Comparative Transcriptome Analysis of Ampelopsis megalophylla for Identifying Genes Involved in Flavonoid Biosynthesis and Accumulation during Different Seasons


Abstract
:1. Introduction
2. Results
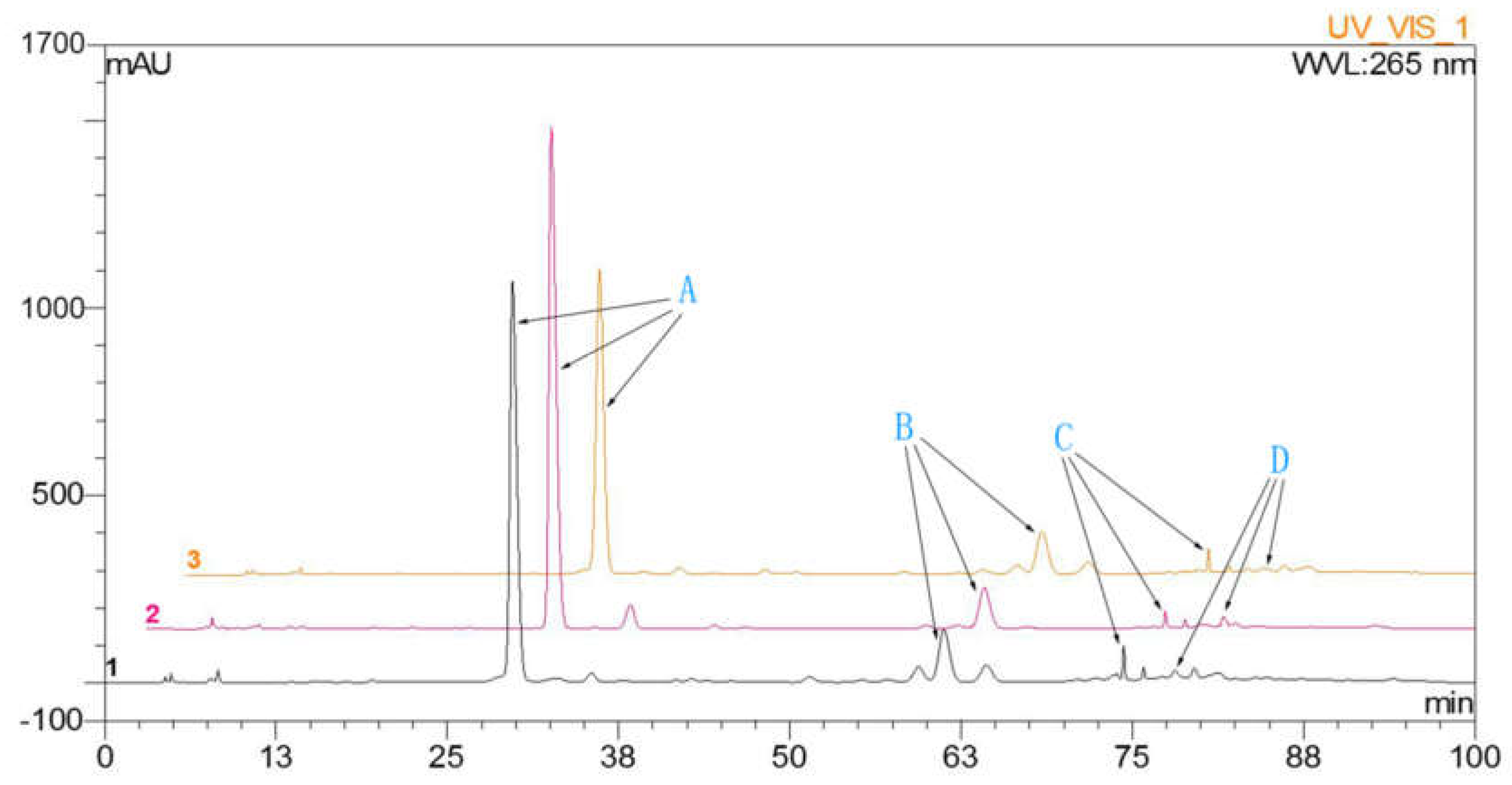
2.1. Determination of Flavonoids in A. megalophylla
2.2. Transcriptome Sequencing and De Novo Assembly
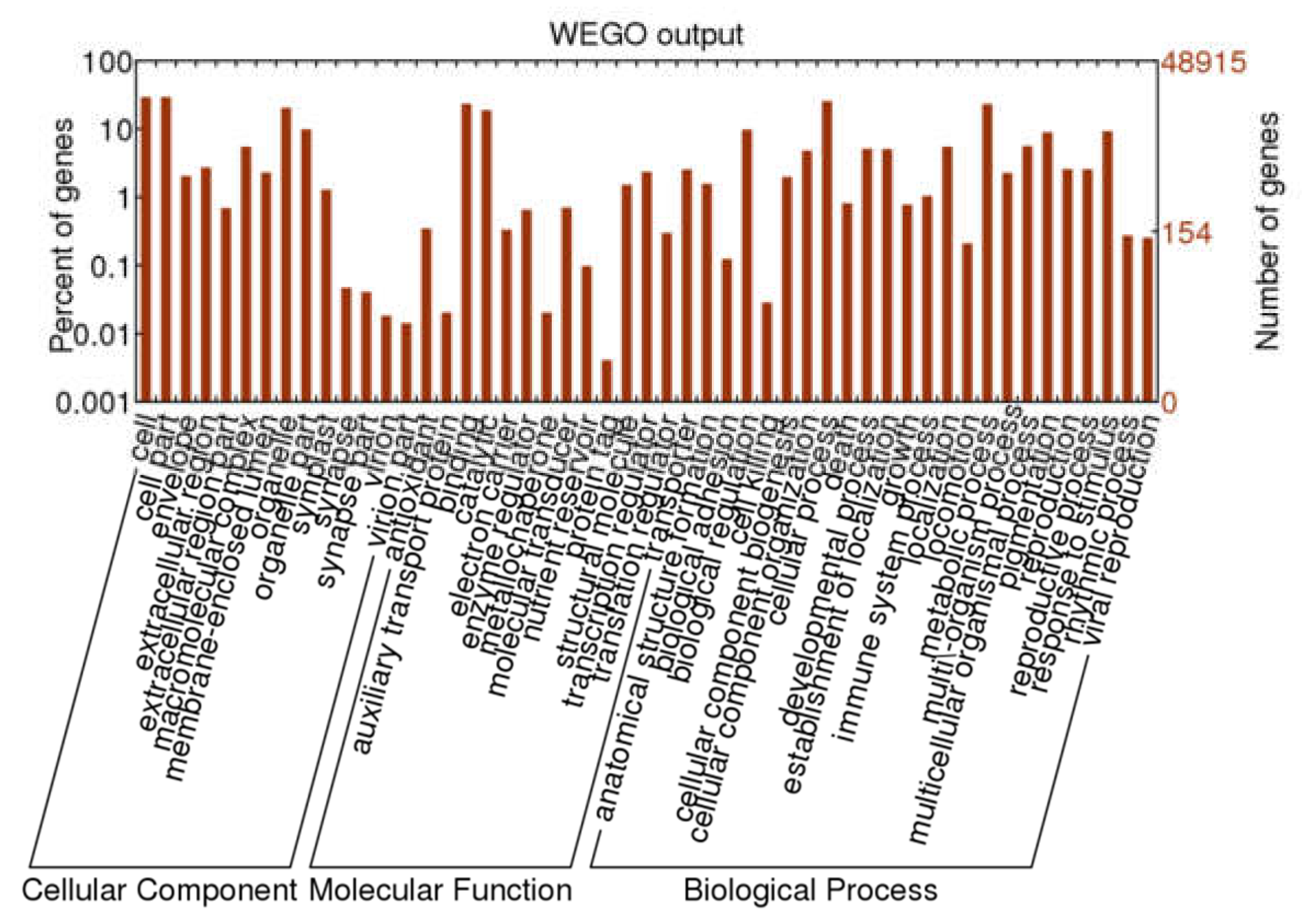
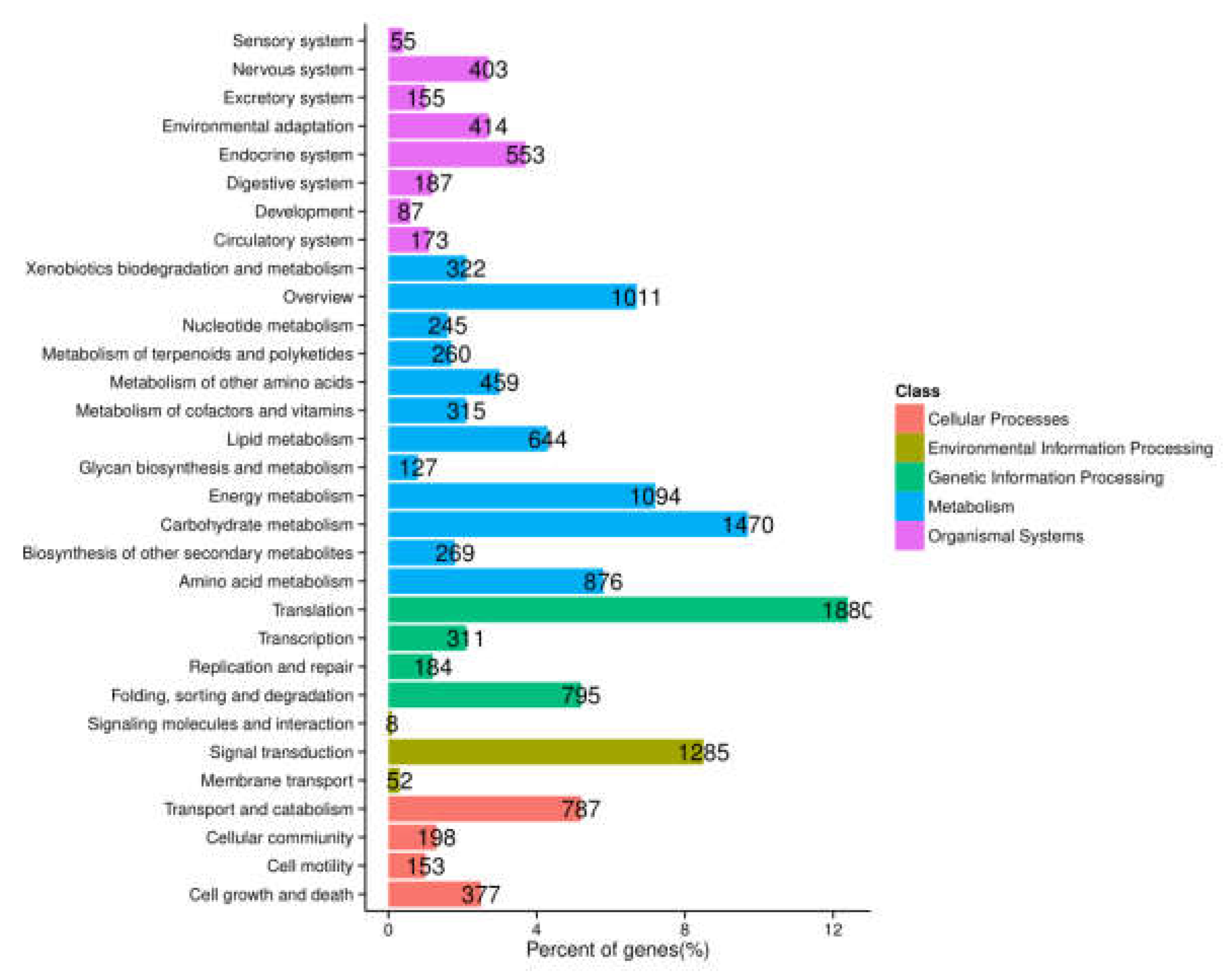
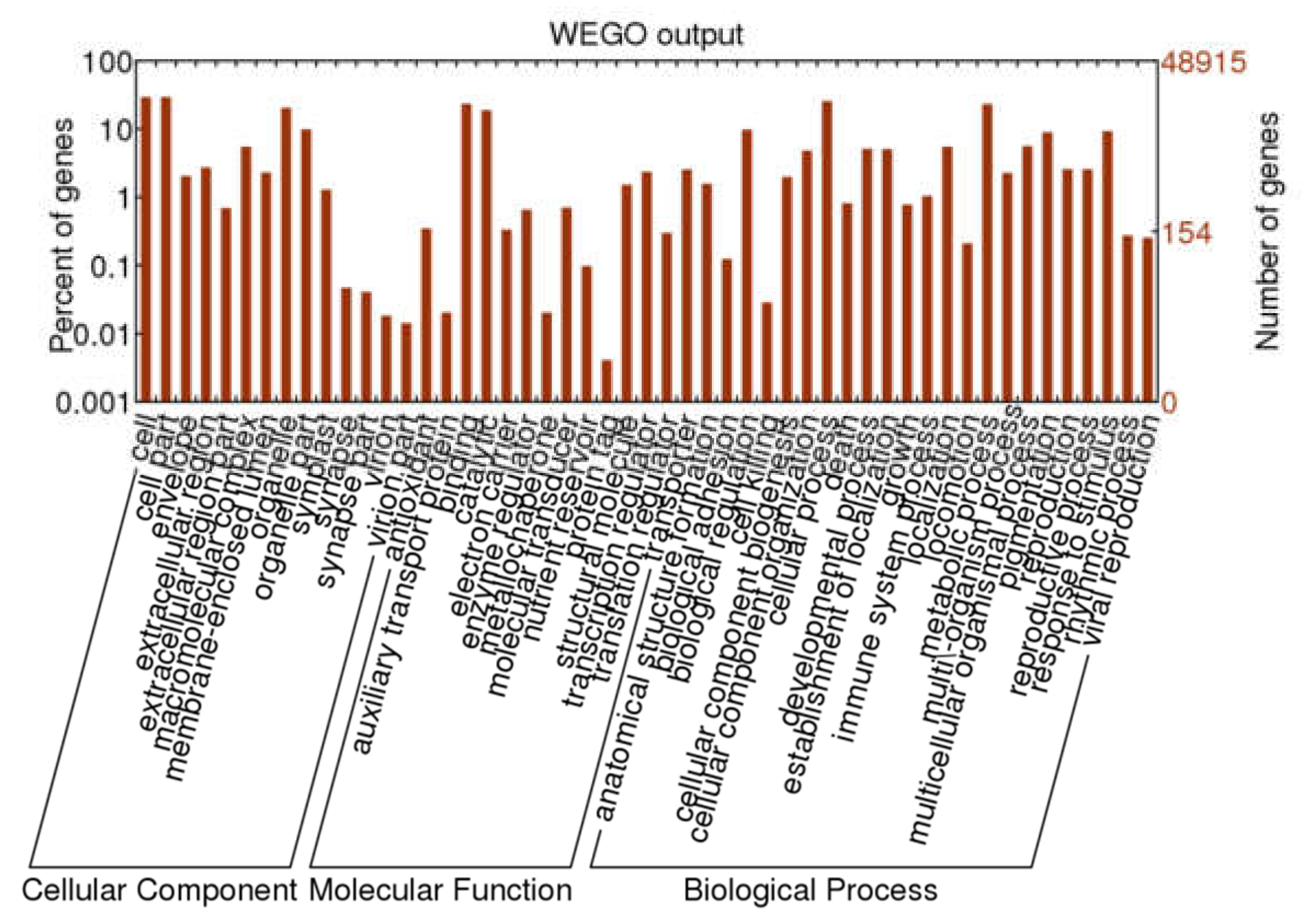
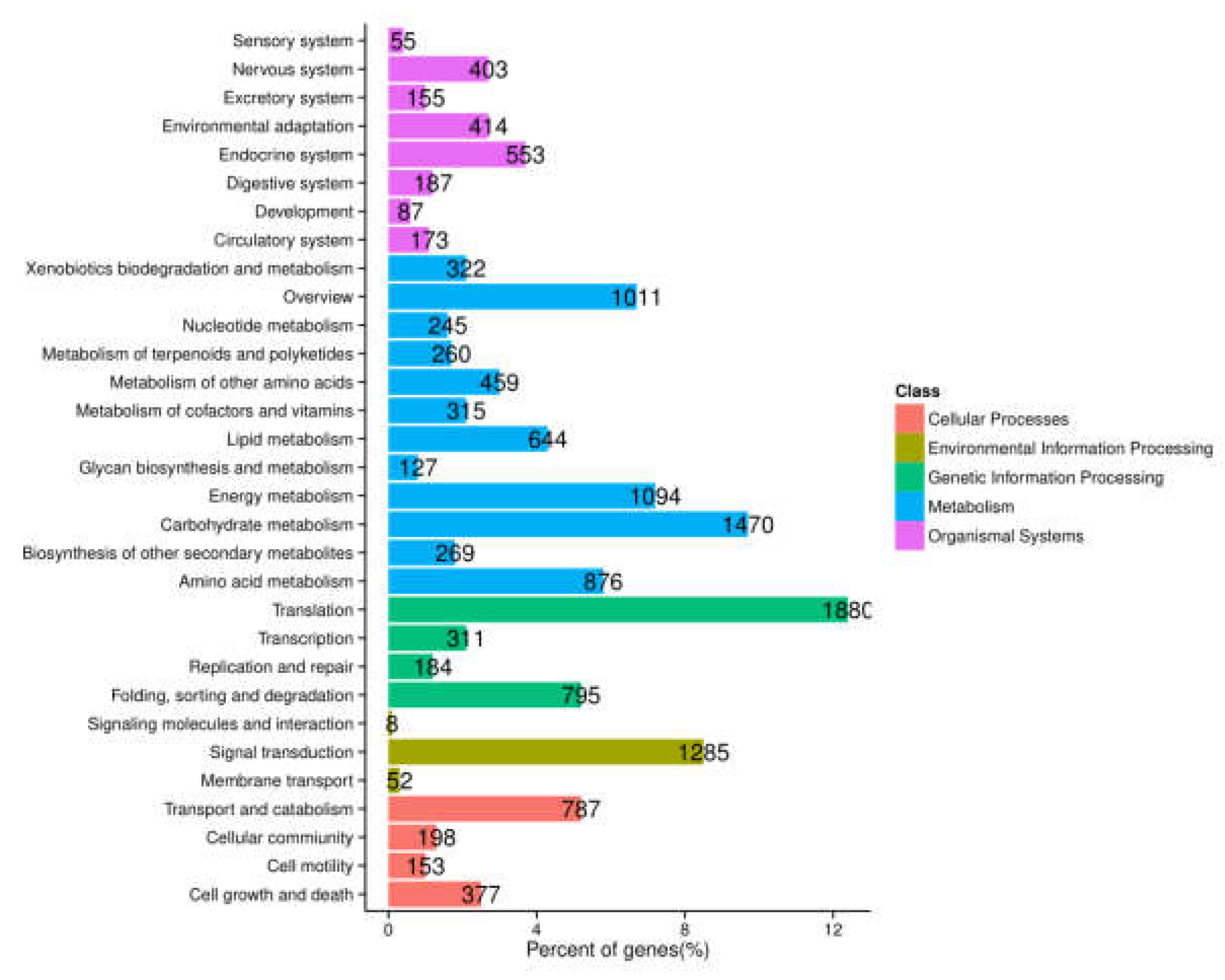
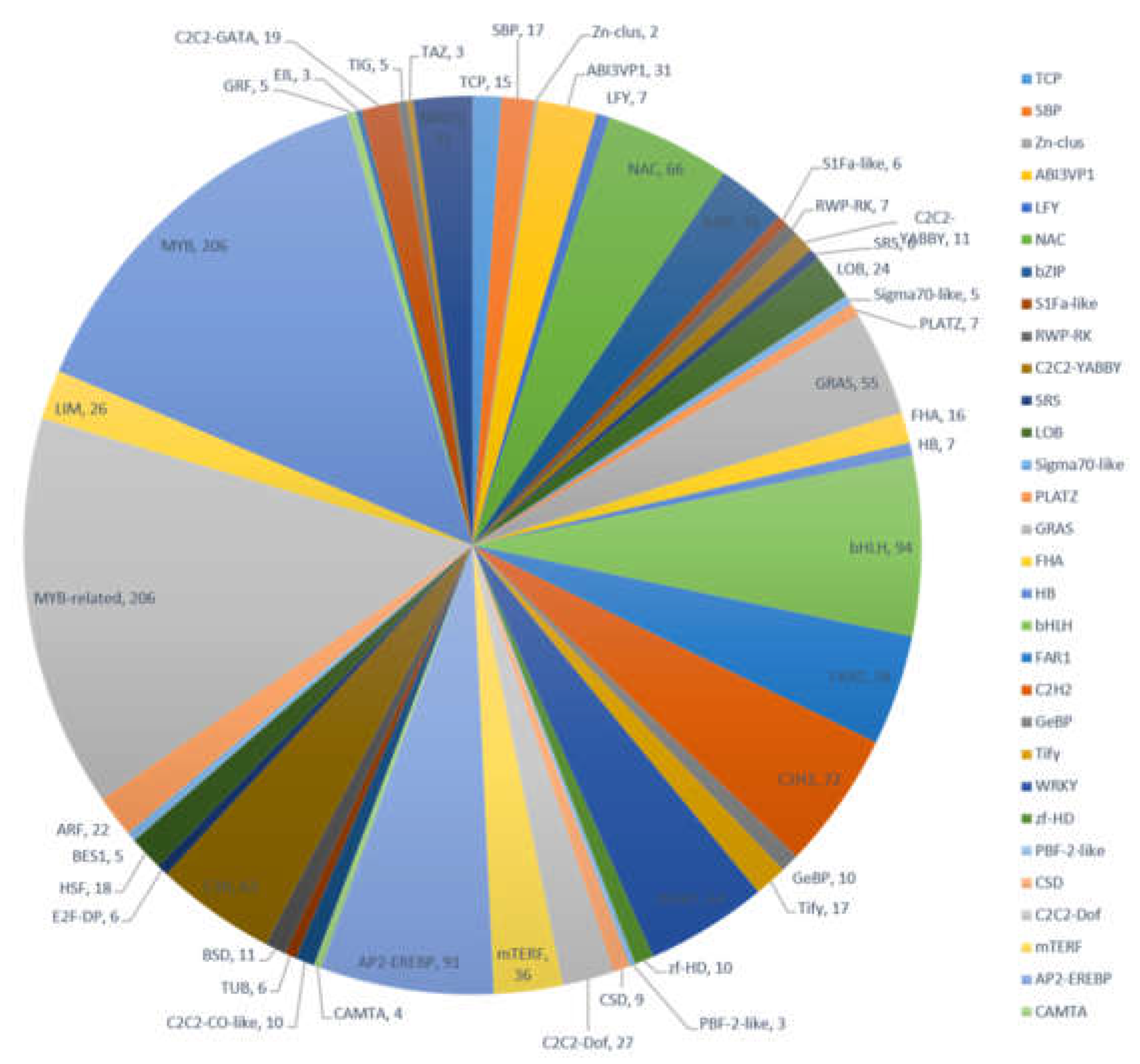
2.3. Functional Annotation and Classification of A. megalophylla Unigenes
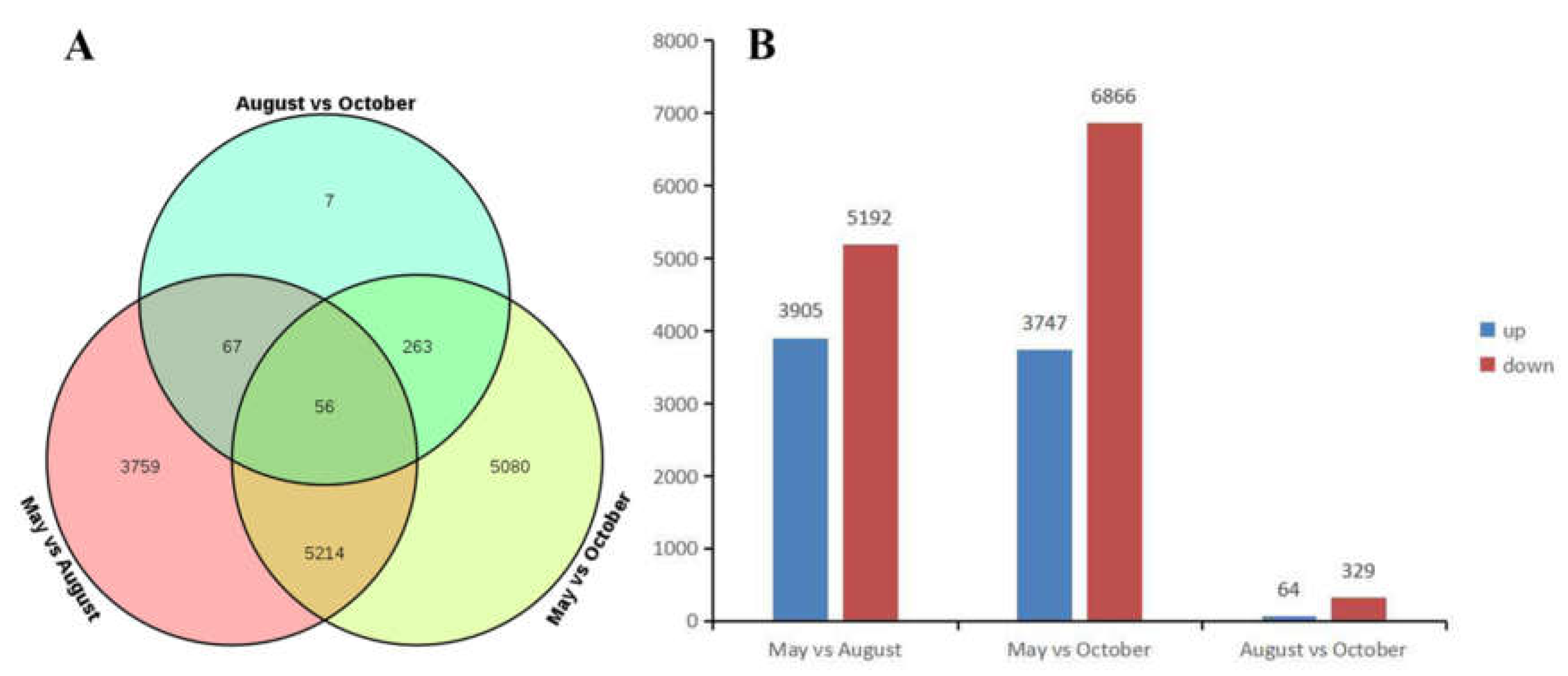
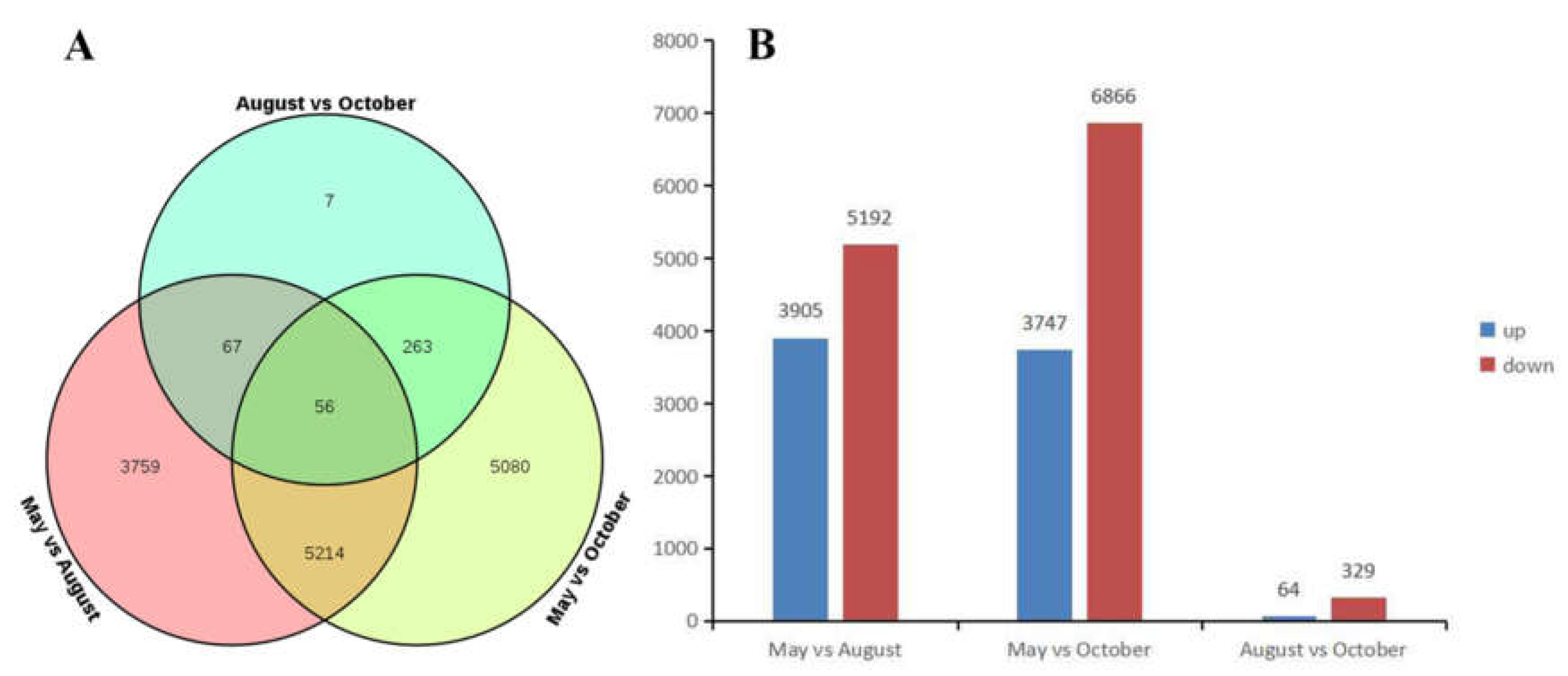
2.4. Differentially Expressed Gene (DEG) Analysis of A. megalophylla
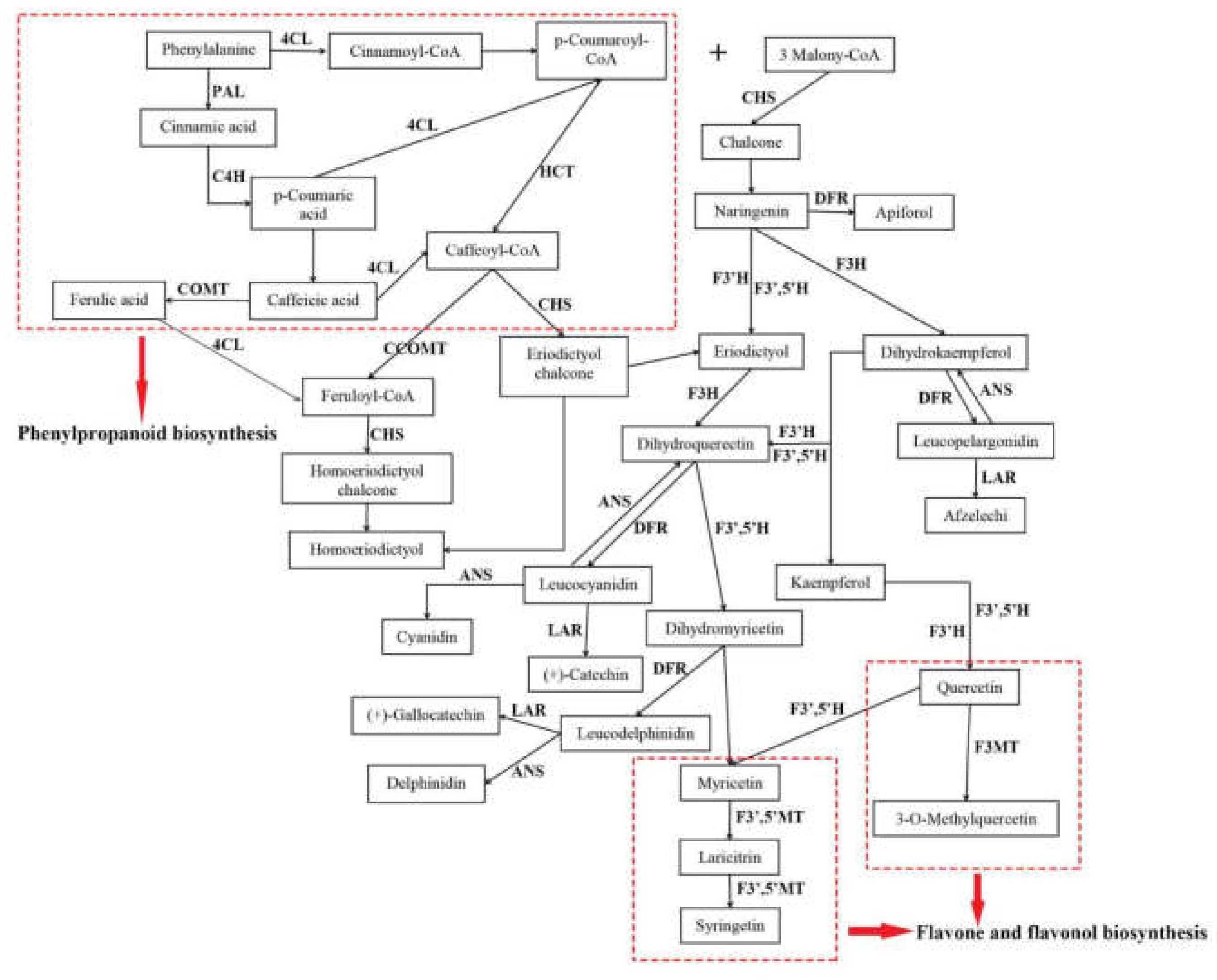
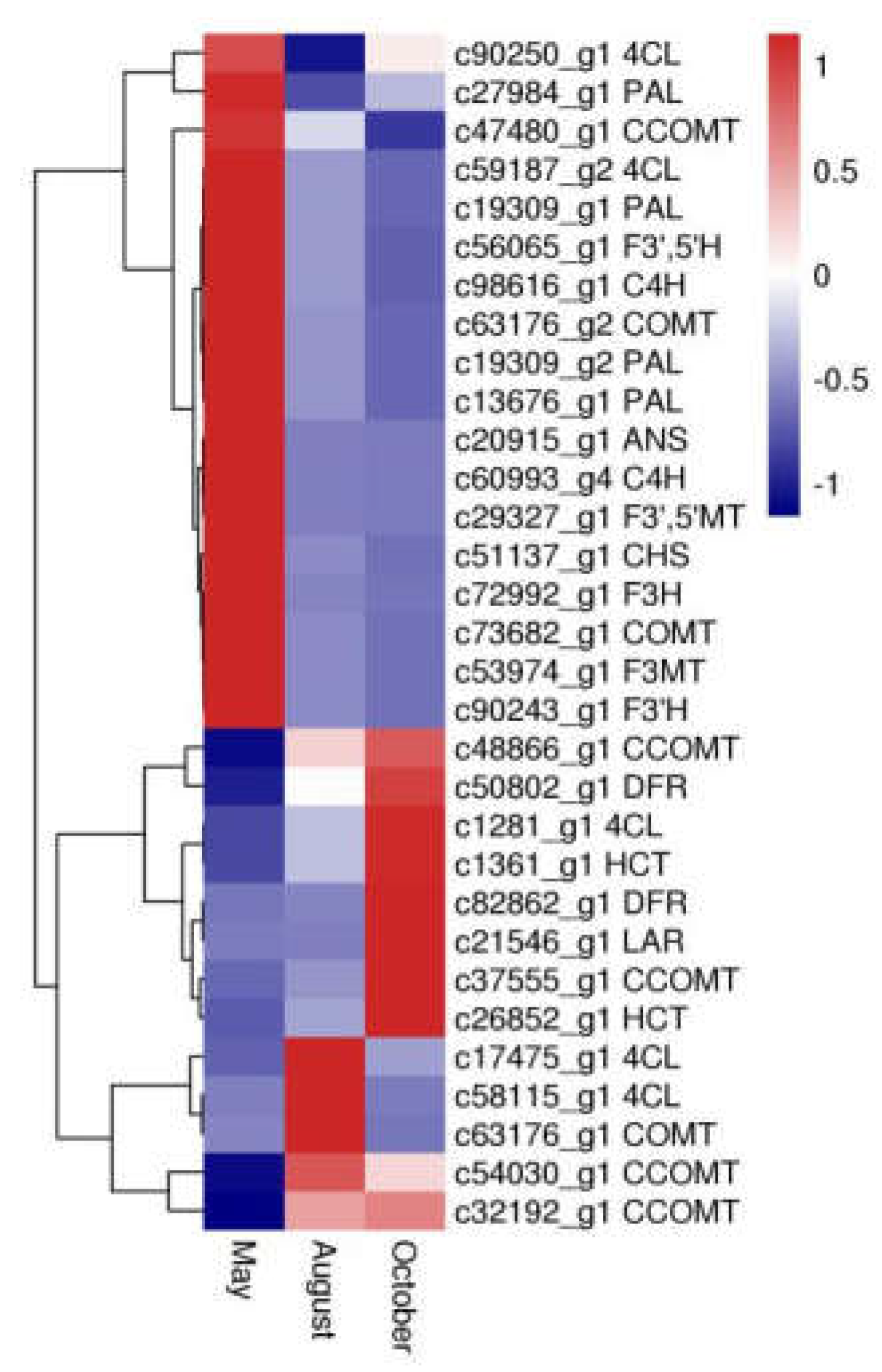
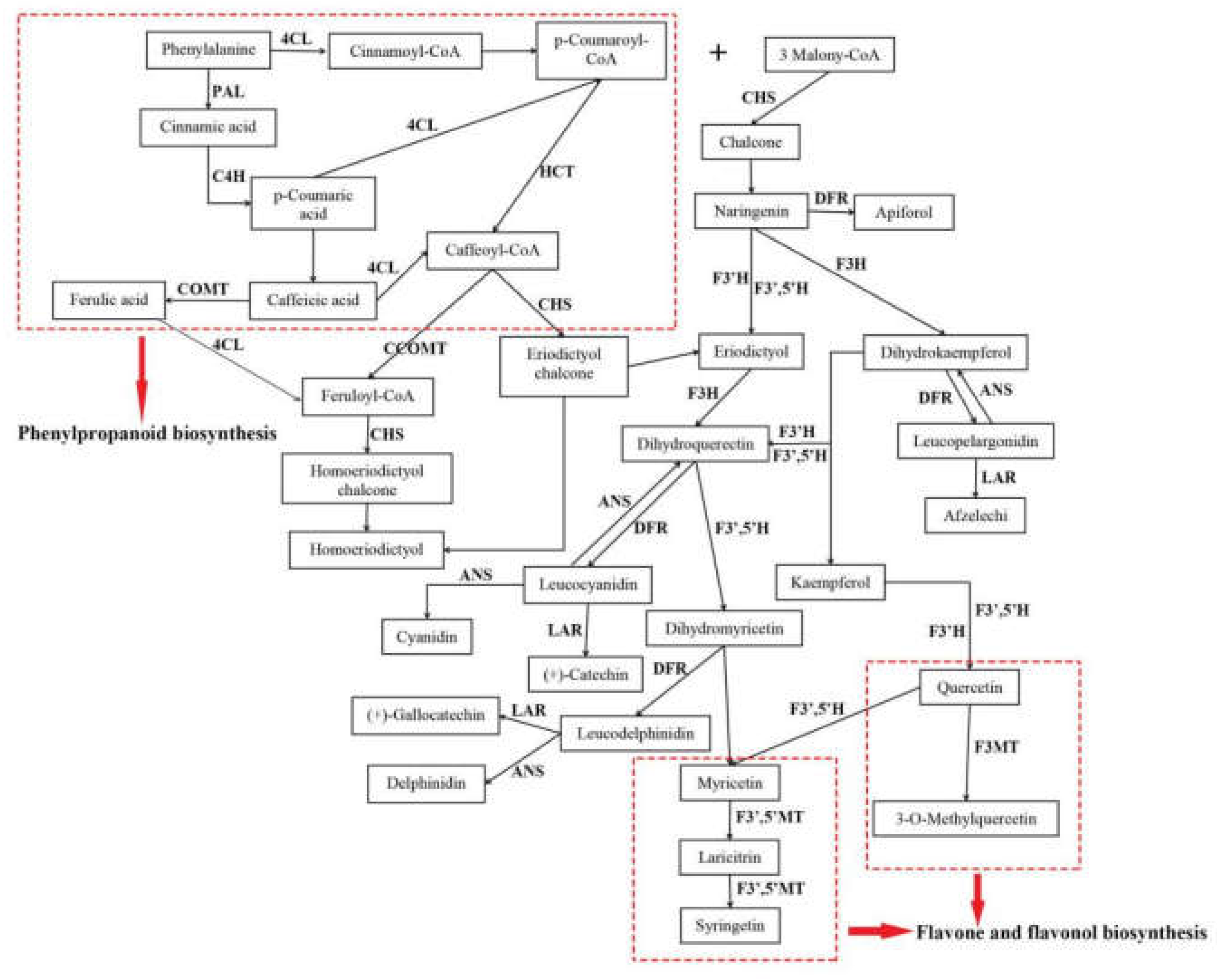
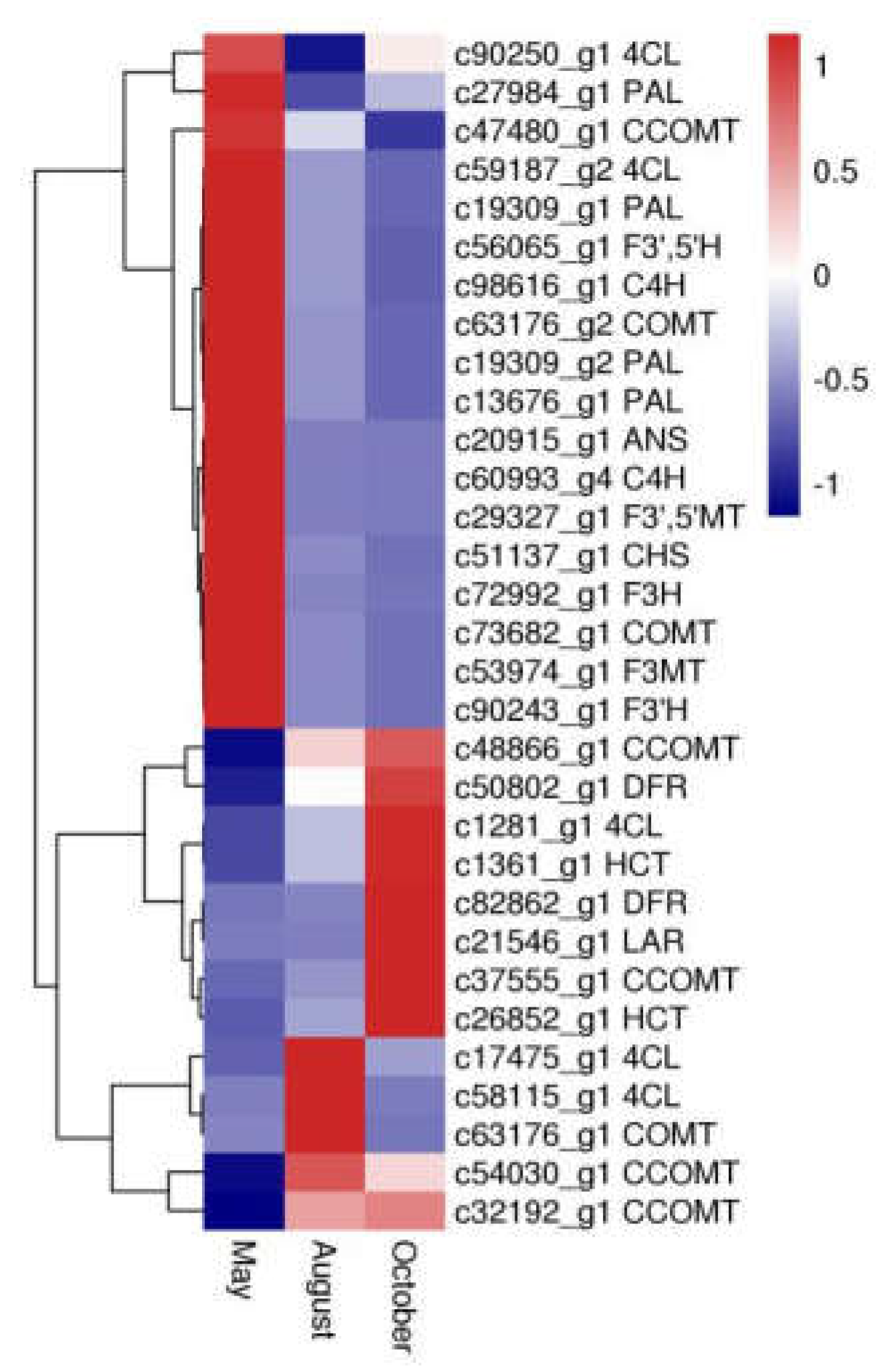
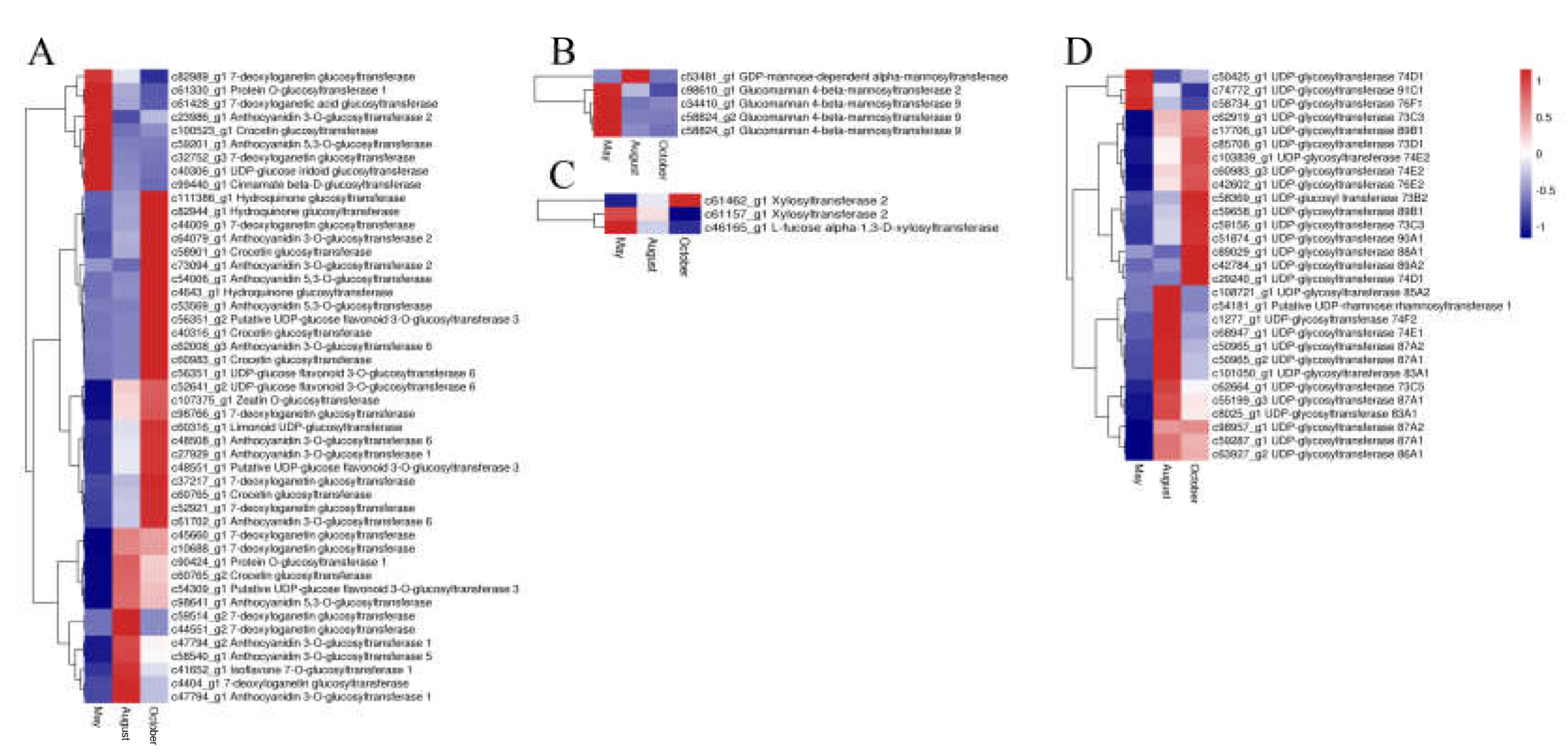
2.5. Candidate Genes Related to Flavonoid Biosynthesis
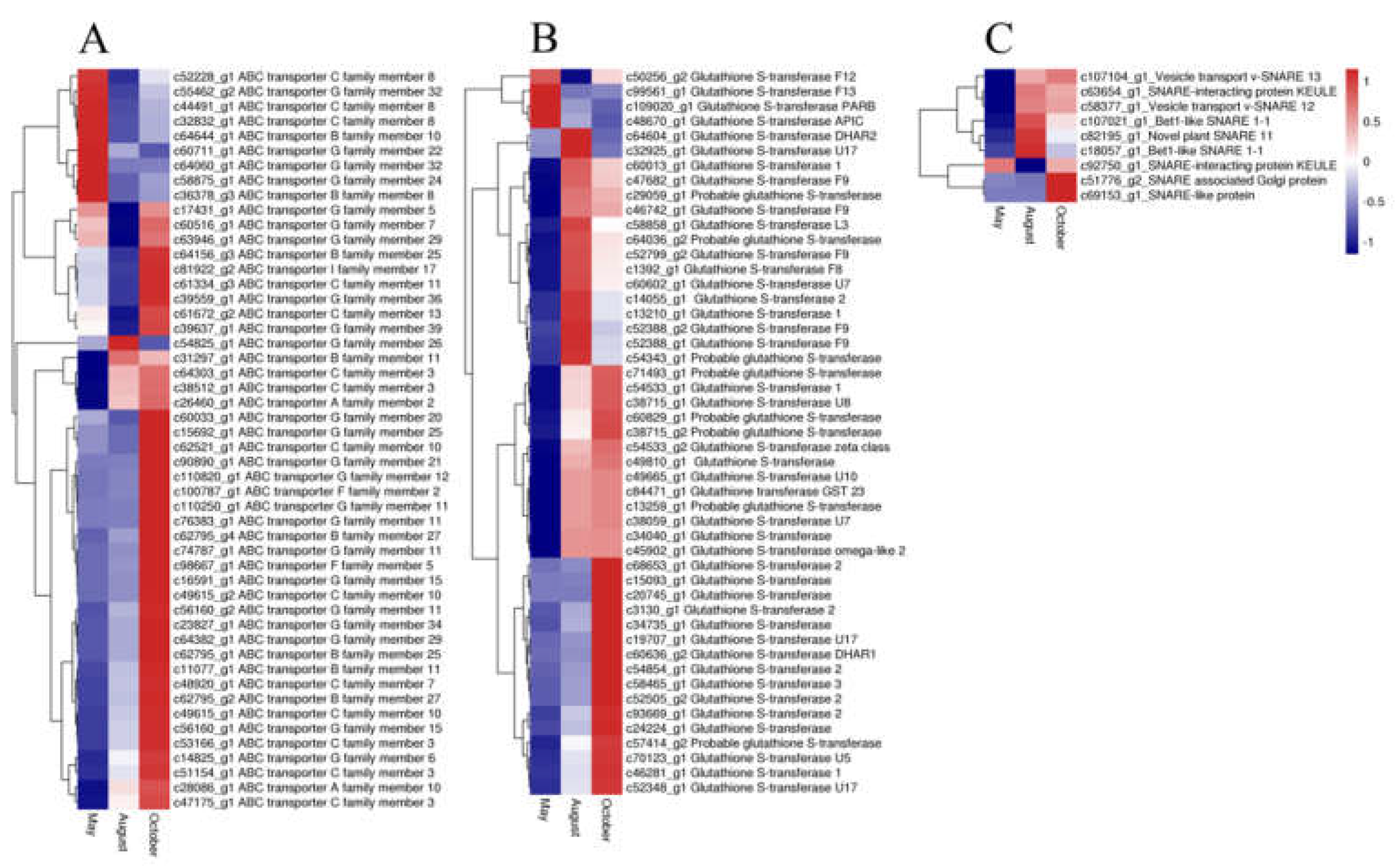
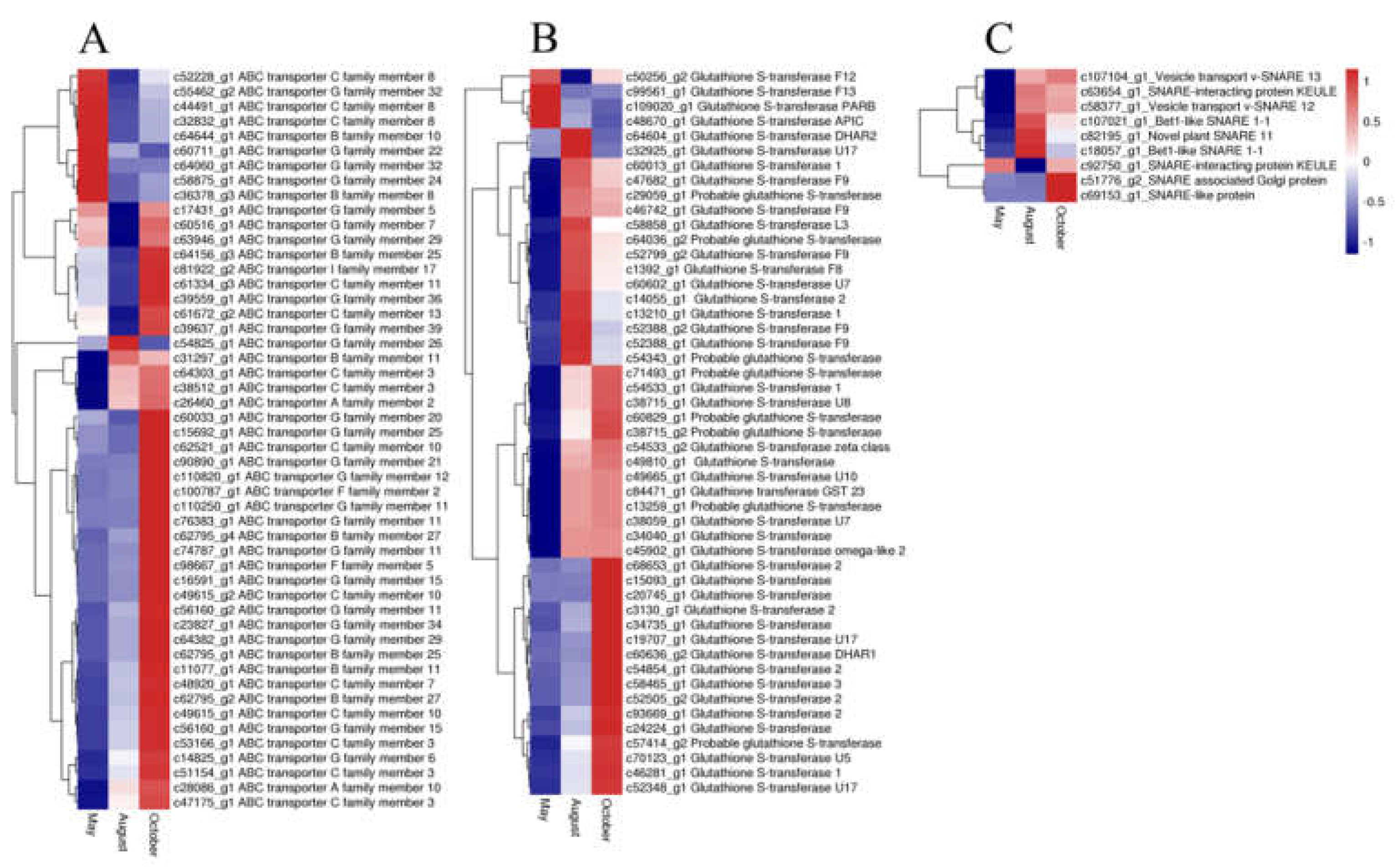
2.6. Candidate Genes Related to Flavonoid Accumulation and Biomodification
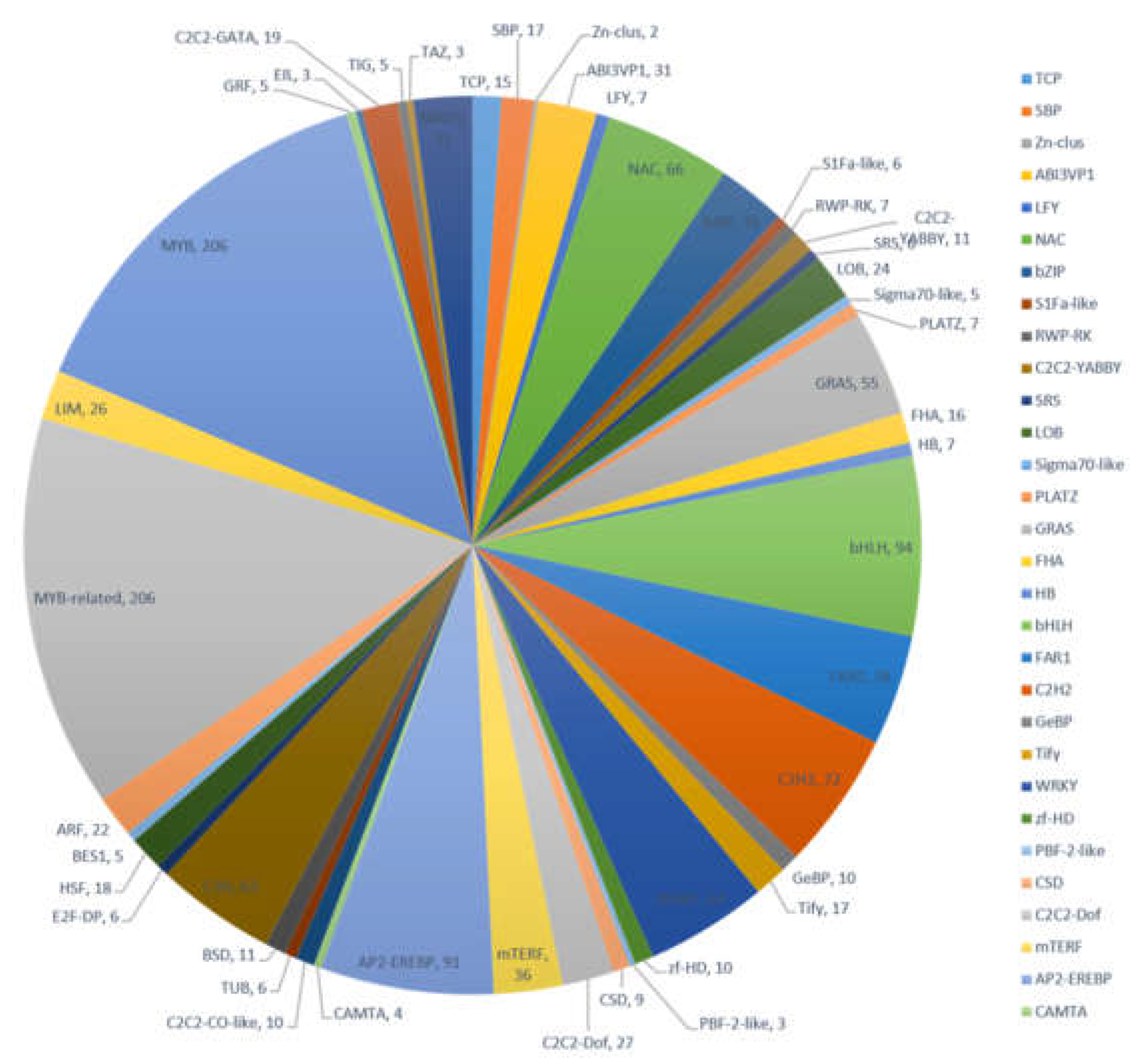
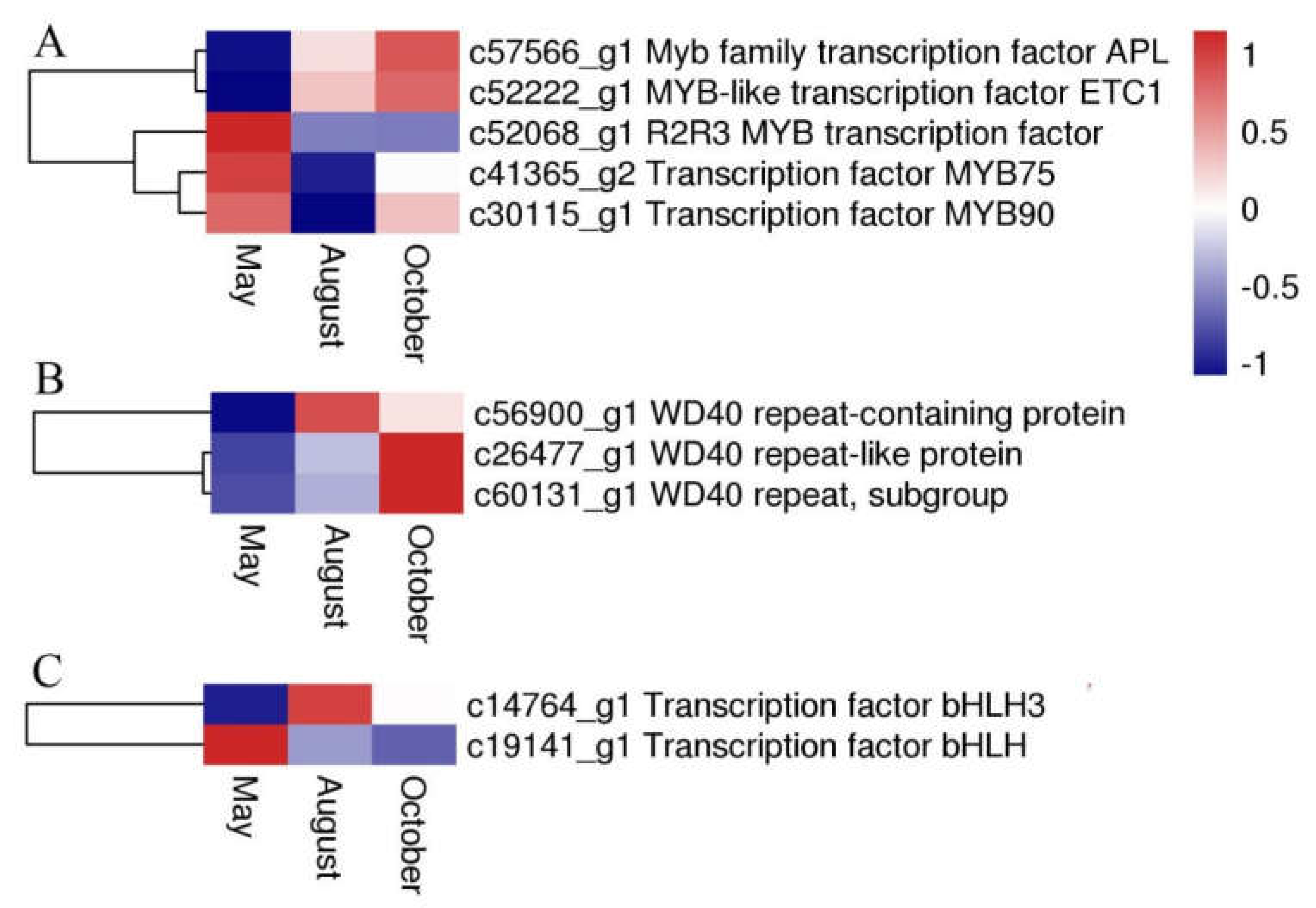
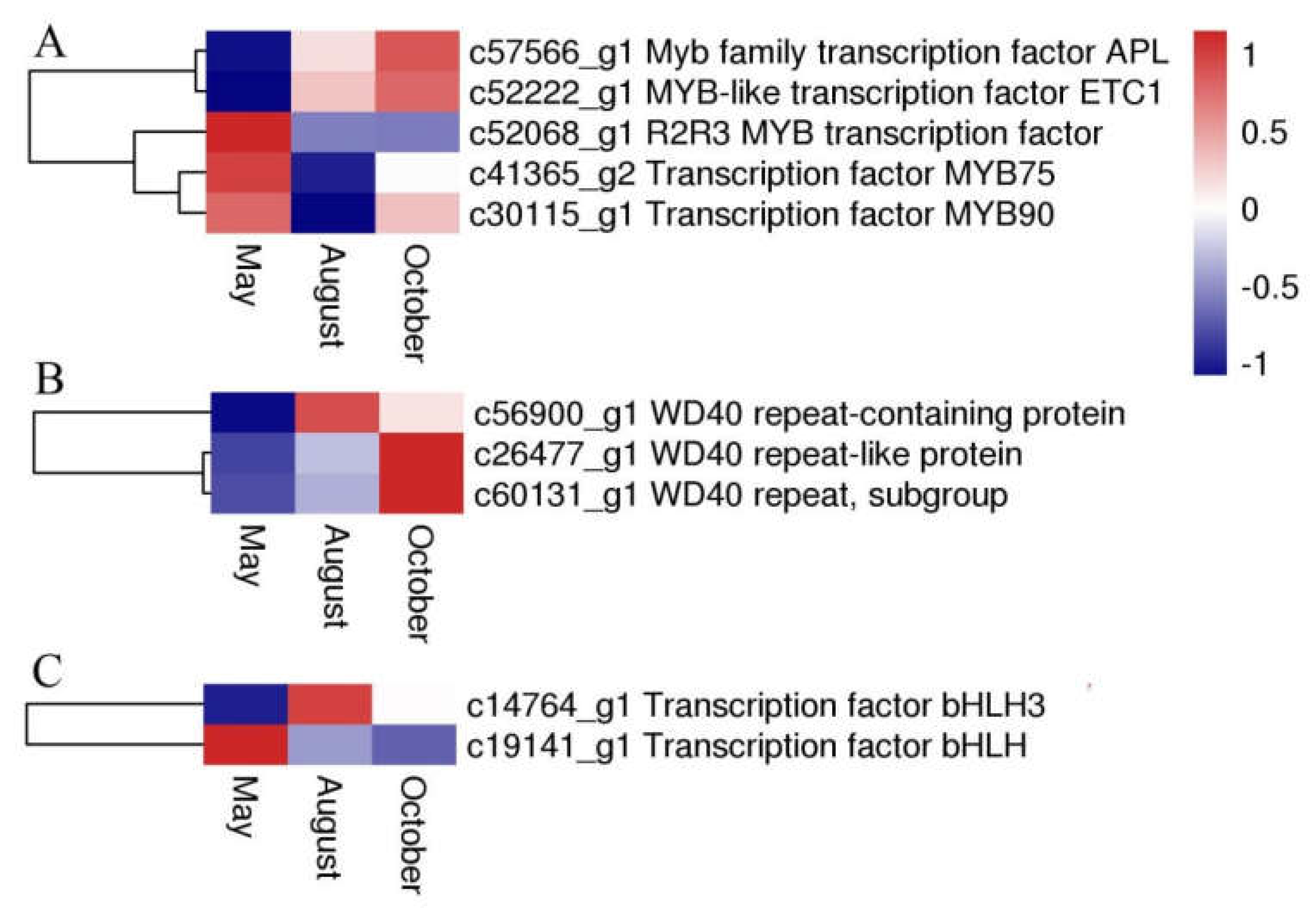
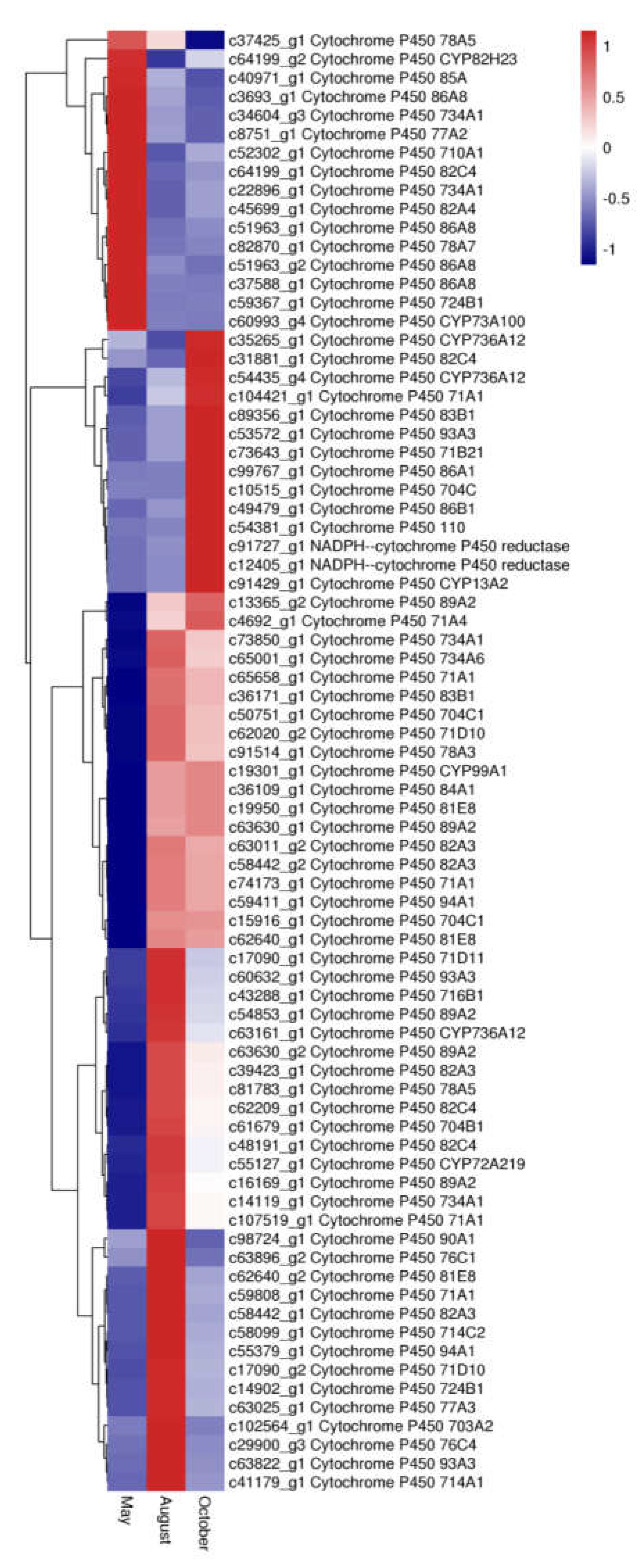
2.7. Expression of Transcription Factors and P450 Family Genes Related Flavonoid Biosynthesis and Transport
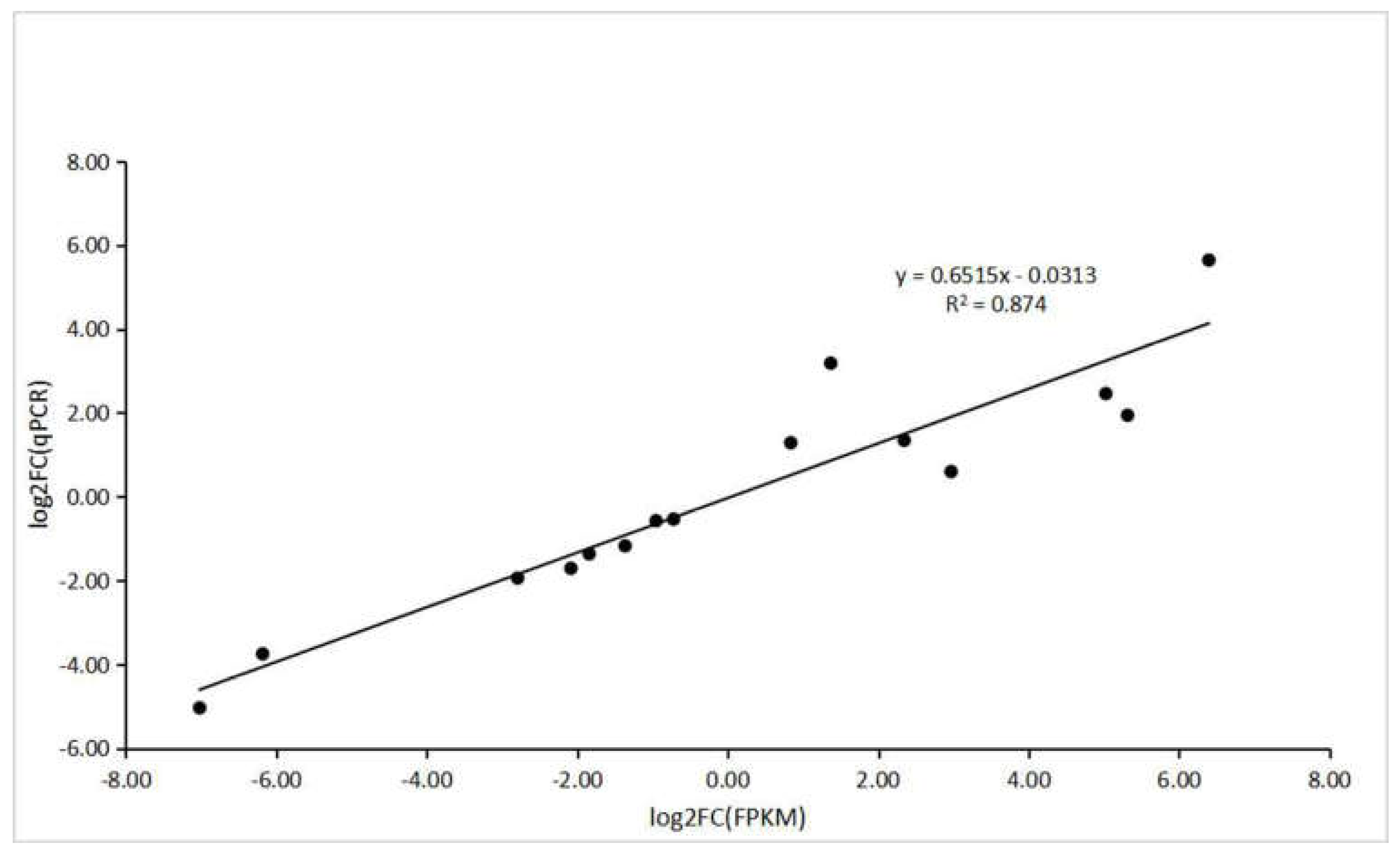
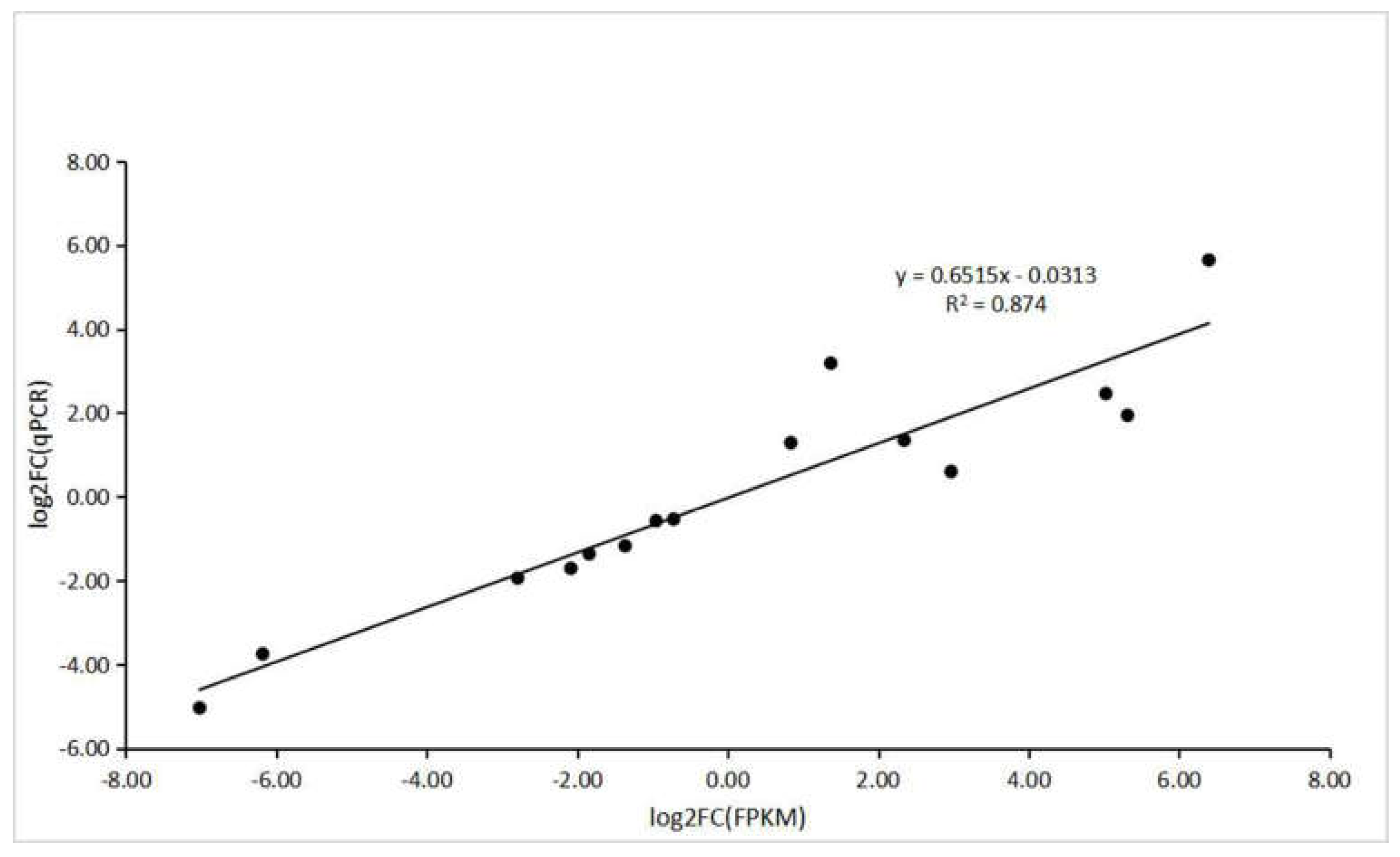
2.8. Validation of DEGs by qPCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Extraction and Determination of Flavonoids in A. megalophylla
4.3. RNA Sequencing and De Novo Assembly
4.4. Functional Annotation and Sequence Analysis
4.5. Analysis of Unigene Differential Expression Genes (DEGs)
4.6. Quantitative Real-Time PCR
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chen, M.L.; Chen, X.J.; Li, G.; Zhang, X.Q. Ampelopsis megalophylla ethanol extract of experimental studies on inhibition effect on expression of HBsAg, HBeAg 2215 cells. Chin. J. Tradit. Med. Sci. Technol. 2014, 19, 329–330. [Google Scholar]
- Yang, R.J.; Chen, X.J.; He, X.Y.; Rao, R.; Zhang, X.Q.; Liu, Y.W. Study on anti-HBV virus effect of petroleum ether extracts of Ampelopsis megalophylla in vitro. Lishizhen Med. Materia Med. Res. 2011, 22, 153–215. [Google Scholar]
- Xie, X.F.; Wang, J.W.; Zhang, H.P.; Li, Q.X.; Chen, B.Y. Chemical composition, antimicrobial and antioxidant activities of essential oil from Ampelopsis megalophylla. Nat. Prod. Res. 2014, 28, 853–860. [Google Scholar] [CrossRef]
- Cheng, P.; Gui, C.; Huang, J.; Xia, Y.; Fang, Y.; Da, G.Z.; Zhang, X.Q. Molecular mechanisms of ampelopsin from Ampelopsis megalophylla induces apoptosis in HeLa cells. Oncol. Lett. 2017, 14, 2691–2698. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.F.; Wang, J.W.; Zhang, H.P.; Li, Q.X.; Chen, B.Y. Characterization and antitumor activities of a water-soluble polysaccharide from Ampelopsis megalophylla. Carbohydr. Polym. 2015, 129, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Zhang, X.J.; Zhang, X.Q.; Zhang, Y.W. Study on chemical constituents of Ampelopsis megalophylla. Lishizhen Med. Mater. Med. Res. 2010, 22, 866–867. [Google Scholar]
- Zheng, G.H.; Wu, J.; Liu, D.; Qiao, M. HPLC for Fingerprint Analysis of Flavones from Meicha from Different Harvest Time. Chin. Arch. Tradit. Chin. Med. 2009, 27, 980–982. [Google Scholar]
- Katsuyama, Y.; Funa, N.; Miyahisa, I.; Horinouchi, S. Synthesis of unnatural flavonoids and stilbenes by exploiting the plant biosynthetic pathway in Escherichia coli. Chem Biol. 2007, 14, 613–621. [Google Scholar] [CrossRef]
- Nabavi, M.S.; Samec, D.; Tomczyk, M.; Milella, L.; Russo, D.; Habtemariam, S.; Suntar, I.; Rastrelli, L.; Daglia, M.; Xiao, J.; et al. Flavonoid biosynthetic pathways in plants: Versatile targets for metabolic engineering. Biotechnol. Adv. 2018. [Google Scholar] [CrossRef]
- Lei, Z.; Zhou, C.; Ji, X.; Wei, G.; Huang, Y.; Yu, W.; Luo, Y.; Qiu, Y. Transcriptome Analysis Reveals genes involved in flavonoid biosynthesis and accumulation in Dendrobium catenatum from Different Locations. Sci. Rep. 2018, 8, 6373. [Google Scholar] [CrossRef]
- Liu, X.; Lu, Y.; Yuan, Y.; Liu, S.; Guan, C.; Chen, S.; Liu, Z. De novo transcriptome of Brassica juncea seed coat and identification of genes for the biosynthesis of flavonoids. PLoS ONE 2013, 8, e71110. [Google Scholar] [CrossRef] [PubMed]
- Lepiniec, L.; Debeaujon, I.; Routaboul, J.M.; Baudry, A.; Pourcel, L.; Nesi, N.; Caboche, M. Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 2006, 57, 405–430. [Google Scholar] [CrossRef]
- Zifkin, M.; Jin, A.; Ozga, J.A.; Zaharia, L.I.; Schernthaner, J.P.; Gesell, A.; Abrams, S.R.; Kennedy, J.A.; Constabel, C.P. Gene expression and metabolite profiling of developing highbush blueberry fruit indicates transcriptional regulation of flavonoid metabolism and activation of abscisic acid metabolism. Plant Physiol. 2012, 158, 200–224. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y. Study of Medicinal Plants of Ampelopsis by DNA Molecular Identification and Chemical Composition Analysis. Master’s Thesis, Hubei University of Chinese Medicine, Wuhan, China, 2016. [Google Scholar]
- Li, C.F.; Zhu, Y.; Yu, Y.; Zhao, Q.Y.; Wang, S.J.; Wang, X.C.; Yao, M.Z.; Luo, D.; Li, X.; Chen, L.; et al. Global transcriptome and gene regulation network for secondary metabolite biosynthesis of tea plant (Camellia sinensis). BMC Genom. 2015, 16, 560. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Zhu, C.; Zhou, Y.; Niu, X.; Miao, M.; Tang, X.; Chen, F.; Zhao, W.; Liu, Y. Transcriptome analysis of differentially expressed unigenes involved in flavonoid biosynthesis during flower development of Chrysanthemum morifolium ‘Chuju’. Sci. Rep. 2018, 8, 13414. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.B.; Datir, S.S.; Bhonde, Y.; Kelkar, N.; Samdani, P.; Tamhane, V.A. De novo root transcriptome of a medicinally important rare tree Oroxylum indicum for characterization of the flavonoid biosynthesis pathway. Phytochemistry 2018, 156, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Fang, Y.; Huang, M.J.; Xiao, Y.; Liu, Y.; Ma, X.R.; Zhao, H. High flavonoid accompanied with high starch accumulation triggered by nutrient starvation in bioenergy crop duckweed (Landoltia punctata). BMC Genom. 2017, 18, 166. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Yonekura-Sakakibara, K.; Niida, R.; Watanabe-Takahashi, A.; Saito, K. Phytochemical genomics in Arabidopsis thaliana: A case study for functional identification of flavonoid biosynthesis genes. Pure Appl. Chem. 2007, 4, 811–823. [Google Scholar] [CrossRef]
- Han, Y.; Huang, K.; Liu, Y.; Jiao, T.; Ma, G.; Qian, Y.; Wang, P.; Dai, X.; Gao, L.; Xia, T. Functional Analysis of Two Flavanone-3-Hydroxylase Genes from Camellia sinensis: A Critical Role in Flavonoid Accumulation. Genes 2017, 8, 300. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, H.; Meng, L.; Hu, K.; Dai, S.L. Isolation and functional analysis of a homolog of flavonoid 3′,5′-hydroxylase gene from Pericallis × hybrida. Physiol. Plant 2013, 149, 151–159. [Google Scholar] [CrossRef]
- Sugiyama, A.; Shitan, N.; Yazaki, K. Involvement of a soybean ATP-binding cassette-type transporter in the secretion of genistein, a signal flavonoid in legume-Rhizobium symbiosis. Plant Physiol. 2007, 144, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Lipka, V.; Kwon, C.; Panstruga, R. SNARE-ware: The role of SNARE-domain proteins in plant biology. Annu. Rev. Cell Dev. Biol. 2007, 23, 147–174. [Google Scholar] [CrossRef]
- Klein, M.; Martinoia, E.; Hoffmann-Thoma, G.; Weissenbock, G. A membrane-potential dependent ABC-like transporter mediates the vacuolar uptake of rye flavone glucuronides: Regulation of glucuronide uptake by glutathione and its conjugates. Plant J. 2000, 21, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, S.D.; Matthews, M.A.; Di Gaspero, G.; Gambetta, G.A. Water deficits accelerate ripening and induce changes in gene expression regulating flavonoid biosynthesis in grape berries. Planta 2007, 227, 101–112. [Google Scholar] [CrossRef]
- Yoshida, K.; Toyama, Y.; Kameda, K.; Kondo, T. Contribution of each caffeoyl residue of the pigment molecule of gentiodelphin to blue color development. Phytochemistry 2000, 54, 85–92. [Google Scholar] [CrossRef]
- Cos, P.; Calomme, M.; Sindambiwe, J.B.; De Bruyne, T.; Cimanga, K.; Pieters, L.; Vlietinck, A.J.; Vanden, B.D. Cytotoxicity and lipid peroxidation-inhibiting activity of flavonoids. Planta Med. 2001, 67, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, A.G.; Liu, F.; Hosomi, Y.; Shii, K.; Roth, R.A. Activation of protein kinase C alpha inhibits signaling by members of the insulin receptor family. J. Biol. Chem. 1995, 270, 21600–21605. [Google Scholar] [CrossRef]
- Moreira, M.R.; Kanashiro, A.; Kabeya, L.M.; Polizello, A.C.; Azzolini, A.E.; Curti, C.; Oliveira, C.A.; T-do Amaral, A.; Lucisano-Valim, Y.M. Neutrophil effector functions triggered by Fc-gamma and/or complement receptors are dependent on B-ring hydroxylation pattern and physicochemical properties of flavonols. Life Sci. 2007, 81, 317–326. [Google Scholar] [CrossRef]
- Li, Q.; Yu, H.M.; Meng, X.F.; Lin, J.S.; Li, Y.J.; Hou, B.K. Ectopic expression of glycosyltransferase UGT76E11 increases flavonoid accumulation and enhances abiotic stress tolerance in Arabidopsis. Plant Biol. 2018, 20, 10–19. [Google Scholar] [CrossRef]
- Nagatomo, Y.; Usui, S.; Ito, T.; Kato, A.; Shimosaka, M.; Taguchi, G. Purification, molecular cloning and functional characterization of flavonoid C-glucosyltransferases from Fagopyrum esculentum M. (buckwheat) cotyledon. Plant J. 2014, 80, 437–448. [Google Scholar] [CrossRef]
- Yonekura-Sakakibara, K.; Fukushima, A.; Nakabayashi, R.; Hanada, K.; Matsuda, F.; Sugawara, S.; Inoue, E.; Kuromori, T.; Ito, T.; Shinozaki, K.; et al. Two glycosyltransferases involved in anthocyanin modification delineated by transcriptome independent component analysis in Arabidopsis thaliana. Plant J. 2012, 69, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Bak, S.; Beisson, F.; Bishop, G.; Hamberger, B.; Hofer, R.; Paquette, S.; Werck-Reichhart, D. Cytochromes p450. Arab. Book 2011, 9, e0144. [Google Scholar] [CrossRef] [PubMed]
- Fujino, N.; Tenma, N.; Waki, T.; Ito, K.; Komatsuzaki, Y.; Sugiyama, K.; Yamazaki, T.; Yoshida, S.; Hatayama, M.; Yamashita, S.; et al. Physical interactions among flavonoid enzymes in snapdragon and torenia reveal the diversity in the flavonoid metabolon organization of different plant species. Plant J. 2018, 94, 372–392. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, P.; Gupta, A.P.; Bedi, Y.S.; Vishwakarma, R.A.; Gandhi, S.G. Mannitol Stress Directs Flavonoid Metabolism toward Synthesis of Flavones via Differential Regulation of Two Cytochrome P450 Monooxygenases in Coleus forskohlii. Front Plant Sci. 2016, 7, 985. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Forkmann, G. Cloning and expression of flavone synthase II from Gerbera hybrids. Plant J. 1999, 20, 611–618. [Google Scholar] [CrossRef]
- Ayabe, S.; Akashi, T. Cytochrome P450s in flavonoid metabolism. Phytochem. Rev. 2006, 5, 271–282. [Google Scholar] [CrossRef]
- Jin, M.; Zhu, Z.B.; Guo, Q.S.; Shen, H.J.; Wang, Y.R. Growth and accumulation of bioactive compounds in medicinal Chrysanthemum morifolium Ramat. cv. ‘Chuju’ under different colored shade polyethylene. J. Med. Plants Res. 2012, 6, 398–404. [Google Scholar] [CrossRef]
- Schuler, M.A. The role of cytochrome P450 monooxygenases in plant-insect interactions. Plant Physiol. 1996, 112, 1411–1419. [Google Scholar] [CrossRef]
- Peng, Y.; Lin-Wang, K.; Cooney, J.M.; Wang, T.; Espley, R.V.; Allan, A.C. Differential regulation of the anthocyanin profile in purple kiwifruit (Actinidia species). Hortic. Res. 2019, 6, 3. [Google Scholar] [CrossRef]
Sample Availability: Not available. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Dihydromyricetin | Myricitrin | Quercitrin | Myricetin |
|---|---|---|---|---|
| May | 331.226 ± 1.976 | 20.889 ± 0.518 | 2.334 ± 0.025 | 2.455 ± 0.353 |
| August | 277.761 ± 4.908 | 15.836 ± 0.127 | 1.202 ± 0.0319 | 2.843 ± 0.482 |
| October | 220.605 ± 1.652 | 18.308 ± 0.328 | 1.659 ± 0.100 | 1.786 ± 0.152 |
| Item | Sample | Read Number | Base Number (bp) | GC Content | Q20 | Q30 |
|---|---|---|---|---|---|---|
| Raw data | S-5 | 44,211,695 | 5,526,461,833 | 45.83% | 95.31% | 91.25% |
| S-8 | 58,464,607 | 7,308,075,917 | 46.19% | 95.75% | 92.07% | |
| S-10 | 47,325,290 | 5,915,661,250 | 46.17% | 95.10% | 90.86% | |
| Clean data | S-5 | 43,135,219 | 5,391,902,417 | 45.88% | 95.74% | 91.71% |
| S-8 | 57,752,125 | 7,219,015,583 | 46.23% | 96.13% | 92.46% | |
| S-10 | 45,535,950 | 5,691,993,750 | 46.15% | 95.51% | 91.32% |
| Category | Account | Mapped Ratio (%) |
|---|---|---|
| Annotated | 50,054 | 59.67% |
| Swiss-Prot | 41,139 | 49.04% |
| Blast-NR | 38,096 | 45.41% |
| GO | 22,788 | 27.17% |
| Pfam | 19,906 | 23.73% |
| KEGG | 12,693 | 15.07% |
| EggNOG | 10,863 | 12.95% |
| TmHMM | 8860 | 10.56% |
| SignalP | 2967 | 3.54% |
| All | 83,886 | 100.00% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Zhou, P.; Gui, C.; Da, G.; Gong, L.; Zhang, X. Comparative Transcriptome Analysis of Ampelopsis megalophylla for Identifying Genes Involved in Flavonoid Biosynthesis and Accumulation during Different Seasons. Molecules 2019, 24, 1267. https://doi.org/10.3390/molecules24071267
Yang M, Zhou P, Gui C, Da G, Gong L, Zhang X. Comparative Transcriptome Analysis of Ampelopsis megalophylla for Identifying Genes Involved in Flavonoid Biosynthesis and Accumulation during Different Seasons. Molecules. 2019; 24(7):1267. https://doi.org/10.3390/molecules24071267
Chicago/Turabian StyleYang, Min, Peina Zhou, Chun Gui, Guozheng Da, Ling Gong, and Xiuqiao Zhang. 2019. "Comparative Transcriptome Analysis of Ampelopsis megalophylla for Identifying Genes Involved in Flavonoid Biosynthesis and Accumulation during Different Seasons" Molecules 24, no. 7: 1267. https://doi.org/10.3390/molecules24071267
APA StyleYang, M., Zhou, P., Gui, C., Da, G., Gong, L., & Zhang, X. (2019). Comparative Transcriptome Analysis of Ampelopsis megalophylla for Identifying Genes Involved in Flavonoid Biosynthesis and Accumulation during Different Seasons. Molecules, 24(7), 1267. https://doi.org/10.3390/molecules24071267