1. Introduction
Genetics plays a crucial role in oral health in numerous ways. Several oral health conditions have been linked to genetic mutations and chromosomal abnormalities. The most common oral disease, dental caries, is known to be influenced by a genetic makeup that may increase an individual’s susceptibility by impacting the structure and compromising the strength of the hard tissue of their teeth [
1,
2]. The risk of developing periodontal diseases like gingivitis or periodontitis may be increased by genetic variations that affect the body’s immune response to oral bacteria [
3,
4,
5]. Often, variables such as food, oral hygiene, and systemic diseases could be thought to have a larger impact on the etiology of the most common oral diseases if the supporting evidence for the role of genetic factors in oral conditions has not been systematically analyzed. Today, research has shown that some genetic mutations, when present, can increase the risk of developing oral cancers [
6], while other genetic factors can influence the size, shape, and position of teeth, impacting esthetics, function, and overall oral health. Genetic influences can also affect the development and function of salivary glands, including the amount of saliva produced and the quality of the saliva, both of which can impact oral health. Genetic impacts can even be manifested at birth with the development of craniofacial anomalies, such as cleft lip with/or without cleft palate, which are caused by a combination of genetic and environmental factors [
7].
It is essential to understand that while genetics determines the predisposition or risk of contracting a disease, it does not provide certainty that the individual will eventually develop the disease. Most oral diseases have multifactorial causes, including genetics, epigenetics, and environmental factors, that are interlinked. While genetics can contribute to oral health conditions, environmental and lifestyle factors, such as diet, oral hygiene habits, and the use of substances such as tobacco, alcohol, and recreational drugs, also play a significant role and can influence cellular function. Human DNA or genes could be considered analogous to a computer motherboard, which is the main component that controls all operations of a computing device. However, the intensity of the electricity, exposure to heat and cold, the hard disk space, and viral infections are external factors that can influence the “health” of the computer—i.e., its performance and efficiency. Similarly, an individual’s genetics is usually responsible for host factors such as salivary composition, immune responses, the structure of the enamel, dentin, and cementum (hard tissues of a tooth), as well as the sizes and shapes of the jaw, throat, teeth, and tongue. Additionally, environmental influences, such as fluoride exposure, oral hygiene habits, drug exposure, occupational exposure, oxygen levels, and radiation therapy, can influence optimal oral function and health. As such, disease risk and severity may be exacerbated or mitigated by the interactions of intrinsic/genetic and extrinsic/environmental factors. The goal of this study is to systematically review the literature to methodically categorize oral diseases and conditions based on their genetic or environmental linkages based on the number of supported peer-reviewed articles to affirm the genetic contribution to oral conditions.
2. Materials and Methods
2.1. Standardized Criteria and PRISMA Flowchart
This systematic review followed the Cochrane Collaboration guidelines and the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) criteria. The authors adhered to the current Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) criteria, as well as to the recently published models of systematic review to ensure the standardization of the data inclusion/exclusion criteria for studies investigating the cause or contribution of genetic factors in oral conditions.
2.2. Search Strategy
An electronic search was conducted using the NCBI and Cochrane databases from 1960 up to May 2024 to ensure a comprehensive analysis of all peer-reviewed articles pertaining to genetic contribution or genetic causes and various oral conditions. However, no articles were retrieved from the 1960–1970 decade in our data analysis. The search encompassed randomized controlled trials (RCTs), pilot studies, clinical trials, case reports, and review articles.
The keywords used in the literature search were “genetics”, or “genome” or “DNA variation” or “DNA mutation” and “Dentinogenesis imperfecta” or “Dental caries” or “Gingivitis” or “Dental Attrition”, or the remaining of the oral conditions, for the NCBI database. Similarly, the keywords used for Cochrane were “genetics”, or “genome” or “DNA variation” or “DNA mutation” AND “Dentinogenesis imperfecta” or “Dental caries” or “Gingivitis” or “Dental Attrition”, or the remaining of the oral conditions, for the article title and abstract. We conducted the search using these keywords for each oral condition separately. Relevant abstracts were downloaded from the NCBI and Cochrane databases. The abstracts of the retrieved articles were assessed to determine whether details related to the indication genetic factors or genetic mutations as the etiology of an oral condition or significant association between DNA mutations and an oral condition were included.
2.3. Inclusion Criteria
Articles meeting the following criteria were included: (1) published in English, (2) randomized controlled trails (RCTs), pilot studies, clinical studies, or animal studies, and (3) focused on genetic factors or DNA mutations leading or contributing to oral and dental disorders.
2.4. Exclusion Criteria
Articles meeting the following criteria were excluded: (1) reviews, (2) articles pertaining to epigenetics or environmental factors, (3) case reports not focused on genetic testing and genotyping, (4) duplicates, (5) questions not focused, and (6) published in a language other than English.
2.5. Data Collection and Analysis Process
Two calibrated curators independently reviewed all titles and abstracts for the inclusion and exclusion criteria, followed by a second review in case of any disagreement. The entire retrieved article was evaluated in case of any disagreements between the two curators regarding inclusion or exclusion. A third previously calibrated reviewer reviewed the articles that raised disagreement among the two curators. An evidence strength for the included articles was developed for the data analysis out of the total retrieved articles from the literature search as follows:
Evidence strength 0 for < 10 articles’ focus on genetic contribution or causes;
Evidence strength 1 for ≥ 10 articles’ focus on genetic contribution or causes;
Evidence strength 2 for ≥ 20 articles’ focus on genetic contribution or causes;
Evidence strength 3 for ≥ 30 articles’ focus on genetic contribution or causes;
Evidence strength 4 for ≥ 40 articles’ focus on genetic contribution or causes;
Evidence strength 5 for ≥ 50 articles’ focus on genetic contribution or causes.
3. Results
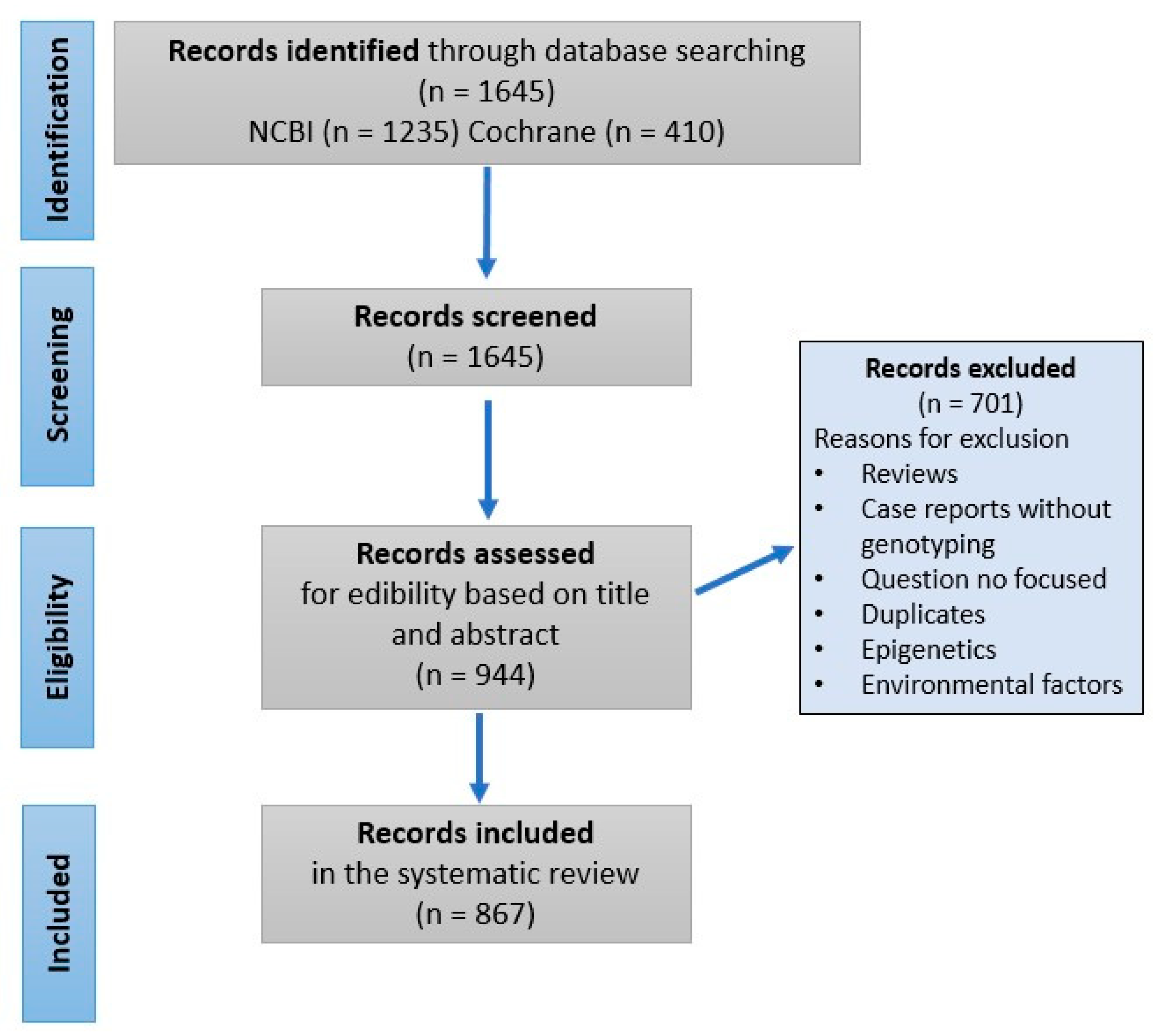
Initially, 1645 articles were included after completing electronic searches of the NCBI and Cochrane databases. Following screening, we excluded 701 articles based on the exclusion criteria mentioned above. The final systematic review included 860 and 7 publications from the NCBI and Cochrane databases, respectively (
Figure 1). A review of these publications resulted in categorizing orofacial disorders into those that have strong, moderate, or weak genetic influence (
Table 1). We describe briefly below some of the oral and dental anomalies caused primarily by genetic alterations.
3.1. Orofacial Disorders with Strong to Moderate Genetic Components
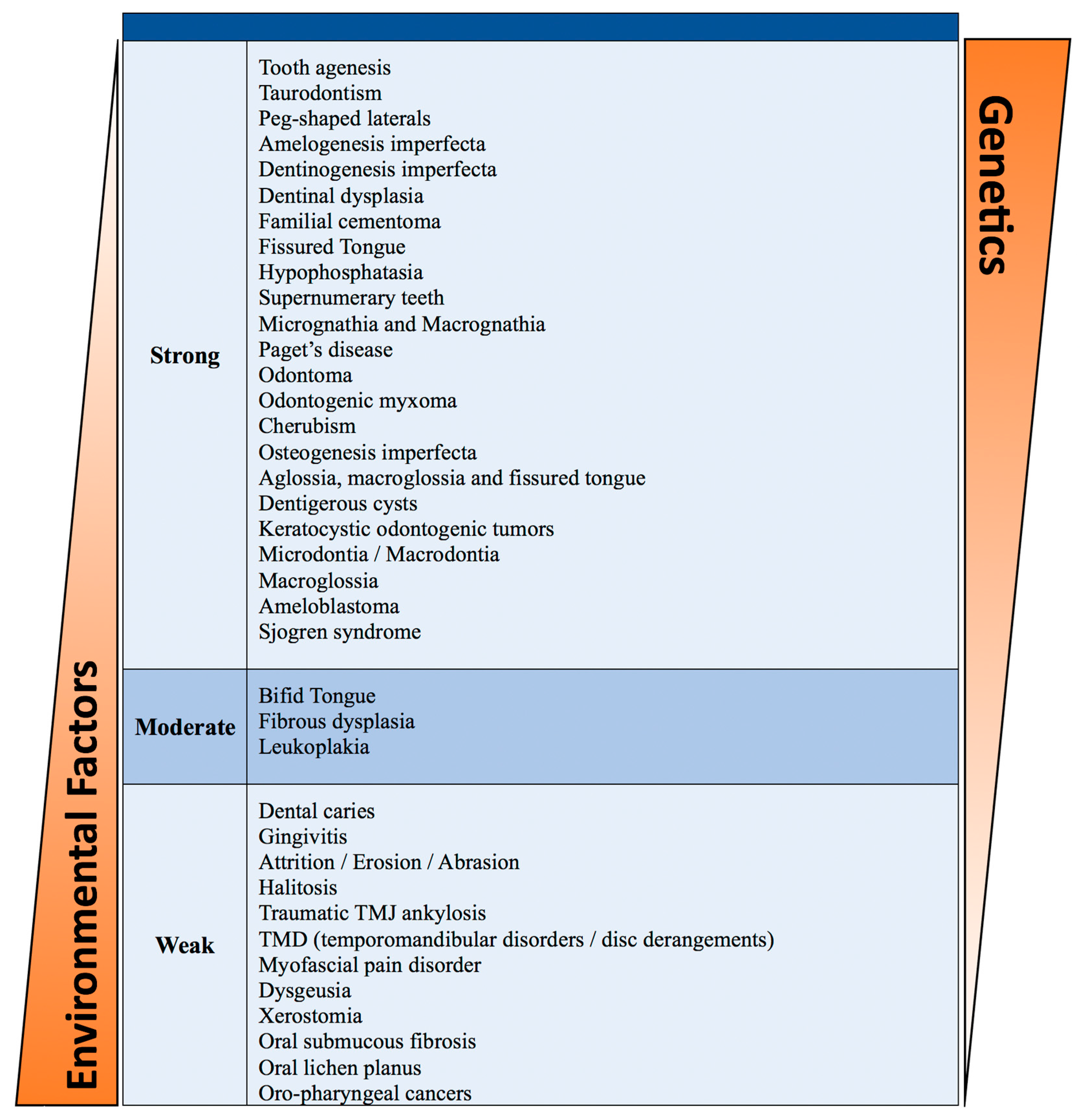
Oral disorders and diseases that occur at birth or in young children have a vital genetic component. These conditions may be attributed to a single gene disruption or chromosomal abnormality. Environmental factors can influence the incidence and severity of these conditions (
Table 1). The limitations of our data analysis are the potential biases in genetic studies due to population diversity and/or environmental interplay.
3.1.1. Tooth Agenesis
Hypodontia
Hypodontia is a condition in which a person has fewer teeth than normal, either because some teeth are missing or fail to develop properly (usually < 6 missing teeth). It is a relatively common condition that affects 5% (95% CI: 4.1–5.9) of the population in North America [
8]. In the permanent dentition, teeth that typically exhibit tooth agenesis are third molars (>20%), followed by mandibular second premolars (4.2%), maxillary lateral incisors (2.3%), and maxillary second premolars (2.2%) [
9]. Unilateral agenesis is more frequent than bilateral except for the maxillary lateral incisors [
10]. Hypodontia can occur as a nonsyndromic, isolated condition or as part of a congenital syndrome. It has been linked to mutations in several key genes, including
PAX9,
MSX1, and
AXIN2 [
11,
12,
13,
14,
15,
16,
17,
18,
19].
PAX9 mutations are particularly significant as they disrupt tooth development through interference with epithelial–mesenchymal interactions crucial for tooth germ and enamel organ formation [
12].
MSX1 is involved in the early stages of dental development, and its mutations can lead to a failure in the development of certain teeth [
19].
AXIN2, a gene that plays a role in the Wnt signaling pathway, affects the differentiation of dental mesenchymal cells [
20]. The etiology of this condition is multifactorial, with strong genetic components and contributions. Twin studies demonstrated that the loss of lateral incisors and premolars is inherited in an autosomal-dominant fashion with variable expressivity and incomplete penetrance [
10].
Oligodontia
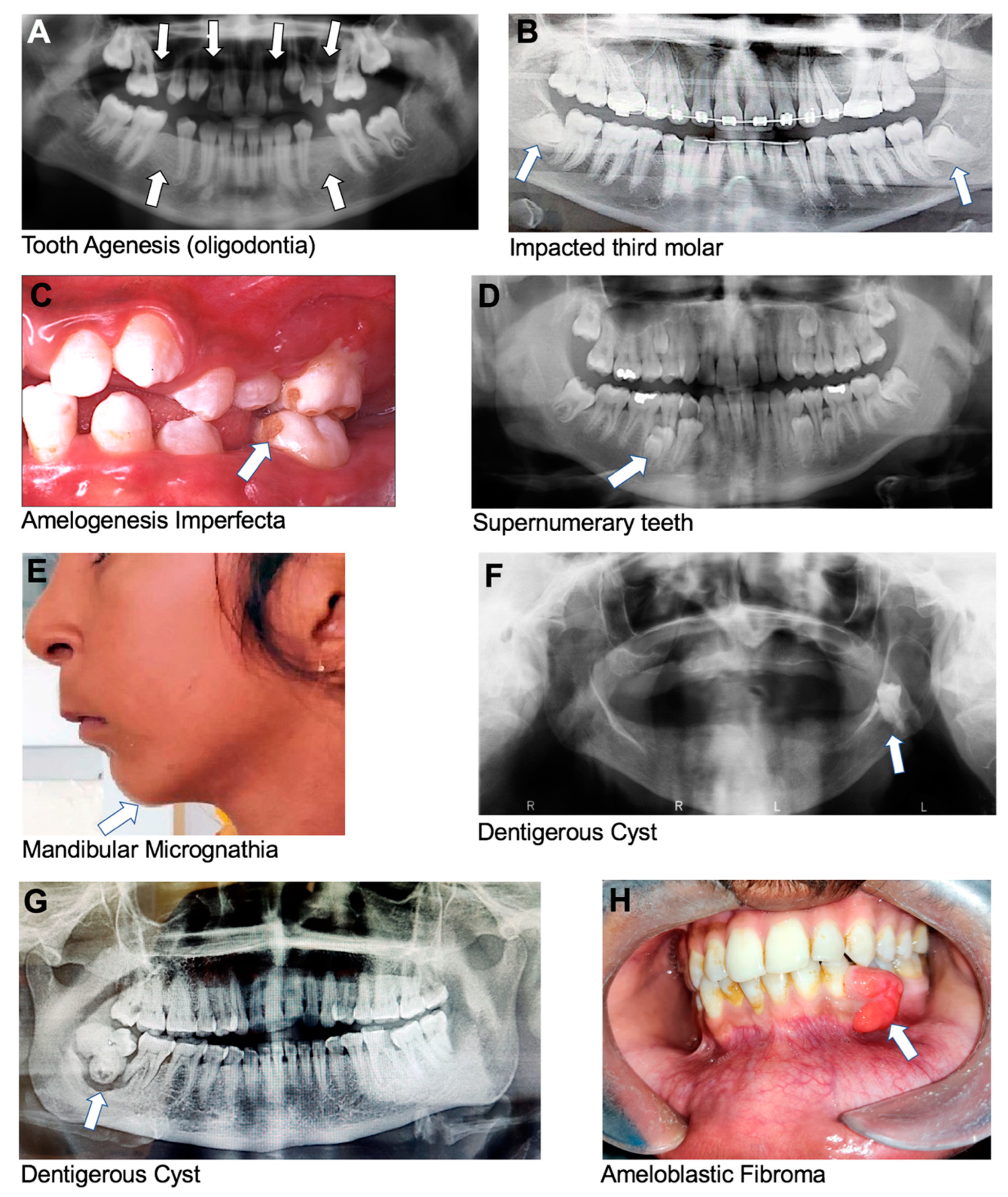
Oligodontia is a rare genetic condition that affects the development of teeth during childhood, characterized by six or more missing permanent teeth (
Figure 2A), and is similar to hypodontia. Oligodontia can be caused by mutations in various genes involved in tooth development, like
AXIN2,
MSX1, and
PAX9 [
11,
12,
13,
14,
16,
20,
21,
22,
23].
MSX1 mutations impair the initial stages of dental development, while
PAX9 mutations affect epithelial–mesenchymal interactions necessary for early tooth formation [
12,
19]. The condition can also be associated with other congenital syndromes, such as ectodermal dysplasia, Down syndrome, or familial cleft lip and palate, which are inherited in an autosomal-dominant pattern [
20,
21,
22,
23].
Anodontia
Anodontia is a rare genetic condition characterized by the complete absence of teeth, with individuals not developing primary or permanent teeth. Manifestations include a lack of alveolar ridge development, which reduces the lower face’s vertical dimension and causes the lips’ vermilion border to vanish, giving an elderly look [
24,
25].
Tooth agenesis is typically caused by genetic mutations in
MSX1,
PAX9, and
AXIN2 that affect tooth development [
26,
27]. In some cases, it can be associated with other syndromic disorders, such as derivatives of ectodermal dysplasia, Van der Woude syndrome, Rieger’s syndrome, and Witkop’s tooth and nail syndrome [
28,
29,
30,
31].
3.1.2. Tooth Shape Alterations
Taurodontism
Taurodontism is a condition that affects the shape and size of teeth, particularly molars. It is characterized by the elongation of the pulp chamber and the apical displacement of the furcation, resulting in a rectangular or cylindrical tooth shape. The roots of taurodont teeth are often shorter and thicker than those of normal teeth [
32]. Taurodontism can be a hereditary condition or associated with certain genetic syndromes, such as Klinefelter syndrome (extra copy of X chromosome) and Down syndrome (trisomy 21), which are inherited in a dominant pattern [
32,
33,
34]. It can also occur due to certain environmental factors, such as radiation exposure. Individuals with taurodontism may not experience any symptoms, but it can make dental procedures more challenging due to the altered shape and size of the teeth [
34].
Peg-shaped Laterals
Peg laterals, also known as peg-shaped or small lateral incisors, are a dental condition where the lateral incisors are smaller than usual and have a conical shape, instead of the usual rectangular shape, and peg laterals are more pointed and tapered [
35]. This condition is typically a result of genetics and can affect the lateral incisors unilaterally or bilaterally [
36].
3.1.3. Tooth Size Abnormalities
Microdontia
Microdontia is characterized by the presence of abnormally small teeth, which are smaller than the normal size for a person’s age and sex. Microdontia can be classified as localized/isolated, true generalized, or relatively generalized [
37], and it most commonly affects the upper lateral incisors. Microdontia is multifactorial with a strong genetic contribution and environmental modifications during tooth development, or it can be caused due to certain medical conditions, such as pituitary dwarfism (no definitive single genetic cause) [
38] or Down syndrome [
9,
39,
40].
Macrodontia
Macrodontia is characterized by abnormally large teeth, which are larger than the normal size for a person’s age and sex. Macrodontia can be classified as localized/isolated, true generalized, or relatively generalized [
32,
33]. It is most commonly seen in molars and premolars. Macrodontia can be caused by genetic and environmental factors during tooth development. It is associated with certain syndromes and medical conditions, including otodental syndrome, insulin-resistant diabetes, pituitary gigantism, and hemifacial hyperplasia [
9,
41,
42,
43].
3.1.4. Tooth Eruption Discrepancies
Impacted or Embedded Teeth
Impacted teeth are teeth that cannot emerge fully into the mouth due to a lack of space, an obstruction in their eruption path, and/or a misalignment resulting in a lack of an eruptive path; all these factors are influenced by genetics (
Figure 2B). This can occur with any tooth in the mouth, but it most commonly affects the third molars, or wisdom teeth, and maxillary canines [
44,
45].
3.1.5. Tooth Structure Abnormalities
Amelogenesis Imperfecta
Amelogenesis imperfecta (AI) is a genetic disorder that affects the development of tooth enamel, which is the hard, protective, outer layer of teeth. This condition can affect both primary and permanent teeth, and it can result in teeth that are discolored, weak, and prone to fracture [
46]. There are several types of AI, each with its own specific pattern of inheritance and symptoms. The disorder can be inherited in an autosomal-dominant, autosomal-recessive, or X-linked manner, depending on the specific genetic mutation involved [
28]. Mutations in the genes
AMELX,
ENAM,
MMP20,
KLK4, and
DLX3 are reported to be associated with the development of AI [
9,
28,
46,
47]. Clinically, teeth affected by AI may be yellow or brown, have rough or pitted surfaces, or be abnormally small or misshapen (
Figure 2C). Teeth affected by this condition may also be sensitive to temperature and pressure.
Dentinogenesis Imperfecta
Dentinogenesis imperfecta (DI) is a genetic disorder that affects the development of dentin that forms the bulk of the tooth structure beneath the enamel [
28]. The prevalence of DI is 1 in 8000 people [
48]. This condition can affect both primary and permanent teeth, resulting in teeth that have brown to blue discoloration, are weak, and are prone to attrition and breakage [
49]. There are three types of DI, and the specific symptoms and severity of the condition can vary widely depending on the type. Type I is characterized by abnormal dentin with concurrent osteogenesis imperfecta, when the primary teeth are more severely affected. Osteogenesis imperfecta is usually associated with mutations in the genes encoding collagen type 1,
COL1A1, and
COL1A2 [
28,
48,
50]. In type II of DI, patients have dentin abnormalities, but without bone disorders. Mutations in the gene
DSPP have been associated with the development of DI type II [
28,
48]. Type III of DI is a rare variant found in the tri-racial southern Maryland population known as the “Brandywine isolate”. Its clinical characteristics vary and are similar to those observed in DI-I and -II. The primary teeth show multiple pulp exposures and radiographically show a “shell”-like appearance [
48,
51].
In some cases, teeth affected by DI may be gray or brown in color and have a bulbous shape and a translucent appearance. Like AI, the mutations that cause DI affect the quality and quantity of the dentin produced, leading to the characteristic symptoms of the condition. It is associated with various syndromes, such as Ehlers–Danlos syndrome, Goldblatt syndrome, Schimke immuno-osseous dysplasia, and Brachio-Skeleto-Genital syndrome [
48,
52,
53].
Dentinal Dysplasia
Dentinal dysplasia (DD) is caused by mutations in genes that are involved in the development of dentin, specifically in odontogenesis [
54]. While DD-1 is a genetically heterogenous disease, DD-2 appears to result from dentine sialophosphoprotein (DSPP) mutations [
48,
55]. There are two types of dentinal dysplasia, type I and type II, and the specific symptoms and severity of the condition can vary widely depending on the type. In type I (radicular DD) dentinal dysplasia, affected teeth have a characteristic “shell-like” and “rootless tooth” appearance due to the lack of root development [
9]. In type II dentinal dysplasia, affected teeth have a similar appearance to DI type II, where the primary dentition is more severely affected. Characteristic features involve amorphous, tubule-less dentin, pulpal obliteration, and the opalescent appearance of the tooth [
9].
Hypophosphatasia
Hypophosphatasia (HPP) is an uncommon metabolic disorder induced by a mutation in the alkaline phosphatase (ALPL) gene affecting the liver, bones, and/or kidneys [
9,
56]. Dental features associated with HPP are acellular cementum aplasia or hypoplasia, enlarged pulp chambers linked with faulty dentin mineralization, and early exfoliation of both primary and secondary teeth [
9].
3.1.6. Supernumerary Teeth
Supernumerary teeth are extra teeth that develop in the dental arch beyond the normal set of teeth. Supernumerary teeth can be impacted or can erupt into any part of the dental arch, but they are most commonly found in the anterior maxillary region (
Figure 2D) [
28]. A mesiodens is a type of supernumerary tooth that is located in the midline between the two maxillary central incisors and is the most common type of supernumerary tooth, and it can occur in both primary and permanent dentitions. A mesiodens can cause problems, such as the delayed eruption of permanent incisors, the crowding of teeth, diastema (gaps between teeth), and the misalignment of teeth [
57]. In rare cases, a mesiodens can also lead to cyst formation and infection. A paramolar is a supernumerary tooth that forms towards the buccal or palatal of maxillary molars. The most common issue caused by paramolars is the crowding of existing teeth, which can lead to misalignment and improper bite. Paramolars can also cause pain and discomfort if they impinge upon adjacent teeth or if they cause gingival inflammation [
58]. In many cases, supernumerary teeth have been reported in patients with different types of disorders and syndromes, including cleidocranial dysplasia [
28,
57,
59,
60], Ehlers–Danlos syndrome type III [
28,
59,
60], Ellis–Van Creveld syndrome [
28,
59,
60], Gardner’s syndrome [
28,
57,
59,
60], orofaciodigital syndrome type I [
28,
60], and cleft lip and/or palate [
28,
57,
60]. Supernumerary teeth are associated with mutation in the genes
RUNX2 and
APC [
28,
61].
3.1.7. Abnormalities of Jaw Size and Structure
Micrognathia
Micrognathia is a congenital medical condition characterized by an abnormally small mandible or maxilla (
Figure 2E). Micrognathia can be caused by various factors, including genetic abnormalities, or problems with the growth and development of the jaw bones during fetal development [
62]. It can also occur due to environmental factors, such as exposure to certain medications or toxins during pregnancy [
62]. Mutations in more than fifteen different groups of genes have been associated with the development of micrognathia [
62]. Micrognathia can occur in isolation or as part of a symptom of other craniofacial conditions. For example, micrognathia is often seen as part of the Pierre Robin sequence that is mostly caused by mutations in
SOX9 [
62,
63]. The Pierre Robin sequence occurs in about 1 per 8500 live births [
64]. It is called a sequence due to a sequence of events that occur during fetal development—the mandible does not grow enough, which causes the tongue to be pushed back, preventing the secondary palatal shelves from developing, leading to a failure of the palatal bones to close and remain separated in the midline. Babies born with the Pierre Robin sequence may have difficulty breathing, feeding, and/or sleeping. Symptoms can range from being very mild to quite severe, including an underdeveloped mandible, cleft palate, glossoptosis, and airway obstruction [
64].
Macrognathia
Macrognathia is a rare condition characterized by an abnormally large mandible or maxilla and can be congenital or acquired. Congenital macrognathia is usually caused by genetic abnormalities or developmental disorders, whereas acquired macrognathia may result from conditions such as acromegaly, trauma, or tumor growth [
65]. A classic example of macrognathia is seen in the Hapsburg royal family, who exhibited a severe underbite (class III malocclusion) that was inherited over multiple generations in an autosomal-dominant fashion [
65].
3.1.8. Bone Disorders
Paget’s Disease of Bone
Paget’s disease (PD), also known as osteitis deformans, is a rare, chronic bone disorder that affects the normal formation and breakdown of bone tissue. It is characterized by the abnormal growth of bone tissue, which can cause bones to become enlarged, weak, and deformed [
66,
67]. The exact cause of Paget’s disease is unknown, but it is thought to be related to a combination of genetic and environmental factors. Symptoms of Paget’s disease can vary widely but may include bone pain, joint pain and stiffness, bone deformities, hearing loss, and headaches. In some cases, the initial stage of the condition may be asymptomatic and may only be discovered through routine medical imaging [
66]. Treatment for Paget’s disease typically involves medications to control bone turnover and pain. Bisphosphonates [
67], calcitonin, and other drugs that inhibit bone resorption are commonly used to manage the symptoms, which increase the risk for osteonecrosis of the jaw. In some cases, surgery may be necessary to correct bone deformities or replace joints that have been damaged by the disease. Without treatment, Paget’s disease can lead to complications such as fractures, arthritis, and nerve compression [
66]. The occurrence of PD has a familial tendency, and it is linked to polymorphisms in DNA coding in the centrosome structure. As a result, genetics may play a significant part in deciding the prevalence of Paget’s disease [
66]. Mutations in the
SQSTM gene have been strongly associated with the occurrence of Paget’s disease [
67,
68].
Fibrous Dysplasia
Fibrous dysplasia is a form of benign fibro-osseous disorder characterized by altered osteogenesis. It can be categorized into monostotic (affects a single bone) or polyostotic (affects multiple bones) [
67]. The maxilla is more commonly affected when compared to the mandible [
67,
69]. The disorder is manifested as a painless bone enlargement. The monostotic variant can occur in the jaw, as well as in the ethmoidal or calvarial bones. The polyostotic variants frequently constitute McCune–Albright syndrome (MAS) [
67,
70]. MAS is a classic triad of café-au-lait skin pigmentation, polyostotic fibrous dysplasia, and peripheral precocious puberty [
71]. Most of the features of MAS can be attributed to mutations in
GNAS [
67]. Several mutations can lead to gain of function by causing overactivity in the target tissues, as well as a variety of clinical symptoms that vary in magnitude and age of onset [
72].
Cherubism
Cherubism is an extremely rare bone disorder where bone is resorbed only in the jaw bones (mandible and maxilla). The resulting cavities in bone are always symmetrical and fill up with expansive fibro-osseous tissues, giving patients a cherub-like appearance. Cherubism is caused by mutations in the
SH3BP2 gene, which plays a role in regulating bone growth and remodeling [
73,
74,
75]. Symptoms of cherubism usually appear during childhood and may include facial swelling, pain, deformities, and dental problems, such as delayed tooth eruption or displacement of teeth. In some cases, cherubism can also affect the eyes, leading to vision problems or eye movement abnormalities [
73,
76].
Osteogenesis Imperfecta
Osteogenesis imperfecta (OI) is a rare heritable bone disease that affects 8 in 100,000 people [
77]. It is also known as “brittle bone disease”. A dominant mutation in the genes
COL1A1 and
COL1A2 has been associated with OI [
77,
78,
79,
80]. Aside from bone fragility, the conventional description of OI manifestation includes blue or grey scleral discoloration and tooth structural defects known as dentinogenesis imperfecta (DI) [
78]. OI can be broadly classified into four types, where type 1 is mild, type 2 is neonatal lethal, type 3 is severe, and type 4 is moderately severe [
78]. Characteristic features of OI include a reduction in bone mass, skeletal deformities, growth defects, and fragility fractures.
3.1.9. Tongue Anomalies
Ankyloglossia
Ankyloglossia, also known as tongue-tie, is a condition in which the tongue is tethered to the floor of the mouth by a short and tight lingual frenulum, restricting tongue movements [
81]. Possible complications associated with ankyloglossia are difficulty in breastfeeding, speech impairment from breastfeeding, speech impairment, or social or mechanical issues, such as the inability to lick the lips or play wind instruments [
82]. The milder forms of ankyloglossia might resolve with age, or affected people may learn to adapt effectively to their restricted lingual mobility, while in more impactful cases, frenotomy, frenectomy, or frenuloplasty can be beneficial [
83,
84,
85].
Aglossia
Aglossia is a rare congenital condition denoting an absence of the tongue or a severely underdeveloped tongue. This can result in significant difficulties with speaking, eating, and swallowing. Aglossia may occur on its own or as part of a syndromic condition, such as aglossia–adactylia syndrome or oromandibular limb hypogenesis syndrome [
86,
87,
88].
Bifid Tongue
Bifid tongue, also known as cleft tongue or forked tongue, is a rare condition in which the tongue is split into two distinct sections, giving it a “forked” appearance. Bifid tongue occurs during fetal development when the tongue fails to fuse properly at the lingual septum. In most cases, bifid tongue does not cause any significant health problems, and patients with this condition can eat, speak, and swallow normally. However, in some cases, bifid tongue may be associated with cleft palate or other genetic disorders [
89].
Macroglossia
Macroglossia is a condition in which the tongue is abnormally large in proportion to the oral cavity and may cause functional and/or esthetic concerns. It can be congenital or acquired. In the pediatric population, macroglossia can be a sign of an underlying condition, such as Down syndrome [
90,
91], Beckwith–Wiedemann syndrome (BWS) [
90,
92], or mucopolysaccharidosis (Maroteaux–Lamy syndrome) [
90,
93]. In adults, macroglossia can be a symptom of conditions such as hypothyroidism, amyloidosis, or acromegaly [
90,
94].
Fissured Tongue
Fissured tongue, also known as lingua plicata, is a benign condition in which the surface of the tongue appears to be grooved or furrowed. The fissures or grooves can vary in depth and can run parallel or perpendicular to each other. Fissured tongue is a relatively common condition with an incidence of 5–10% in the global population [
95]. It is usually not painful or associated with any significant symptoms, but there may be some discomfort or sensitivity when eating certain foods. Fissured tongue is often seen in families and is more common in people with certain conditions, such as Down syndrome [
96,
97,
98], Melkersson–Rosenthal syndrome [
96,
99], or psoriasis [
96,
100,
101].
3.1.10. Odontogenic Tumors and Cysts
Odontogenic tumors are a heterogeneous group of tumors that originate from the tissues that form teeth and other structures related to teeth, such as the jawbone and oral mucosa [
102]. These tumors can be benign or malignant and can occur at any age, although they are most commonly seen in adults [
103]. Although the etiology of most odontogenic tumors is unknown, there have been significant studies identifying the genetic underpinnings of particular odontogenic tumors [
104]. Some common types of odontogenic lesions are as follows:
Ameloblastoma
This is a slow-growing, locally invasive, benign tumor that arises from the cells that form the enamel of the teeth. It has a global incidence rate of 0.92 cases per million person-years [
105]. Ameloblastoma constitutes approximately 10% of the odontogenic tumors [
105]. It is most commonly found in the mandibular molar region [
106]. The exact etiology of ameloblastoma is unknown, but mutations in genes involved in the MAPK pathway, such as
BRAFV600E, are associated with ameloblastomas [
105,
107,
108]. It usually causes a painless swelling resulting in facial asymmetry, malocclusion, or the loosening of teeth;
Odontoma
These are the most common odontogenic tumors, and the exact cause is unknown [
109,
110]. They are benign tumors composed of dental tissues such as enamel, dentin, the cementum, and pulp. They are classified into two types: complex odontomas and compound odontomas [
111]. Odontomas are generally evident radiographically as multiple unerupted teeth. It can cause tooth displacement and malocclusion [
112]. Odontomas are often associated with Gardner’s syndrome and Herman’s syndrome [
109,
113]. A few cases have also been seen in association with Rubinstein–Taybi syndrome [
114,
115];
Cementoma
This is a benign tumor that arises from the cementoblasts that form the cementum. The cementum is anatomically and functionally connected to the periodontal ligament, which helps anchor the root of the tooth to the adjacent alveolar bone. Cementomas can cause pain and swelling [
116]. A probable relation between mutations in
ANO5 and familial gigantiform cementoma has been reported by a few studies [
117,
118,
119];
Dentigerous Cyst
This is one of the most common odontogenic developmental cysts that originates from remnants of the reduced enamel epithelium [
120]. It is a fluid-filled sac that develops around an unerupted tooth (
Figure 2F,G). It is usually benign but can cause damage to surrounding teeth and bones [
121]. Mutations in the gene
PTCH and polymorphism in chromosome 1qh+ have been found in patients with dentigerous cysts [
121,
122];
Ameloblastic Fibroma
This is a benign tumor that arises from the cells that form the enamel and dentin of teeth. Ameloblastic fibromas are uncommon, accounting for approximately 2% of all odontogenic tumors [
123,
124]. The tumors have been designated as childhood and adolescent tumors because they occur virtually exclusively in the first and second decades of life [
124]. It is usually represented as a painless swelling, more commonly seen in the mandible compared to the maxilla (
Figure 2H) [
125];
Odontogenic Myxoma
This is a benign tumor that arises from the connective tissue that surrounds the teeth. It is locally invasive and has a high recurrence rate [
126]. It is rare and can cause bone destruction [
127]. Although myxomas are observed in conjunction with mutations in
PRKAR1, only a few cases of odontogenic myxomas have showed this mutation, and additional research is required to investigate the relationship [
128,
129];
Keratocystic Odontogenic Tumor (KCOT):
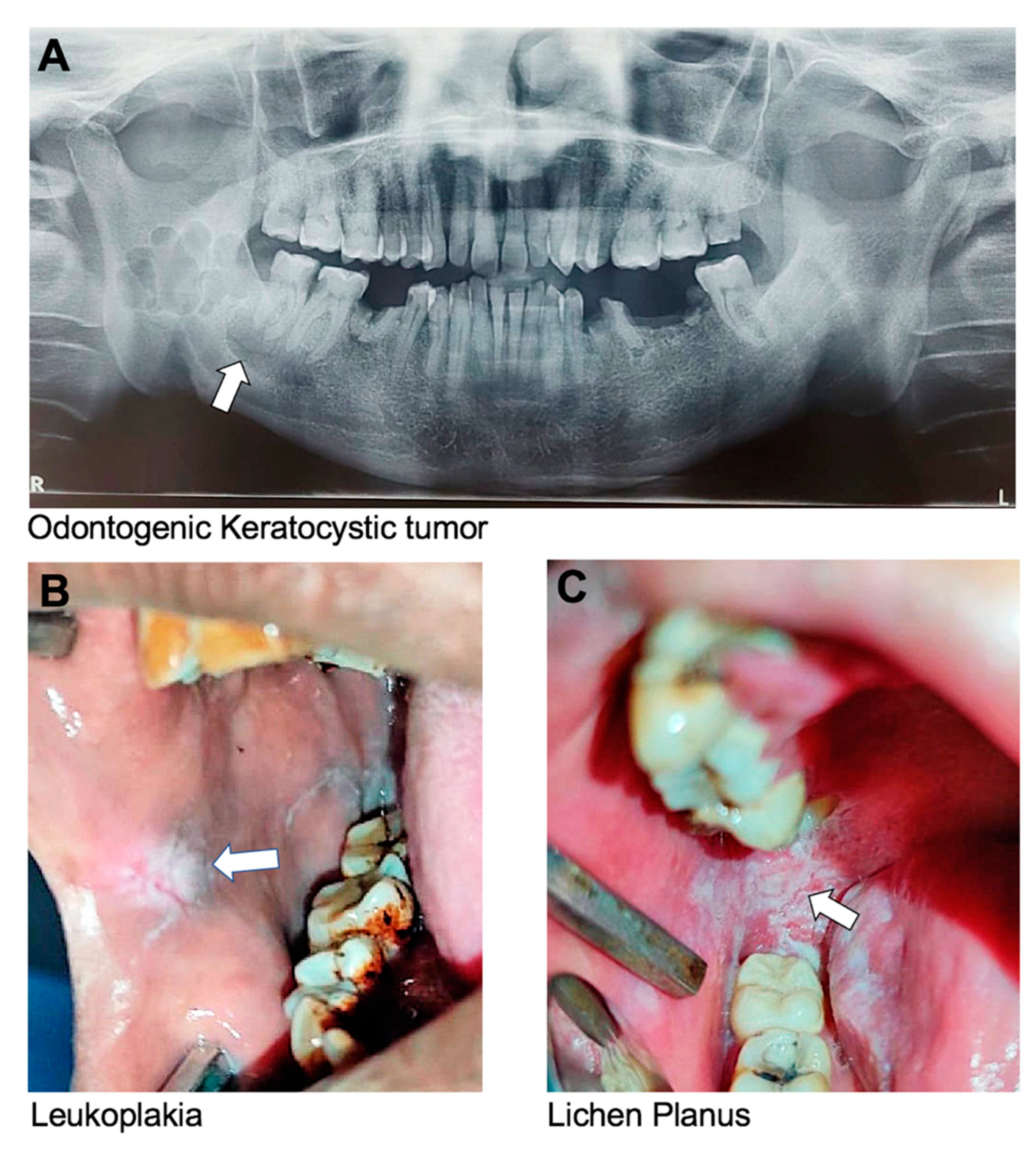
Previously known as an odontogenic keratocyst, a KCOT is a benign but locally aggressive cystic tumor that originates from the epithelial cells of the dental lamina. It most commonly affects the mandible and can cause bone destruction if left untreated (
Figure 3A). In some cases, genetic testing may be recommended for individuals with multiple or recurrent KCOTs, as there is an association with a genetic disorder called nevoid basal cell carcinoma syndrome [
106]. A few studies have associated mutations in
PTCH with KCOT [
122,
130,
131].
3.1.11. Premalignant Conditions and Diseases
Leukoplakia
Leukoplakia is a potentially malignant lesion characterized by white or grayish patches having a mud-crack-like appearance that cannot be scraped off. It forms on the mucous membranes of the mouth, including the tongue, buccal mucosa, and gingiva. The patches are typically thick, raised, and have a rough or scaly texture (
Figure 3B) [
132]. Leukoplakia is usually painless but may be sensitive to touch or hot, spicy foods. The exact cause of leukoplakia is unknown, but it is believed to be associated with irritants such as tobacco use, alcohol use, and certain viral infections. Long-term use of tobacco is the most common cause of leukoplakia, particularly in the form of chewing tobacco or snuff. Heavy alcohol consumption may also increase the risk of developing leukoplakia, especially when combined with tobacco use. Despite being a possibly malignant disease, the total malignant development of oral leukoplakia is on the order of 5% or higher [
133].
Oral Submucous Fibrosis
Oral submucous fibrosis (OSF) is a chronic, progressive disease that affects the oral mucosa. It is characterized by the formation of fibrous bands of tissue in the oral mucosa, which can lead to restricted mouth opening and difficulty eating and speaking. The exact cause of OSF is not fully understood, but it is believed to be associated with the use of betel quid, a substance commonly used in South Asian countries for chewing [
134]. Betel quid contains several substances, including areca nut, which is believed to be responsible for fibrosis. Symptoms of OSF may include a burning sensation in the mouth, dryness and a feeling of tightness in the mouth, difficulty opening the mouth, and changes in the texture and color of the oral mucosa. In advanced cases, the fibrous bands may extend into the throat and cause difficulty swallowing [
135].
Oral Lichen Planus
Oral lichen planus is a chronic inflammatory condition that affects the oral mucous membrane. Lichen planus can also affect the skin, nails, and hair. Oral lichen planus is characterized by the appearance of white, lacy patches on the labial and buccal mucosa, gingiva, and tongue (
Figure 3C) [
136,
137]. These patches may be painful or cause a burning sensation, particularly when eating or drinking acidic or spicy foods. In more severe cases, oral lichen planus may cause blistering, ulceration, or thickening of the oral mucosa. Oral lichen planus is considered to be an autoimmune condition. Certain medications, such as nonsteroidal anti-inflammatory drugs (NSAIDs), may also contribute to the development of oral lichen planus [
138].
3.1.12. Oral and Pharyngeal Cancers
Environmental and behavioral factors, such as tobacco use and infections with the human papillomavirus (HPV), have been implicated in various oral and pharyngeal cancers [
139]. Tobacco use is another major cause of several types of cancer, including those affecting the oral cavity, lungs, pharynx, esophagus, bladder, pancreas, and kidneys, among others. Tobacco contains harmful chemicals that can damage DNA and cause mutations, leading to cancer [
140]. The oropharyngeal cancers, collectively referred to as head and neck cancer (HNC), are challenging to manage, especially salivary gland cancers. Major salivary gland cancers comprise almost 11% of the total head and neck cancers in the United States [
141]. Oral squamous cell carcinoma (OSCC) makes up the majority of HNCs. The survival rates of patients with OSCC have not increased over the past few decades despite improvements in surgery, radiation, and chemotherapy. As a result, a whole new strategy for its treatment that makes use of genetic tools has emerged [
142,
143].
3.2. Genetic Testing for Oral Health Conditions
Genetic markers can be used to identify individuals who are more likely to experience congenital birth defects or juvenile oral health issues. The American Dental Association (ADA) does not recommend using genetic testing for prognosis and treatment planning. Nevertheless, the ADA does affirm that genetic testing holds potential for clinical application and future treatment planning. Moreover, there are a few dental clinics in the United States that offer genetic testing for aggressive periodontitis (AP) and Sjogren’s syndrome by testing mutations in Cathepsin C
(CTSC),
LYST,
COL5A1,
FcyIIB, and
FPRI for AP. Genetic risk factors have been newly identified for Sjögren’s syndrome, including
IRF5,
STAT4,
CXCR5,
IL12A,
HLA-DRA, and
BLK, which are linked to autoimmune disorders as well [
144,
145]. In the future, genetic testing for screening and counseling might be recommended to differentiate between oral diseases and conditions that have a strong genetic component and are inherited in a family in a dominant or recessive fashion (
Figure 4). This information could be crucial to identify individuals who are at high risk for developing severe oral diseases and disorders with strong genetic components, and for developing preventative and precision dental care to achieve the optimal treatment outcome [
146]. An encouraging advancement for genetically complex diseases is the development of the Polygenic Risk Score (PRS), which has emerged as a powerful tool that can assess an individual’s heritable risk of developing a disorder depending on the total number of genetic variants identified in a panel of genes involved in a particular condition [
147].
4. Conclusions
This systematic review highlights the pivotal role of genetics in shaping oral health outcomes. Through an in-depth analysis of various oral conditions influenced by genetic factors, our study highlights the necessity of integrating genetic considerations into oral healthcare practices. Our findings confirm that an individual’s susceptibility to oral health conditions is significantly influenced by their genetic makeup (
Figure 4). These results highlight the need to increase awareness of genetic risk factors for oral diseases and to provide adequate resources for patients, parents of pediatric patients, healthcare providers, and dental hygienists. Early detection of potential conditions through genetic education, counseling, and testing could be vital for enhancing oral healthcare delivery. Nonetheless, the feasibility and ethical implications of genetic testing may pose challenges to implementing these advanced approaches for prevention and early detection.
This review emphasizes the potential utility of genetic testing as a routine diagnostic tool for screening and counseling purposes. As we move towards better oral and overall health for our patients, integrating genetic information into healthcare delivery can pave the way for personalized preventive and treatment plans. While genetics plays a significant role, maintaining good oral hygiene habits, regular dental visits, and healthy lifestyle choices remain essential to minimize the risk of developing oral health problems. The future of oral healthcare should consider both genetic and environmental influences to identify individuals at high risk and develop appropriate preventative and timely treatment plans, ultimately promoting better oral health and overall well-being for our patients.
It is evident that an individual’s oral health may be significantly influenced by their genetic makeup. This evidence makes it important to raise awareness and provide adequate resources to patients, or in the case of pediatric patients, their parents, as well as their healthcare providers, and it aids in the early detection of a potential condition. The roles of genetic education, knowledge, experience, and counseling are therefore critical in oral healthcare delivery. Genetic testing for screening and counseling should emerge as a routine diagnostic aid as we progress on the path toward better oral health and better overall health for our patient populations.
Maintaining good oral hygiene habits, visiting a healthcare provider regularly, and making healthy lifestyle choices can help reduce the risk of developing some oral and systemic health conditions. The future of oral healthcare delivery should consider the genetic component and contribution to identifying individuals at high risk for detrimental oral health disorders to develop appropriate preventive and timely treatment plans.
Author Contributions
W.D.F. and A.J.-T. contributed to the conceptualization and preparation of this study; Z.L., A.J.-T. and W.D.F. wrote the original draft of the manuscript; J.V. and L.G. contributed to the writing, review, and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the NIDCR, grant number R21CA267006, and the UTHealth President Excellence Accelerator Program (0017324) to W.D.F.
Institutional Review Board Statement
IRB approval was not required for anonymous, unidentified clinical images for educational and research purposes.
Acknowledgments
The authors thank Ariadne Letra, (University of Pittsburgh), Cleverick (C.D.) Johnson, and Ritu Tiwari (UTHealth Houston School of Dentistry) for providing clinical images for this study. We are grateful to Ritu Tiwari, Oral and Maxillofacial Radiologist, for reviewing the diagnosis of the radiology and panoramic X-rays. We thank Gunjan Bhatia and Ghenwa Alayoubi for their outstanding help with the systematic review analysis.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cogulu, D.; Saglam, C. Genetic aspects of dental caries. Front. Dent. Med. 2022, 3, 1060177. [Google Scholar] [CrossRef]
- Wang, X.; Shaffer, J.; Weyant, R.; Cuenco, K.; DeSensi, R.; Crout, R.; McNeil, D.; Marazita, M. Genes and Their Effects on Dental Caries May Differ between Primary and Permanent Dentitions. Caries Res. 2010, 44, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Esberg, A.; Haworth, S.; Kuja-Halkola, R.; Magnusson, P.K.E.; Johansson, I. Heritability of Oral Microbiota and Immune Responses to Oral Bacteria. Microorganisms 2020, 8, 1126. [Google Scholar] [CrossRef] [PubMed]
- Nibali, L.; Di Iorio, A.; Tu, Y.K.; Vieira, A.R. Host genetics role in the pathogenesis of periodontal disease and caries. J. Clin. Periodontol. 2017, 44, S52–S78. [Google Scholar] [CrossRef]
- Pearn, J.; Gage, J. Genetics and oral health. Aust. Dent. J. 1987, 32, 1–10. [Google Scholar] [CrossRef]
- Jurel, S.K.; Gupta, D.S.; Singh, R.D.; Singh, M.; Srivastava, S. Genes and oral cancer. Indian J. Hum. Genet. 2014, 20, 4–9. [Google Scholar] [CrossRef]
- Kohli, S.S.; Kohli, V.S. A comprehensive review of the genetic basis of cleft lip and palate. J. Oral Maxillofac. Pathol. 2012, 16, 64–72. [Google Scholar] [CrossRef]
- Khalaf, K.; Miskelly, J.; Voge, E.; Macfarlane, T.V. Prevalence of hypodontia and associated factors: A systematic review and meta-analysis. J. Orthod. 2014, 41, 299–316. [Google Scholar] [CrossRef]
- Cobourne, M.T.; Sharpe, P.T. Diseases of the tooth: The genetic and molecular basis of inherited anomalies affecting the dentition. WIREs Dev. Biol. 2013, 2, 183–212. [Google Scholar] [CrossRef]
- Al-Ani, A.H.; Antoun, J.S.; Thomson, W.M.; Merriman, T.R.; Farella, M. Hypodontia: An Update on Its Etiology, Classification, and Clinical Management. Biomed. Res. Int. 2017, 2017, 9378325. [Google Scholar] [CrossRef]
- Alkhatib, R.; Obeidat, B.; AL-Eitan, L.; Abdo, N.; Obeidat, F.; Aman, H. Family-based association study of genetic analysis of paired box gene 9 polymorphisms in the peg-shaped teeth in the Jordanian Arab population. Arch. Oral Biol. 2021, 121, 104966. [Google Scholar] [CrossRef] [PubMed]
- Šerý, O.; Bonczek, O.; Hloušková, A.; Černochová, P.; Vaněk, J.; Míšek, I.; Krejčí, P.; Hollá, L.I. A screen of a large Czech cohort of oligodontia patients implicates a novel mutation in thePAX9gene. Eur. J. Oral Sci. 2015, 123, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.; Bansal, R.; Das, P. Whole Genome Sequencing Reveals Novel Non-Synonymous Mutation in Ectodysplasin A (EDA) Associated with Non-Syndromic X-Linked Dominant Congenital Tooth Agenesis. PLoS ONE 2014, 9, e106811. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, A.; da Fonseca, J.; Grinham, J.; Doudney, K.; Gomes, R.; de Paula, L.; Stanier, P. Autosomal-dominant Ankyloglossia and Tooth Number Anomalies. J. Dent. Res. 2009, 89, 128–132. [Google Scholar] [CrossRef]
- Shahid, M.; Balto, H.A.; Al-Hammad, N.; Joshi, S.; Khalil, H.S.; Somily, A.M.; Sinjilawi, N.A.-A.; Al-Ghamdi, S.; Faiyaz-Ul-Haque, M.; Dhillon, V.S. Mutations in MSX1, PAX9 and MMP20 genes in Saudi Arabian patients with tooth agenesis. Eur. J. Med. Genet. 2016, 59, 377–385. [Google Scholar] [CrossRef]
- Bonczek, O.; Balcar, V.J.; Šerý, O. PAX9 gene mutations and tooth agenesis: A review. Clin. Genet. 2017, 92, 467–476. [Google Scholar] [CrossRef]
- Kirac, D.; Eraydin, F.; Avcilar, T.; Ulucan, K.; Özdemir, F.; Guney, A.I.; Kaspar, E.; Keshi, E.; Isbir, T. Effects of PAX9 and MSX1 gene variants to hypodontia, tooth size and the type of congenitally missing teeth. Cell. Mol. Biol. 2016, 62, 78–84. [Google Scholar] [CrossRef]
- Sarkar, T.; Ranjan, P.; Kanathur, S.; Gupta, A.; Das, P. An in vitro and computational validation of a novel loss-of-functional mutation in PAX9 associated with non-syndromic tooth agenesis. Mol. Genet. Genom. MGG 2023, 298, 183–199. [Google Scholar] [CrossRef]
- Tatematsu, T.; Kimura, M.; Nakashima, M.; Machida, J.; Yamaguchi, S.; Shibata, A.; Goto, H.; Nakayama, A.; Higashi, Y.; Miyachi, H.; et al. An Aberrant Splice Acceptor Site Due to a Novel Intronic Nucleotide Substitution in MSX1 Gene Is the Cause of Congenital Tooth Agenesis in a Japanese Family. PLoS ONE 2015, 10, e0128227. [Google Scholar] [CrossRef]
- Bergendal, B.; Klar, J.; Stecksén-Blicks, C.; Norderyd, J.; Dahl, N. Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am. J. Med. Genet. Part A 2011, 155, 1616–1622. [Google Scholar] [CrossRef]
- Goldenberg, M.; Das, P.; Messersmith, M.; Stockton, D.W.; Patel, P.I.; D’Souza, R.N. Clinical, radiographic, and genetic evaluation of a novel form of autosomal-dominant oligodontia. J. Dent. Res. 2000, 79, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, S.; Keski-Filppula, R.; Alapulli, H.; Nieminen, P.; Anttonen, V. Familial oligodontia and regional odontodysplasia associated with a PAX9 initiation codon mutation. Clin. Oral Investig. 2019, 23, 4107–4111. [Google Scholar] [CrossRef]
- Bural, C.; Oztas, E.; Ozturk, S.; Bayraktar, G. Multidisciplinary treatment of non-syndromic oligodontia. Eur. J. Dent. 2012, 6, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Rathee, M.; Malik, P.; Dua, M.; Yadav, V. Early functional, esthetic, and psychological rehabilitation of preschool child with nonsyndromic oligodontia and anodontia in mixed dentition stage through conservative systematic approach: A case report with 5-year follow-up. Contemp. Clin. Dent. 2016, 7, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Varghese, A.A.; Xavier, A.M.; Ramanarayanan, V. Removable prosthetic management for tooth agenesis in the pediatric population: A systematic review of case reports and case series. J. Prosthet Dent. 2023, 132, 1250–e1. [Google Scholar] [CrossRef]
- Nieminen, P. Genetic basis of tooth agenesis. J. Exp. Zool. B Mol. Dev. Evol. 2009, 312B, 320–342. [Google Scholar] [CrossRef]
- Schonberger, S.; Kadry, R.; Shapira, Y.; Finkelstein, T. Permanent Tooth Agenesis and Associated Dental Anomalies among Orthodontically Treated Children. Children 2023, 10, 596. [Google Scholar] [CrossRef]
- Khan, M.I.; Ahmed, N.; Neela, P.K.; Unnisa, N. The Human Genetics of Dental Anomalies. Glob. Med. Genet. 2022, 9, 76–81. [Google Scholar] [CrossRef]
- Ritwik, P.; Patterson, K.K. Diagnosis of Tooth Agenesis in Childhood and Risk for Neoplasms in Adulthood. Ochsner J. 2018, 18, 345–350. [Google Scholar] [CrossRef]
- De Coster, P.J.; Marks, L.A.; Martens, L.C.; Huysseune, A. Dental agenesis: Genetic and clinical perspectives. J. Oral Pathol. Med. 2009, 38, 1–17. [Google Scholar] [CrossRef]
- AlQarni, M.A.; Togoo, R.A.; AlShahrani, I. A Review of Hypodontia: Classification, Prevalence, Etiology, Associated Anomalies, Clinical Implications and Treatment Options. World J. Dent. 2013, 4, 117–125. [Google Scholar] [CrossRef]
- Jayashankara, C.; Shivanna, A.K.; Sridhara, K.; Kumar, P.S. Taurodontism: A dental rarity. J. Oral Maxillofac. Pathol. 2013, 17, 478. [Google Scholar] [CrossRef] [PubMed]
- Dineshshankar, J.; Sivakumar, M.; Balasubramanium, A.M.; Kesavan, G.; Karthikeyan, M.; Prasad, V.S. Taurodontism. J. Pharm. Bioallied Sci. 2014, 6, S13–S15. [Google Scholar] [CrossRef] [PubMed]
- Chetty, M.; Roomaney, I.A.; Beighton, P. Taurodontism in dental genetics. BDJ Open 2021, 7, 25. [Google Scholar] [CrossRef]
- Omeish, N.; Nassif, A.; Feghali, S.; Vi-Fane, B.; Bosco, J. Esthetic and functional rehabilitation of peg-shaped maxillary lateral incisors: Practical recommendations. Clin. Case Rep. 2022, 10, e05507. [Google Scholar] [CrossRef]
- Devasya, A.; Sarpangala, M. Dracula tooth: A very rare case report of peg-shaped mandibular incisors. J. Forensic Dent. Sci. 2016, 8, 164–166. [Google Scholar] [CrossRef]
- Laverty, D.P.; Thomas, M.B.M. The restorative management of microdontia. Br. Dent. J. 2016, 221, 160–166. [Google Scholar] [CrossRef]
- Bargale, S.D.; Kiran, S.D. Non-syndromic occurrence of true generalized microdontia with mandibular mesiodens—A rare case. Head. Face Med. 2011, 7, 19. [Google Scholar] [CrossRef]
- Desai, S.S. Down syndrome: A review of the literature. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 1997, 84, 279–285. [Google Scholar] [CrossRef]
- Rahul, V.K.; Mathew, C.; Jose, S.; Thomas, G.; Noushad, M.C.; Feroz, T.P.M. Oral Manifestation in Mentally Challenged Children. J. Int. Oral Health 2015, 7, 37–41. [Google Scholar]
- Stolbizer, F.; Cripovich, V.; Paolini, A. Macrodontia associated with growth-hormone therapy: A case report and review of the literature. Eur. J. Paediatr. Dent. 2020, 21, 53–54. [Google Scholar] [CrossRef] [PubMed]
- Dugmore, C.R. Bilateral macrodontia of mandibular second premolars: A case report. Int. J. Paediatr. Dent. 2001, 11, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Kumar Mandal, P.; Ghosh, C. Bilateral Molariform Mandibular Second Premolars. Case Rep. Dent. 2015, 2015, 809463. [Google Scholar] [CrossRef] [PubMed]
- Santosh, P. Impacted Mandibular Third Molars: Review of Literature and a Proposal of a Combined Clinical and Radiological Classification. Ann. Med. Health Sci. Res. 2015, 5, 229–234. [Google Scholar] [CrossRef]
- Adeyemo, W.L.; James, O.; Oladega, A.A.; Adamson, O.O.; Adekunle, A.A.; Olorunsola, K.D.; Busch, T.; Butali, A. Correlation Between Height and Impacted Third Molars and Genetics Role in Third Molar Impaction. J. Maxillofac. Oral Surg. 2021, 20, 149–153. [Google Scholar] [CrossRef]
- Gadhia, K.; McDonald, S.; Arkutu, N.; Malik, K. Amelogenesis imperfecta: An introduction. Br. Dent. J. 2012, 212, 377–379. [Google Scholar] [CrossRef]
- Sabandal, M.M.I.; Schäfer, E. Amelogenesis imperfecta: Review of diagnostic findings and treatment concepts. Odontology 2016, 104, 245–256. [Google Scholar] [CrossRef]
- Barron, M.J.; McDonnell, S.T.; MacKie, I.; Dixon, M.J. Hereditary dentine disorders: Dentinogenesis imperfecta and dentine dysplasia. Orphanet J. Rare Dis. 2008, 3, 31. [Google Scholar] [CrossRef]
- Jindal, M.; Maheshwari, S.; Verma, R.; Khan, M.T. Comparative Study of Dentinogenesis Imperfecta in Different Families of the Same Topographical Region. Int. J. Clin. Pediatr. Dent. 2009, 2, 27–34. [Google Scholar] [CrossRef]
- Cauwels, R.G.E.C.; De Coster, P.J.; Mortier, G.R.; Marks, L.A.M.; Martens, L.C. Dentinogenesis imperfecta associated with short stature, hearing loss and mental retardation: A new syndrome with autosomal recessive inheritance? J. Oral Pathol. Med. 2005, 34, 444–446. [Google Scholar] [CrossRef]
- MacDougall, M.; Jeffords, L.; Gu, T.; Knight, C.; Frei, G.; Reus, B.; Otterud, B.; Leppert, M.; Leach, R. Genetic Linkage of the Dentinogenesis Imperfecta Type III Locus to Chromosome 4q. J. Dent. Res. 1999, 78, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.N. Dentinogenesis imperfecta-associated syndromes. Am. J. Med. Genet. 2001, 104, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.N. A newly recognized syndrome of skeletal dysplasia with opalescent and rootless teeth. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2001, 92, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Putrino, A.; Caputo, M.; Galeotti, A.; Marinelli, E.; Zaami, S. Type I Dentin Dysplasia: The Literature Review and Case Report of a Family Affected by Misrecognition and Late Diagnosis. Medicina 2023, 59, 1477. [Google Scholar] [CrossRef]
- Chen, D.; Li, X.; Lu, F.; Wang, Y.; Xiong, F.; Li, Q. Dentin dysplasia type I—A dental disease with genetic heterogeneity. Oral Dis. 2019, 25, 439–446. [Google Scholar] [CrossRef]
- Mornet, E. Hypophosphatasia. Orphanet J. Rare Dis. 2007, 2, 40. [Google Scholar] [CrossRef]
- Ata-Ali, F.; Ata-Ali, J.; Peñarrocha-Oltra, D.; Peñarrocha-Diago, M. Prevalence, etiology, diagnosis, treatment and complications of supernumerary teeth. J. Clin. Exp. Dent. 2014, 6, e414–e418. [Google Scholar] [CrossRef]
- Sulabha, A.N.; Sameer, C. Unusual Bilateral Paramolars Associated with Clinical Complications. Case Rep. Dent. 2015, 2015, 851765. [Google Scholar] [CrossRef]
- Fleming, P.S.; Xavier, G.M.; DiBiase, A.T.; Cobourne, M.T. Revisiting the supernumerary: The epidemiological and molecular basis of extra teeth. Br. Dent. J. 2010, 208, 25–30. [Google Scholar] [CrossRef]
- Anthonappa, R.P.; King, N.M.; Rabie, A.B.M. Aetiology of supernumerary teeth: A literature review. Eur. Arch. Paediatr. Dent. 2013, 14, 279–288. [Google Scholar] [CrossRef]
- Khambete, N.; Kumar, R. Genetics and presence of non-syndromic supernumerary teeth: A mystery case report and review of literature. Contemp. Clin. Dent. 2012, 3, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhao, Y.; Qian, Y.; Lu, C.; Shen, G.; Dai, J. A genetic-phenotypic classification for syndromic micrognathia. J. Hum. Genet. 2019, 64, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Saint-Jeannet, J.P. Sox9 function in craniofacial development and disease. Genesis 2011, 49, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Gangopadhyay, N.; Mendonca, D.A.; Woo, A.S. Pierre Robin Sequence. Semin. Plast. Surg. 2012, 26, 76–82. [Google Scholar] [CrossRef]
- Peacock, Z.S.; Klein, K.P.; Mulliken, J.B.; Kaban, L.B. The Habsburg Jaw—Re-examined. Am. J. Med. Genet. Part A 2014, 164, 2263–2269. [Google Scholar] [CrossRef]
- Karunakaran, K.; Murugesan, P.; Rajeshwar, G.; Babu, S. Paget’s disease of the mandible. J. Oral Maxillofac. Pathol. 2012, 16, 107–109. [Google Scholar] [CrossRef]
- Eversole, R.; Su, L.; ElMofty, S. Benign Fibro-Osseous Lesions of the Craniofacial Complex A Review. Head. Neck Pathol. 2008, 2, 177–202. [Google Scholar] [CrossRef]
- Daroszewska, A.; Ralston, S.H. Genetics of Paget’s disease of bone. Clin. Sci. 2005, 109, 257–263. [Google Scholar] [CrossRef]
- Waldron, C.A.; Giansanti, J.S. Benign fibro-osseous lesions of the jaws: A clinical-radiologic-histologic review of sixty-five cases. Oral Surg. Oral Med. Oral Pathol. 1973, 35, 190–201. [Google Scholar] [CrossRef]
- Eversole, L.R.; Sabes, W.R.; Rovin, S. Fibrous dysplasia: A nosologic problem in the diagnosis of fibro-osseous lesions of the jaws. J. Oral Pathol. 1972, 1, 189–220. [Google Scholar] [CrossRef]
- Lumbroso, S.; Paris, F.; Sultan, C.; European Collaborative Study. Activating Gsalpha mutations: Analysis of 113 patients with signs of McCune-Albright syndrome—A European Collaborative Study. J. Clin. Endocrinol. Metab. 2004, 89, 2107–2113. [Google Scholar] [CrossRef] [PubMed]
- Javaid, M.K.; Boyce, A.; Appelman-Dijkstra, N.; Ong, J.; Defabianis, P.; Offiah, A.; Arundel, P.; Shaw, N.; Pos, V.D.; Underhil, A.; et al. Best practice management guidelines for fibrous dysplasia/McCune-Albright syndrome: A consensus statement from the FD/MAS international consortium. Orphanet J. Rare Dis. 2019, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lima, G.d.M.G.; Almeida, J.D.; Cabral, L.A.G. Cherubism: Clinicoradiographic Features and Treatment. J. Oral Maxillofac. Res. 2010, 1, e2. [Google Scholar] [CrossRef] [PubMed]
- Reichenberger, E.J.; Levine, M.A.; Olsen, B.R.; Papadaki, M.E.; Lietman, S.A. The role of SH3BP2 in the pathophysiology of cherubism. Orphanet J. Rare Dis. 2012, 7, S5. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Mishra, R. Fibrous dysplasia and cherubism. Indian J. Plast. Surg. 2015, 48, 236–248. [Google Scholar] [CrossRef]
- Papadaki, M.E.; Lietman, S.A.; Levine, M.A.; Olsen, B.R.; Kaban, L.B.; Reichenberger, E.J. Cherubism: Best clinical practice. Orphanet J. Rare Dis. 2012, 7, S6. [Google Scholar] [CrossRef]
- Bbd, M.O.T.; Najirad, M.; Ma, M.S.; Rauch, F.; Sutton, V.R.; Lee, B.; Retrouvey, J.-M.; Esfandiari, S. Oral health-related quality of life in children and adolescents with osteogenesis imperfecta: Cross-sectional study. Orphanet J. Rare Dis. 2018, 13, 1–8. [Google Scholar] [CrossRef]
- Tauer, J.T.; Robinson, M.; Rauch, F. Osteogenesis Imperfecta: New Perspectives from Clinical and Translational Research. JBMR Plus 2019, 3, e10174. [Google Scholar] [CrossRef]
- Rauch, F.; Lalic, L.; Roughley, P.; Glorieux, F.H. Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J. Bone Miner. Res. 2010, 25, 1367–1374. [Google Scholar] [CrossRef]
- Charoenngam, N.; Nasr, A.; Shirvani, A.; Holick, M.F. Hereditary Metabolic Bone Diseases: A Review of Pathogenesis, Diagnosis and Management. Genes 2022, 13, 1880. [Google Scholar] [CrossRef]
- Jamilian, A.; Fattahi, F.H.; Kootanayi, N.G. Ankyloglossia and tongue mobility. Eur. Arch. Paediatr. Dent. 2014, 15, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Messner, A.H.; Lalakea, M.L. Ankyloglossia: Controversies in management. Int. J. Pediatr. Otorhinolaryngol. 2000, 54, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Chaubal, T.V.; Dixit, M.B. Ankyloglossia and its management. J. Indian Soc. Periodontol. 2011, 15, 270–272. [Google Scholar] [CrossRef]
- Marmet, C.; Shell, E.; Marmet, R. Neonatal frenotomy may be necessary to correct breastfeeding problems. J. Hum. Lact. 1990, 6, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Ferrés-Amat, E.; Pastor-Vera, T.; Ferrés-Amat, E.; Mareque-Bueno, J.; Prats-Armengol, J.; Ferrés-Padró, E. Multidisciplinary management of ankyloglossia in childhood. Treatment of 101 cases. A protocol. Med. Oral Patol. Oral Cir. Bucal. 2016, 21, e39–e47. [Google Scholar] [CrossRef]
- Yuan, G.; Singh, G.; Chen, S.; Perez, K.C.; Wu, Y.; Liu, B.; Helms, J.A. Cleft Palate and Aglossia Result from Perturbations in Wnt and Hedgehog Signaling. Cleft Palate-Craniofacial J. 2017, 54, 269–280. [Google Scholar] [CrossRef]
- Johnson, G.F.; Robinow, M. Aglossia-adactylia. Radiology 1978, 128, 127–132. [Google Scholar] [CrossRef]
- Gupta, S.R. Isolated aglossia congenita: A rare case of oromandibular limb hypogenesis syndrome type I B. J. Oral Maxillofac. Pathol. 2012, 16, 414–419. [Google Scholar] [CrossRef]
- Nutalapati, R.; Jayasuriya, N. Salivary hamartoma with a bifid tongue in an adult patient. Natl. J. Maxillofac. Surg. 2018, 9, 61–63. [Google Scholar] [CrossRef]
- Mueller, D.T.; Callanan, V.P. Congenital Malformations of the Oral Cavity. Otolaryngol. Clin. North Am. 2007, 40, 141–160. [Google Scholar] [CrossRef]
- Topouzelis, N.; Iliopoulos, C.; Kolokitha, O.E. Macroglossia. Int. Dent. J. 2011, 61, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Van Lierde, K.M.; Mortier, G.; Huysman, E.; Vermeersch, H. Long-term impact of tongue reduction on speech intelligibility, articulation and oromyofunctional behaviour in a child with Beckwith–Wiedemann syndrome. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Alpöz, A.R.; Çoker, M.; Çelen, E.; Ersin, N.K.; Gökçen, D.; van Diggelenc, O.P.; Huijmansc, J.G. The oral manifestations of Maroteaux-Lamy syndrome (mucopolysaccharidosis VI): A case report. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontology 2006, 101, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, C.; Shimizu, T.; Nakayama, Y.; Haraguchi, M.; Hakuta, C.; Itagaki, Y.; Ogura, A.; Murata, K.; Taira, M.; Numayama, T.; et al. Macroglossia in advanced amyotrophic lateral sclerosis. Muscle Nerve 2016, 54, 386–390. [Google Scholar] [CrossRef]
- Ferris, W.J.; Mikula, S.; Brown, R.; Farquharson, A. Oral Psoriasis of the Tongue: A Case Report. Cureus 2019, 11, e6318. [Google Scholar] [CrossRef]
- Sudarshan, R.; Sree Vijayabala, G.; Samata, Y.; Ravikiran, A. Newer Classification System for Fissured Tongue: An Epidemiological Approach. J. Trop. Med. 2015, 2015, 262079. [Google Scholar] [CrossRef]
- Kanamori, G.; Witter, M.; Brown, J.; Williams-Smith, L. Otolaryngologic Manifestations of Down Syndrome. Otolaryngol. Clin. North Am. 2000, 33, 1285–1292. [Google Scholar] [CrossRef]
- Roizen, N.J.; Patterson, D. Down’s syndrome. Lancet 2003, 361, 1281–1289. [Google Scholar] [CrossRef]
- Ozgursoy, O.B.; Karatayli Ozgursoy, S.; Tulunay, O.; Kemal, O.; Akyol, A.; Dursun, G. Melkersson-Rosenthal syndrome revisited as a misdiagnosed disease. Am. J. Otolaryngol. 2009, 30, 33–37. [Google Scholar] [CrossRef]
- Zhu, J.F.; Kaminski, M.; Pulitzer, D.; Hu, J.; Thomas, H. Psoriasis: Pathophysiology and oral manifestations. Oral Dis. 1996, 2, 135–144. [Google Scholar] [CrossRef]
- Moccia, R.; Zhang, L. Review of Oral Psoriasis. Psoriasis Forum 2014, 20a, 16–20. [Google Scholar] [CrossRef]
- Dornelles, F.M.L.M.; Wagner, V.P.; Fonseca, F.P.; Ariotti, C.M.; Carrard, V.C.; Vargas, P.A.; Sánchez-Romero, C.; Beovide, V.; Bologna-Molina, R.; Martins, M.D. BDNF/TrkB/Akt Signaling Pathway Epithelial Odontogenic Tumors and Keratocyst: An Immunohistochemical Study Comparative with Dental Germs. Appl. Immunohistochem. Mol. Morphol. 2020, 29, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Lima-Verde-Osterne, R.; Turatti, E.; Cordeiro-Teixeira, R.; Barroso-Cavalcante, R. The relative frequency of odontogenic tumors: A study of 376 cases in a Brazilian population. Med. Oral Patol. Oral Cir. Bucal. 2017, 22, e193–e200. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, L.M.; Coura, B.P.; Gomez, R.S.; Gomes, C.C. The Molecular Pathology of Odontogenic Tumors: Expanding the Spectrum of MAPK Pathway Driven Tumors. Front. Oral Health 2021, 2, 740788. [Google Scholar] [CrossRef]
- Ghai, S. Ameloblastoma: An Updated Narrative Review of an Enigmatic Tumor. Cureus 2022, 14, e27734. [Google Scholar] [CrossRef]
- Martín-Hernán, F.; Campo-Trapero, J.; Cano-Sánchez, J.; García-Martín, R.; Martínez-López, M.; Ballestín-Carcavilla, C. A comparative study of the expression of cyclin D1, COX-2, and KI-67 in odontogenic keratocyst vs. ameloblastoma vs. orthokeratinized odontogenic cyst. Rev. Esp. Patol. 2022, 55, 90–95. [Google Scholar] [CrossRef]
- Sweeney, R.T.; McClary, A.C.; Myers, B.R.; Biscocho, J.; Neahring, L.; A Kwei, K.; Qu, K.; Gong, X.; Ng, T.; Jones, C.D.; et al. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat. Genet. 2014, 46, 722–725. [Google Scholar] [CrossRef]
- Infante-Cossio, P.; Prats-Golczer, V.; Gonzalez-Perez, L.-M.; Belmonte-Caro, R.; Martinez-De-Fuentes, R.; Torres-Carranza, E.; Gacto-Sanchez, P.; Gomez-Cia, T. Treatment of recurrent mandibular ameloblastoma. Exp. Ther. Med. 2013, 6, 579–583. [Google Scholar] [CrossRef]
- Satish, V.; Prabhadevi, M.C.; Sharma, R. Odontome: A Brief Overview. Int. J. Clin. Pediatr. Dent. 2011, 4, 177–185. [Google Scholar] [CrossRef]
- Yadav, M.; Godge, P.; Meghana, S.M.; Kulkarni, S.R. Compound odontoma. Contemp. Clin. Dent. 2012, 3, S13–S15. [Google Scholar] [CrossRef]
- Nelson, B.L.; Thompson, L.D.R. Compound Odontoma. Head Neck Pathol. 2010, 4, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Kikuiri, T.; Mishima, H.; Imura, H.; Suzuki, S.; Matsuzawa, Y.; Nakamura, T.; Fukumoto, S.; Yoshimura, Y.; Watanabe, S.; Kinoshita, A.; et al. Patients with SATB2-associated syndrome exhibiting multiple odontomas. Am. J. Med. Genet. Part. A 2018, 176, 2614–2622. [Google Scholar] [CrossRef] [PubMed]
- Shekar, S.; Rao, R.S.; Gunasheela, B.; Supriya, N. Erupted compound odontome. J. Oral Maxillofac. Pathol. 2009, 13, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.W.; Rubinstein, J.H. Tumors in Rubinstein-Taybi syndrome. Am. J. Med. Genet. 1995, 56, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Tirali, R.E.; Sar, C.; Cehreli, S.B. Oro-Facio-Dental Findings of Rubinstein-Taybi Syndrome as a Useful Diagnostic Feature. J. Clin. Diagn. Res. 2014, 8, 276–278. [Google Scholar] [CrossRef]
- Punde, P.; Thorat, A.J.; Jangam, A.G.; Subhash, N.R.; Haleem, S.; Vadane, A.K. Cementum Malformations-Diagnostic Dilemma: Study in Western Maharashtra Population. J. Pharm. Bioallied Sci. 2021, 13, S620–S623. [Google Scholar] [CrossRef]
- Andreeva, T.V.; Tyazhelova, T.V.; Rykalina, V.N.; Gusev, F.E.; Goltsov, A.Y.; Zolotareva, O.I.; Aliseichik, M.P.; Borodina, T.A.; Grigorenko, A.P.; Reshetov, D.A.; et al. Whole exome sequencing links dental tumor to an autosomal-dominant mutation in ANO5 gene associated with gnathodiaphyseal dysplasia and muscle dystrophies. Sci. Rep. 2016, 6, 26440. [Google Scholar] [CrossRef]
- Zeng, B.; Liao, J.; Zhang, H.; Fu, S.; Chen, W.; Pan, G.; Li, Q.; Chen, W.; Ferrone, S.; Wu, B.; et al. Novel ANO5 mutation c.1067G>T (p.C356F) identified by whole genome sequencing in a big family with atypical gnathodiaphyseal dysplasia. Head Neck 2018, 41, 230–238. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, Y.; Zhu, L.; Cui, Y.; Gao, Y.; Zhou, C.X. Familial gigantiform cementoma with recurrent ANO5 p. Cys356Tyr mutations: Clinicopathological and genetic study with literature review. Mol. Genet. Genom. Med. 2024, 12, e2277. [Google Scholar] [CrossRef]
- McKinney, S.L.; Lukes, S.M. Dentigerous cyst in a young child: A case report. Can. J. Dent. Hyg. 2021, 55, 177–181. [Google Scholar]
- Ghafouri-Fard, S.; Atarbashi-Moghadam, S.; Taheri, M. Genetic factors in the pathogenesis of ameloblastoma, dentigerous cyst and odontogenic keratocyst. Gene 2021, 771, 145369. [Google Scholar] [CrossRef] [PubMed]
- Pavelić, B.; Levanat, S.; Crnić, I.; Kobler, P.; Anić, I.; Manojlović, S.; Šutalo, J. PTCH gene altered in dentigerous cysts. J. Oral Pathol. Med. 2001, 30, 569–576. [Google Scholar] [CrossRef]
- Cohen, D.M.; Bhattacharyya, I. Ameloblastic fibroma, ameloblastic fibro-odontoma, and odontoma. Oral Maxillofac. Surg. Clin. North Am. 2004, 16, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.L.; Folk, G.S. Ameloblastic Fibroma. Head and Neck Pathol. 2009, 3, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Dallera, P.; Bertonl, F.; Marchetti, C.; Bacchinl, P.; Campobassl, A. Ameloblastic fibroma: A follow-up of six cases. Int. J. Oral Maxillofac. Surg. 1996, 25, 199–202. [Google Scholar] [CrossRef]
- Leiser, Y.; Abu-El-Naaj, I.; Peled, M. Odontogenic myxoma—A case series and review of the surgical management. J. Cranio-Maxillofac. Surg. 2009, 37, 206–209. [Google Scholar] [CrossRef]
- Ghazali, A.B.; Arayasantiparb, R.; Juengsomjit, R.; Lam-Ubol, A. Central Odontogenic Myxoma: A Radiographic Analysis. Int. J. Dent. 2021, 2021, 1093412. [Google Scholar] [CrossRef]
- Gomes, C.C.; Diniz, M.G.; Duarte, A.P.; Bernardes, V.F.; Gomez, R.S. Molecular review of odontogenic myxoma. Oral Oncol. 2011, 47, 325–328. [Google Scholar] [CrossRef]
- Perdigão, P.F.; Stergiopoulos, S.G.; De Marco, L.; Matyakhina, L.; Boikos, S.A.; Gomez, R.S.; Pimenta, F.J.G.S.; Stratakis, C.A. Molecular and immunohistochemical investigation of protein kinase a regulatory subunit type 1A (PRKAR1A) in odontogenic myxomas. Genes, Chromosom. Cancer 2005, 44, 204–211. [Google Scholar] [CrossRef]
- Barreto, D.C.; Gomez, R.S.; Bale, A.E.; Boson, W.L.; De Marco, L. PTCH gene mutations in odontogenic keratocysts. J. Dent. Res. 2000, 79, 1418–1422. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, G.; Weng, T.; Du, J.; Wang, X.; Zhou, J.; Wang, S.; Yang, X. Disruption of Smad4 in Odontoblasts Causes Multiple Keratocystic Odontogenic Tumors and Tooth Malformation in Mice. Mol. Cell. Biol. 2009, 29, 5941–5951. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Udaltsova, N.; A Engels, E.; A Katzel, J.; Yanik, E.L.; A Katki, H.; Lingen, M.W.; Silverberg, M.J. Oral Leukoplakia and Risk of Progression to Oral Cancer: A Population-Based Cohort Study. JNCI J. Natl. Cancer Inst. 2019, 112, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Parlatescu, I.; Gheorghe, C.; Coculescu, E.; Tovaru, S. Oral Leukoplakia—An Update. Maedica 2014, 9, 88–93. [Google Scholar] [PubMed]
- Athukorala, I.A.; Tilakaratne, W.M.; Jayasinghe, R.D. Areca Nut Chewing: Initiation, Addiction, and Harmful Effects Emphasizing the Barriers and Importance of Cessation. J. Addict. 2021, 2021, 9967097. [Google Scholar] [CrossRef]
- Shah, J.S.; Lunagariya, N. Hearing Efficiency in Oral Submucous Fibrosis: A Clinical Study. Indian J. Otolaryngol. Head Neck Surg. 2022, 74, 3626–3630. [Google Scholar] [CrossRef]
- Chiang, C.P.; Yu-Fong Chang, J.; Wang, Y.P.; Wu, Y.H.; Lu, S.Y.; Sun, A. Oral lichen planus—Differential diagnoses, serum autoantibodies, hematinic deficiencies, and management. J. Formos. Med. Assoc. 2018, 117, 756–765. [Google Scholar] [CrossRef]
- Al-Hashimi, I.; Schifter, M.; Lockhart, P.B.; Wray, D.; Brennan, M.; Migliorati, C.A.; Axéll, T.; Bruce, A.J.; Carpenter, W.; Eisenberg, E.; et al. Oral lichen planus and oral lichenoid lesions: Diagnostic and therapeutic considerations. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2007, 103, S25.e1–S25.e12. [Google Scholar] [CrossRef]
- Potts, A.J.; Hamburger, J.; Scully, C. The medication of patients with oral lichen planus and the association of nonsteroidal anti-inflammatory drugs with erosive lesions. Oral Surg. Oral Med. Oral Pathol. 1987, 64, 541–543. [Google Scholar] [CrossRef]
- Howard, J.D.; Chung, C.H. Biology of Human Papillomavirus–Related Oropharyngeal Cancer. Semin. Radiat. Oncol. 2012, 22, 187–193. [Google Scholar] [CrossRef]
- Muthukrishnan, A.; Warnakulasuriya, S. Oral health consequences of smokeless tobacco use. Indian J. Med. Res. 2018, 148, 35–40. [Google Scholar] [CrossRef]
- Boukheris, H.; Curtis, R.E.; Land, C.E.; Dores, G.M. Incidence of carcinoma of the major salivary glands according to the World Health Organization (WHO) Classification, 1992–2006: A population-based study in the United States. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2899–2906. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Masthan, K.M.K.; Babu, N.A.; Dash, K.C. Gene Therapy in Oral Cancer: A Review. J. Clin. Diagn. Res. 2013, 7, 1261–1263. [Google Scholar] [CrossRef]
- Xi, S.; Grandis, J.R. Gene therapy for the treatment of oral squamous cell carcinoma. J. Dent. Res. 2003, 82, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg-Kreuz, J.; Rasmussen, A.; Sivils, K.; Nordmark, G. Genetics and epigenetics in primary Sjögren’s syndrome. Rheumatology 2021, 60, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius, G.E.; Björk, A.; Wahren-Herlenius, M. Genetics and epigenetics of primary Sjögren syndrome: Implications for future therapies. Nat. Rev. Rheumatol. 2023, 19, 288–306. [Google Scholar] [CrossRef]
- Hart, T.C.; Ferrell, R.E. Genetic testing considerations for oral medicine. J. Dent. Educ. 2002, 66, 1185–1202. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}