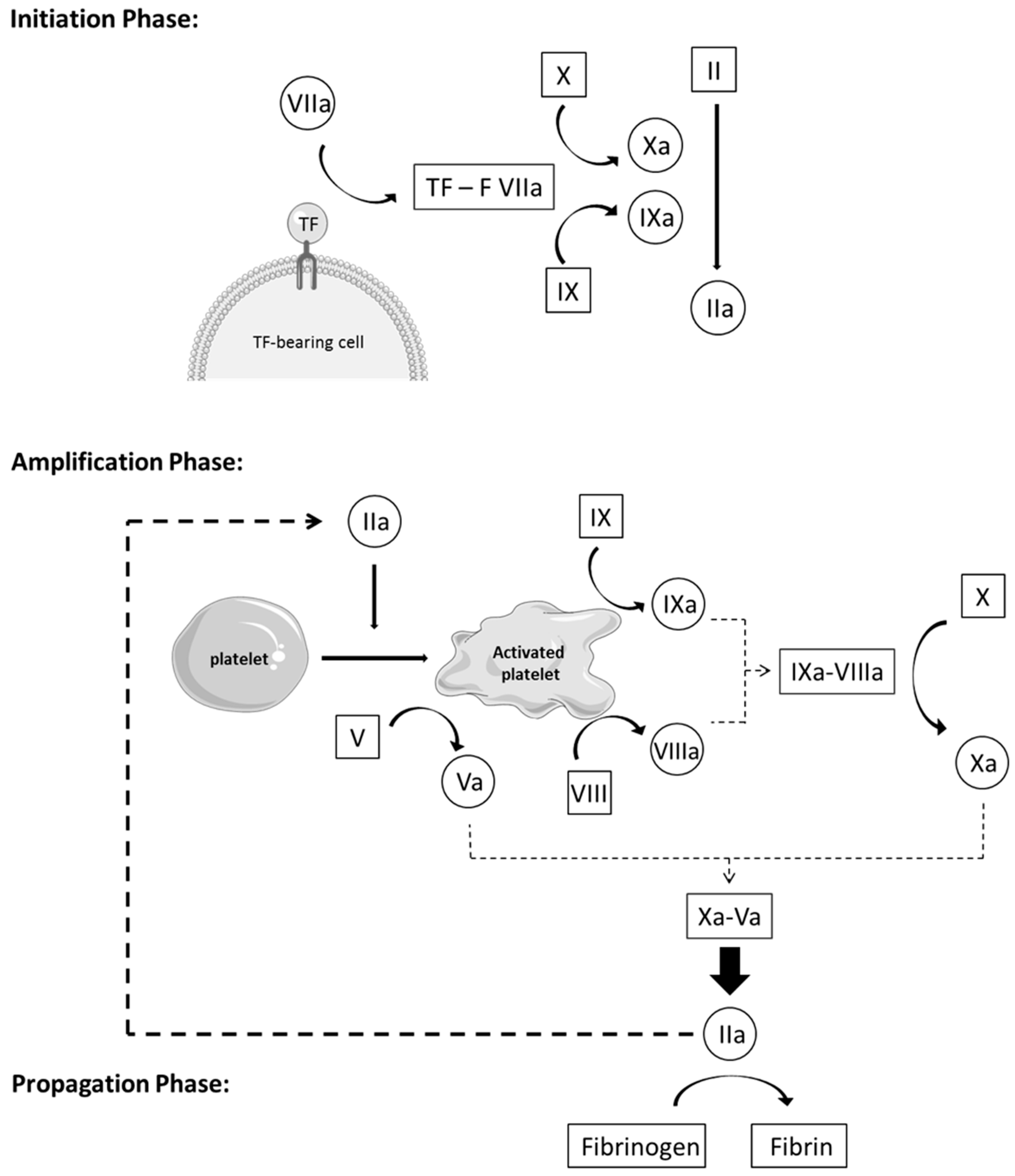
- Initiation phase: Coagulation is activated by TF through the extrinsic pathway, forming, with factor VIIa, an essential enzymatic complex, factor VIIa-TF. This complex is the fuse that triggers coagulation, through activation of factor IX and X, generating only a small amount of thrombin (factor IIa). This initiation phase, nevertheless, is inefficient because pro-cofactor V and VIII are not yet in their most active form.
- Amplification phase: The small amount of thrombin formed during the initiation phase induces an important amplification loop via two distinct mechanisms. Thrombin increases platelet activation and adhesion through platelet agonist properties and induces the conversion of cofactor V and VIII in their active form, Va and VIIIa. Factor V is released from the alpha granules and factor VIII is released from its carrier, vWF, to be activated on the negatively charged platelet surface. Thrombin activates factor IX as well, present on the platelet surface, which forms, with cofactor VIIIa, a tenase complex (IXa-VIIIa), which converts inactivated factor X into its active form. In turn, activated factor Xa forms, with cofactor Va, the prothrombinase complex (Xa-Va), which converts prothrombin [18] into a large amount of thrombin (IIa), leading to an efficient burst of all enzymatic reactions of the whole cascade.
- Propagation phase: Activation of platelets and the intrinsic pathway leads to the formation of massive amounts of thrombin, which is the principal driving force of coagulation. In turn, these vast amounts of thrombin lead to the production of large quantities of fibrin strains—the final product of coagulation—which consolidates the blood clot [11,19,20,21].
3. Tissue Factor
4. Cross-Talk Pathways between Coagulation and Inflammation
5. Cell-Based Therapy
6. Procoagulant Activity
6.1. Islet and Hepatocyte Transplantation
6.2. Mesenchymal Stem Cell Transplantation
6.2.1. In vitro Expression of TF and Procoagulant Activity
6.2.2. MSCs Infusion in Animal Models
6.2.3. MSCs Infusion in Patients
6.2.4. MSCs and IBMIR
7. Procoagulant Activity Modulation
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moll, G.; Ankrum, J.A.; Kamhieh-Milz, J.; Bieback, K.; Ringden, O.; Volk, H.D.; Geissler, S.; Reinke, P. Intravascular Mesenchymal Stromal/Stem Cell Therapy Product Diversification: Time for New Clinical Guidelines. Trends Mol. Med. 2019, 25, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Moll, G.; Rasmusson-Duprez, I.; von Bahr, L.; Connolly-Andersen, A.M.; Elgue, G.; Funke, L.; Hamad, O.A.; Lonnies, H.; Magnusson, P.U.; Sanchez, J.; et al. Are therapeutic human mesenchymal stromal cells compatible with human blood? Stem Cells (Dayt. Ohio) 2012, 30, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.W.; Kwon, M.; Choi, J.C.; Shin, J.W.; Park, I.W.; Choi, B.W.; Kim, J.Y. Familial occurrence of pulmonary embolism after intravenous, adipose tissue-derived stem cell therapy. Yonsei Med. J. 2013, 54, 1293–1296. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, S.; Zhou, L.; Cai, J.; Tan, J.; Gao, X.; Zeng, Z.; Li, D. Thromboembolism Induced by Umbilical Cord Mesenchymal Stem Cell Infusion: A Report of Two Cases and Literature Review. Transplant. Proc. 2017, 49, 1656–1658. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M.; Stephenne, X.; Ottolenghi, C.; Jazouli, N.; Clapuyt, P.; Lacaille, F.; Najimi, M.; de Lonlay, P.; Smets, F. Liver engraftment and repopulation by in vitro expanded adult derived human liver stem cells in a child with ornithine carbamoyltransferase deficiency. JIMD Rep. 2014, 13, 65–72. [Google Scholar] [CrossRef]
- Melmed, G.Y.; Pandak, W.M.; Casey, K.; Abraham, B.; Valentine, J.; Schwartz, D.; Awais, D.; Bassan, I.; Lichtiger, S.; Sands, B.; et al. Human Placenta-derived Cells (PDA-001) for the Treatment of Moderate-to-severe Crohn’s Disease: A Phase 1b/2a Study. Inflamm. Bowel Dis. 2015, 21, 1809–1816. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Strange, C.; Nietert, P.J.; Wang, J.; Turnbull, T.L.; Cloud, C.; Owczarski, S.; Shuford, B.; Duke, T.; Gilkeson, G.; et al. Autologous Mesenchymal Stem Cell and Islet Cotransplantation: Safety and Efficacy. Stem Cells Transl. Med. 2018, 7, 11–19. [Google Scholar] [CrossRef]
- Perlee, D.; van Vught, L.A.; Scicluna, B.P.; Maag, A.; Lutter, R.; Kemper, E.M.; van ’t Veer, C.; Punchard, M.A.; Gonzalez, J.; Richard, M.P.; et al. Intravenous Infusion of Human Adipose Mesenchymal Stem Cells Modifies the Host Response to Lipopolysaccharide in Humans: A Randomized, Single-Blind, Parallel Group, Placebo Controlled Trial. Stem Cells (Dayt. Ohio) 2018, 36, 1778–1788. [Google Scholar] [CrossRef]
- Dubois, C.; Panicot-Dubois, L.; Merrill-Skoloff, G.; Furie, B.; Furie, B.C. Glycoprotein VI-dependent and -independent pathways of thrombus formation in vivo. Blood 2006, 107, 3902–3906. [Google Scholar] [CrossRef]
- Mangin, P.; Yap, C.L.; Nonne, C.; Sturgeon, S.A.; Goncalves, I.; Yuan, Y.; Schoenwaelder, S.M.; Wright, C.E.; Lanza, F.; Jackson, S.P. Thrombin overcomes the thrombosis defect associated with platelet GPVI/FcRgamma deficiency. Blood 2006, 107, 4346–4353. [Google Scholar] [CrossRef]
- Furie, B.; Furie, B.C. Mechanisms of thrombus formation. N. Engl. J. Med. 2008, 359, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.H.; Sim, E.H.; Goh, R.Y.; Park, J.I.; Han, J.Y. Platelet Activation: The Mechanisms and Potential Biomarkers. Biomed Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.M.; Samy, K.P.; Lowe, M.C.; Thompson, P.W.; Cano, J.; Farris, A.B.; Song, M.; Dove, C.R.; Leopardi, F.V.; Strobert, E.A.; et al. Dual islet transplantation modeling of the instant blood-mediated inflammatory reaction. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2015, 15, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Fager, A.M.; Wood, J.P.; Bouchard, B.A.; Feng, P.; Tracy, P.B. Properties of Procoagulant Platelets. Defin. Charact. Subpopul. Bind. A Funct. Prothrombinase 2010, 30, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Lhermusier, T.; Chap, H.; Payrastre, B. Platelet membrane phospholipid asymmetry: From the characterization of a scramblase activity to the identification of an essential protein mutated in Scott syndrome. J. Thromb. Haemost. JTH 2011, 9, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.P.R.; Royston, D. Thrombin generation and its inhibition: A review of the scientific basis and mechanism of action of anticoagulant therapies. BJA Br. J. Anaesth. 2002, 88, 848–863. [Google Scholar] [CrossRef]
- Levi, M. Diagnosis and treatment of disseminated intravascular coagulation. Int. J. Lab. Hematol. 2014, 36, 228–236. [Google Scholar] [CrossRef]
- Tatsumi, K.; Ohashi, K.; Matsubara, Y.; Kohori, A.; Ohno, T.; Kakidachi, H.; Horii, A.; Kanegae, K.; Utoh, R.; Iwata, T.; et al. Tissue factor triggers procoagulation in transplanted mesenchymal stem cells leading to thromboembolism. Biochem. Biophys. Res. Commun. 2013, 431, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.; Monroe, D.M., 3rd. A cell-based model of hemostasis. Thromb. Haemost. 2001, 85, 958–965. [Google Scholar]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Bhaskar, A. Classic theory of haemostasis Cell based model of haemostasis. Curr. Med. Issues J. Cmc Vellore 2016, 14, 5. [Google Scholar]
- Butenas, S. Tissue factor structure and function. Scientifica 2012, 2012, 964862. [Google Scholar] [CrossRef] [PubMed]
- Lwaleed, B.A.; Cooper, A.J.; Voegeli, D.; Getliffe, K. Tissue factor: A critical role in inflammation and cancer. Biol. Res. Nurs. 2007, 9, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Spronk, H.M.; ten Cate, H.; van der Meijden, P.E. Differential roles of tissue factor and phosphatidylserine in activation of coagulation. Thromb. Res. 2014, 133 (Suppl. 1), S54–S56. [Google Scholar] [CrossRef]
- Chu, A.J. Tissue factor, blood coagulation, and beyond: An overview. Int. J. Inflamm. 2011, 2011, 367284. [Google Scholar] [CrossRef]
- Foley, J.H.; Conway, E.M. Cross Talk Pathways Between Coagulation and Inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M. From hepatocytes to stem and progenitor cells for liver regenerative medicine: Advances and clinical perspectives. Cell Prolif. 2011, 44 (Suppl. 1), 39–43. [Google Scholar] [CrossRef]
- Stephenne, X.; Najimi, M.; Ngoc, D.K.; Smets, F.; Hue, L.; Guigas, B.; Sokal, E.M. Cryopreservation of human hepatocytes alters the mitochondrial respiratory chain complex 1. Cell Transplant. 2007, 16, 409–419. [Google Scholar] [CrossRef]
- Kojayan, G.G.; Alexander, M.; Imagawa, D.K.; Lakey, J.R.T. Systematic review of islet cryopreservation. Islets 2018, 10, 40–49. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Barry, F.P.; Murphy, J.M. Mesenchymal stem cells: Clinical applications and biological characterization. Int. J. Biochem. Cell Biol. 2004, 36, 568–584. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shen, S.; Fu, H.; Wang, Z.; Li, X.; Sui, X.; Yuan, M.; Liu, S.; Wang, G.; Guo, Q. Immunomodulatory Functions of Mesenchymal Stem Cells in Tissue Engineering. Stem Cells Int. 2019, 2019, 9671206. [Google Scholar] [CrossRef] [PubMed]
- Ankrum, J.A.; Ong, J.F.; Karp, J.M. Mesenchymal stem cells: Immune evasive, not immune privileged. Nat. Biotechnol. 2014, 32, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, S.; Shi, X.; Cao, H.; Li, L. A pooled analysis of mesenchymal stem cell-based therapy for liver disease. Stem Cell Res. Ther. 2018, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Wu, D.B.; Chen, B.; Chen, E.Q.; Tang, H. Progress in mesenchymal stem cell-based therapy for acute liver failure. Stem Cell Res. Ther. 2018, 9, 227. [Google Scholar] [CrossRef] [PubMed]
- Frederik, N.; Thierry, G.; Pierre-François, L.; Luc, L.; Enev, H.L.; Victor, V.; Virginie, B.; Clerget-Chossat, N.; Sokal, E. GS-16-Safety and tolerability of liver-derived stem cells (HepaStem) infused in patients with acute-on-chronic liver failure or acute decompensation: A European phase I/IIa open-labelled study. J. Hepatol. 2019, 70, e83. [Google Scholar] [CrossRef]
- Kurtz, A. Mesenchymal stem cell delivery routes and fate. Int. J. Stem Cells 2008, 1, 1–7. [Google Scholar] [CrossRef]
- Smets, F.; Dobbelaere, D.; McKiernan, P.; Dionisi-Vici, C.; Broue, P.; Jacquemin, E.; Lopes, A.I.; Goncalves, I.; Mandel, H.; Pawlowska, J.; et al. Phase I/II Trial of Liver Derived Mesenchymal Stem Cells in Pediatric Liver Based Metabolic Disorders: A Prospective, Open Label, Multicenter, Partially Randomized, Safety Study of One Cycle of Heterologous Human Adult Liver-Derived Progenitor Cells (HepaStem(R)) in Urea Cycle Disorders and Crigler-Najjar Syndrome patients. Transplantation 2019, 103, 1903–1915. [Google Scholar] [CrossRef]
- De Becker, A.; Riet, I.V. Homing and migration of mesenchymal stromal cells: How to improve the efficacy of cell therapy? World J. Stem Cells 2016, 8, 73–87. [Google Scholar] [CrossRef]
- Brown, C.; McKee, C.; Bakshi, S.; Walker, K.; Hakman, E.; Halassy, S.; Svinarich, D.; Dodds, R.; Govind, C.K.; Chaudhry, G.R. Mesenchymal stem cells: Cell therapy and regeneration potential. J. Tissue Eng. Regen. Med. 2019, 13, 1738–1755. [Google Scholar] [CrossRef]
- Walsh, T.J.; Eggleston, J.C.; Cameron, J.L. Portal hypertension, hepatic infarction, and liver failure complicating pancreatic islet autotransplantation. Surgery 1982, 91, 485–487. [Google Scholar] [PubMed]
- Shapiro, A.M.; Lakey, J.R.; Rajotte, R.V.; Warnock, G.L.; Friedlich, M.S.; Jewell, L.D.; Kneteman, N.M. Portal vein thrombosis after transplantation of partially purified pancreatic islets in a combined human liver/islet allograft. Transplantation 1995, 59, 1060–1063. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; Senior, P.; Salam, A.; Kin, T.; Imes, S.; Dinyari, P.; Malcolm, A.; Toso, C.; Nilsson, B.; Korsgren, O.; et al. Insulin-heparin infusions peritransplant substantially improve single-donor clinical islet transplant success. Transplantation 2010, 89, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Kin, T.; Kashkoush, S.; Gala-Lopez, B.; Bigam, D.L.; Kneteman, N.M.; Koh, A.; Senior, P.A.; Shapiro, A.M. Portal vein thrombosis is a potentially preventable complication in clinical islet transplantation. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2011, 11, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Bennet, W.; Groth, C.G.; Larsson, R.; Nilsson, B.; Korsgren, O. Isolated human islets trigger an instant blood mediated inflammatory reaction: Implications for intraportal islet transplantation as a treatment for patients with type 1 diabetes. Upsala J. Med. Sci. 2000, 105, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Liuwantara, D.; Chew, Y.V.; Favaloro, E.J.; Hawkes, J.M.; Burns, H.L.; O’Connell, P.J.; Hawthorne, W.J. Characterizing the Mechanistic Pathways of the Instant Blood-Mediated Inflammatory Reaction in Xenogeneic Neonatal Islet Cell Transplantation. Transplant. Direct 2016, 2, e77. [Google Scholar] [CrossRef] [PubMed]
- Moberg, L.; Johansson, H.; Lukinius, A.; Berne, C.; Foss, A.; Kallen, R.; Ostraat, O.; Salmela, K.; Tibell, A.; Tufveson, G.; et al. Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet (Lond. Engl.) 2002, 360, 2039–2045. [Google Scholar] [CrossRef]
- Naziruddin, B.; Iwahashi, S.; Kanak, M.A.; Takita, M.; Itoh, T.; Levy, M.F. Evidence for instant blood-mediated inflammatory reaction in clinical autologous islet transplantation. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2014, 14, 428–437. [Google Scholar] [CrossRef]
- Cabric, S.; Eich, T.; Sanchez, J.; Nilsson, B.; Korsgren, O.; Larsson, R. A new method for incorporating functional heparin onto the surface of islets of Langerhans. Tissue Eng. Part C Methods 2008, 14, 141–147. [Google Scholar] [CrossRef]
- Ozmen, L.; Ekdahl, K.N.; Elgue, G.; Larsson, R.; Korsgren, O.; Nilsson, B. Inhibition of thrombin abrogates the instant blood-mediated inflammatory reaction triggered by isolated human islets: Possible application of the thrombin inhibitor melagatran in clinical islet transplantation. Diabetes 2002, 51, 1779–1784. [Google Scholar] [CrossRef]
- Akima, S.; Hawthorne, W.J.; Favaloro, E.; Patel, A.; Blyth, K.; Mudaliar, Y.; Chapman, J.R.; O’Connell, P.J. Tirofiban and activated protein C synergistically inhibit the Instant Blood Mediated Inflammatory Reaction (IBMIR) from allogeneic islet cells exposure to human blood. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2009, 9, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Beuneu, C.; Vosters, O.; Ling, Z.; Pipeleers, D.; Pradier, O.; Goldman, M.; Verhasselt, V. N-Acetylcysteine derivative inhibits procoagulant activity of human islet cells. Diabetologia 2007, 50, 343–347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, J.; Sun, Z.; Gou, W.; Adams, D.B.; Cui, W.; Morgan, K.A.; Strange, C.; Wang, H. alpha-1 Antitrypsin Enhances Islet Engraftment by Suppression of Instant Blood-Mediated Inflammatory Reaction. Diabetes 2017, 66, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.; Ekdahl, K.N.; Korsgren, O. Control of instant blood-mediated inflammatory reaction to improve islets of Langerhans engraftment. Curr. Opin. Organ Transplant. 2011, 16, 620–626. [Google Scholar] [CrossRef]
- Berman, D.M.; Cabrera, O.; Kenyon, N.M.; Miller, J.; Tam, S.H.; Khandekar, V.S.; Picha, K.M.; Soderman, A.R.; Jordan, R.E.; Bugelski, P.J.; et al. Interference with tissue factor prolongs intrahepatic islet allograft survival in a nonhuman primate marginal mass model. Transplantation 2007, 84, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Lukinius, A.; Moberg, L.; Lundgren, T.; Berne, C.; Foss, A.; Felldin, M.; Kallen, R.; Salmela, K.; Tibell, A.; et al. Tissue factor produced by the endocrine cells of the islets of Langerhans is associated with a negative outcome of clinical islet transplantation. Diabetes 2005, 54, 1755–1762. [Google Scholar] [CrossRef]
- Gustafson, E.K.; Elgue, G.; Hughes, R.D.; Mitry, R.R.; Sanchez, J.; Haglund, U.; Meurling, S.; Dhawan, A.; Korsgren, O.; Nilsson, B. The instant blood-mediated inflammatory reaction characterized in hepatocyte transplantation. Transplantation 2011, 91, 632–638. [Google Scholar] [CrossRef]
- Gustafson, E.; Hamad, O.A.; Deckmyn, H.; Barbu, A.; Ekdahl, K.N.; Nilsson, B. Exposure of von Willebrand Factor on Isolated Hepatocytes Promotes Tethering of Platelets to the Cell Surface. Transplantation 2019, 103, 1630–1638. [Google Scholar] [CrossRef]
- Gustafson, E.; Asif, S.; Kozarcanin, H.; Elgue, G.; Meurling, S.; Ekdahl, K.N.; Nilsson, B. Control of IBMIR Induced by Fresh and Cryopreserved Hepatocytes by Low Molecular Weight Dextran Sulfate Versus Heparin. Cell Transplant. 2017, 26, 71–81. [Google Scholar] [CrossRef]
- Hammel, J.M.; Elfeki, S.K.; Kobayashi, N.; Ito, M.; Cai, J.; Fearon, D.T.; Graham, F.L.; Fox, I.J. Transplanted hepatocytes infected with a complement receptor type 1 (CR1)-containing recombinant adenovirus are resistant to hyperacute rejection. Transplant. Proc. 1999, 31, 939. [Google Scholar] [CrossRef]
- Stephenne, X.; Nicastro, E.; Eeckhoudt, S.; Hermans, C.; Nyabi, O.; Lombard, C.; Najimi, M.; Sokal, E. Bivalirudin in combination with heparin to control mesenchymal cell procoagulant activity. PLoS ONE 2012, 7, e42819. [Google Scholar] [CrossRef] [PubMed]
- Moll, G.; Alm, J.J.; Davies, L.C.; von Bahr, L.; Heldring, N.; Stenbeck-Funke, L.; Hamad, O.A.; Hinsch, R.; Ignatowicz, L.; Locke, M.; et al. Do cryopreserved mesenchymal stromal cells display impaired immunomodulatory and therapeutic properties? Stem Cells (Dayt. Ohio) 2014, 32, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Moll, G.; Ignatowicz, L.; Catar, R.; Luecht, C.; Sadeghi, B.; Hamad, O.; Jungebluth, P.; Dragun, D.; Schmidtchen, A.; Ringden, O. Different Procoagulant Activity of Therapeutic Mesenchymal Stromal Cells Derived from Bone Marrow and Placental Decidua. Stem Cells Dev. 2015, 24, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Christy, B.A.; Herzig, M.C.; Montgomery, R.K.; Delavan, C.; Bynum, J.A.; Reddoch, K.M.; Cap, A.P. Procoagulant activity of human mesenchymal stem cells. J. Trauma Acute Care Surg. 2017, 83, S164–S169. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, B.M.; Martin, K.; Ali, M.T.; Kumar, A.H.; Pillai, M.G.; Kumar, S.P.; O’Sullivan, J.F.; Whelan, D.; Stocca, A.; Khider, W.; et al. Bone Marrow-Derived Mesenchymal Stem Cells Have Innate Procoagulant Activity and Cause Microvascular Obstruction Following Intracoronary Delivery: Amelioration by Antithrombin Therapy. Stem Cells (Dayt. Ohio) 2015, 33, 2726–2737. [Google Scholar] [CrossRef] [PubMed]
- George, M.J.; Prabhakara, K.; Toledano-Furman, N.E.; Wang, Y.W.; Gill, B.S.; Wade, C.E.; Olson, S.D.; Cox, C.S., Jr. Clinical Cellular Therapeutics Accelerate Clot Formation. Stem Cells Transl. Med. 2018, 7, 731–739. [Google Scholar] [CrossRef]
- Netsch, P.; Elvers-Hornung, S.; Uhlig, S.; Kluter, H.; Huck, V.; Kirschhofer, F.; Brenner-Weiss, G.; Janetzko, K.; Solz, H.; Wuchter, P.; et al. Human mesenchymal stromal cells inhibit platelet activation and aggregation involving CD73-converted adenosine. Stem Cell Res. Ther. 2018, 9, 184. [Google Scholar] [CrossRef]
- Liao, L.; Shi, B.; Chang, H.; Su, X.; Zhang, L.; Bi, C.; Shuai, Y.; Du, X.; Deng, Z.; Jin, Y. Heparin improves BMSC cell therapy: Anticoagulant treatment by heparin improves the safety and therapeutic effect of bone marrow-derived mesenchymal stem cell cytotherapy. Theranostics 2017, 7, 106–116. [Google Scholar] [CrossRef]
- Coppin, L.; Najimi, M.; Bodart, J.; Rouchon, M.S.; van der Smissen, P.; Eeckhoudt, S.; Dahlqvist, G.; Castanares-Zapatero, D.; Komuta, M.; Brouns, S.L.; et al. Clinical Protocol to Prevent Thrombogenic Effect of Liver-Derived Mesenchymal Cells for Cell-Based Therapies. Cells 2019, 8, 846. [Google Scholar] [CrossRef]
- Vulliet, P.R.; Greeley, M.; Halloran, S.M.; MacDonald, K.A.; Kittleson, M.D. Intra-coronary arterial injection of mesenchymal stromal cells and microinfarction in dogs. Lancet (Lond. Engl.) 2004, 363, 783–784. [Google Scholar] [CrossRef]
- Oeller, M.; Laner-Plamberger, S.; Hochmann, S.; Ketterl, N.; Feichtner, M.; Brachtl, G.; Hochreiter, A.; Scharler, C.; Bieler, L.; Romanelli, P.; et al. Selection of Tissue Factor-Deficient Cell Transplants as a Novel Strategy for Improving Hemocompatibility of Human Bone Marrow Stromal Cells. Theranostics 2018, 8, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Moll, G.; Jitschin, R.; von Bahr, L.; Rasmusson-Duprez, I.; Sundberg, B.; Lonnies, L.; Elgue, G.; Nilsson-Ekdahl, K.; Mougiakakos, D.; Lambris, J.D.; et al. Mesenchymal stromal cells engage complement and complement receptor bearing innate effector cells to modulate immune responses. PLoS ONE 2011, 6, e21703. [Google Scholar] [CrossRef] [PubMed]
- Ettelaie, C.; Fountain, D.; Collier, M.E.; Elkeeb, A.M.; Xiao, Y.P.; Maraveyas, A. Low molecular weight heparin downregulates tissue factor expression and activity by modulating growth factor receptor-mediated induction of nuclear factor-kappaB. Biochim. Et Biophys. Acta 2011, 1812, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Coppin, L.; Smets, F.; Ambroise, J.; Sokal, E.; Stéphenne, X. Control of the thrombogenic risk with a combination of anticoagulant drugs during Liver Derived Mesenchymal Stem Cells infusions in 11 patients with Crigler-Najjar syndrome or Urea Cycle Disorders. J. Stem Cells Res. Dev. Ther. Under revision.
- Baygan, A.; Aronsson-Kurttila, W.; Moretti, G.; Tibert, B.; Dahllof, G.; Klingspor, L.; Gustafsson, B.; Khoein, B.; Moll, G.; Hausmann, C.; et al. Safety and Side Effects of Using Placenta-Derived Decidual Stromal Cells for Graft-versus-Host Disease and Hemorrhagic Cystitis. Front. Immunol. 2017, 8, 795. [Google Scholar] [CrossRef]
- Seeger, F.H.; Rasper, T.; Fischer, A.; Muhly-Reinholz, M.; Hergenreider, E.; Leistner, D.M.; Sommer, K.; Manavski, Y.; Henschler, R.; Chavakis, E.; et al. Heparin disrupts the CXCR4/SDF-1 axis and impairs the functional capacity of bone marrow-derived mononuclear cells used for cardiovascular repair. Circ. Res. 2012, 111, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Groeneveld, D.; Pereyra, D.; Veldhuis, Z.; Adelmeijer, J.; Ottens, P.; Kopec, A.K.; Starlinger, P.; Lisman, T.; Luyendyk, J.P. Intrahepatic fibrin(ogen) deposition drives liver regeneration after partial hepatectomy in mice and humans. Blood 2019, 133, 1245–1256. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coppin, L.; Sokal, E.; Stéphenne, X. Thrombogenic Risk Induced by Intravascular Mesenchymal Stem Cell Therapy: Current Status and Future Perspectives. Cells 2019, 8, 1160. https://doi.org/10.3390/cells8101160
Coppin L, Sokal E, Stéphenne X. Thrombogenic Risk Induced by Intravascular Mesenchymal Stem Cell Therapy: Current Status and Future Perspectives. Cells. 2019; 8(10):1160. https://doi.org/10.3390/cells8101160
Chicago/Turabian StyleCoppin, Louise, Etienne Sokal, and Xavier Stéphenne. 2019. "Thrombogenic Risk Induced by Intravascular Mesenchymal Stem Cell Therapy: Current Status and Future Perspectives" Cells 8, no. 10: 1160. https://doi.org/10.3390/cells8101160
APA StyleCoppin, L., Sokal, E., & Stéphenne, X. (2019). Thrombogenic Risk Induced by Intravascular Mesenchymal Stem Cell Therapy: Current Status and Future Perspectives. Cells, 8(10), 1160. https://doi.org/10.3390/cells8101160