
Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease: Insights into Pathophysiology and Treatment

 ,
,  ,
,
Abstract

1. Introduction
2. Materials and Methods
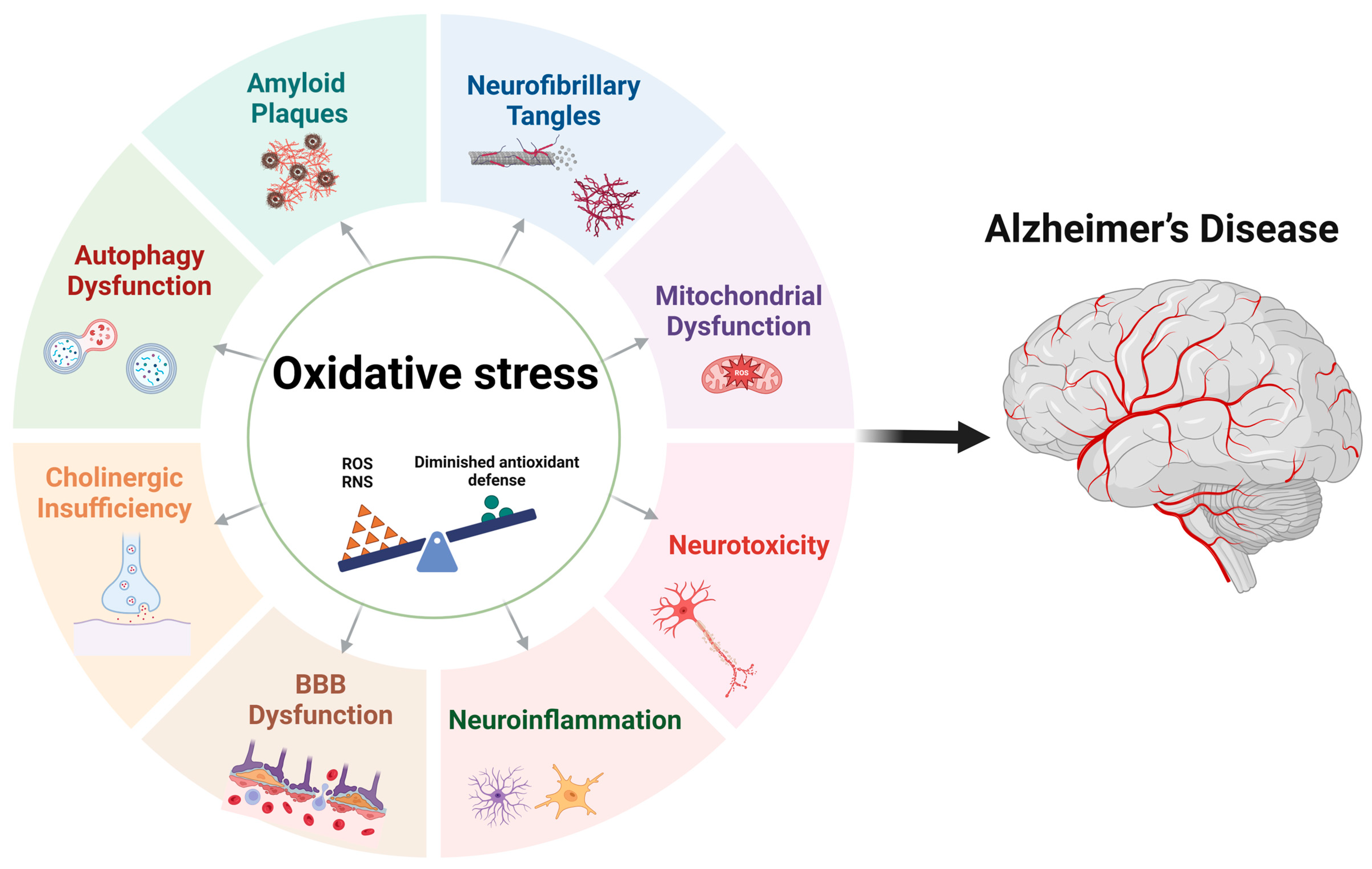
3. Oxidative Stress
4. Oxidative Stress in Alzheimer’s Disease
5. Mitochondrial Dysfunction in Alzheimer’s Disease
6. Oxidative Stress Impact on Cellular Functions
6.1. Protein Oxidation
6.2. Lipid Oxidation
6.3. DNA Oxidation
7. Neurobiological Implications
7.1. Oxidative Stress and Aβ Plaques
7.2. Oxidative Stress and Tau Hyperphosphorylation
7.3. Oxidative Stress and Glutamatergic Signaling and Synaptic Dysfunction
7.4. Role of MAO-A and MAO-B in Alzheimer’s Disease
8. Recent Advances in Alzheimer’s Disease Therapeutics Targeting Oxidative Stress

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT Number | The Intervention | Target/Outcome Measured | Status of the Study | Ref |
|---|---|---|---|---|
| NCT06797817 | Tributyrin | Cognitive function, blood markers (such as BDNF and GFAP), and gut health | Not Yet Recruiting | [184] |
| NCT01731093 | AT-001 | Serum and CSF selenium levels; isoprostanes in serum, urine, and CSF | Completed | [185] |
| NCT01388478 | R-pramipexole | Cognitive performance, cerebral glucose metabolism (via PET scan), and oxidative stress (CSF isoprostane levels) | Completed | [186] |
| NCT01731093 | Tebofortan | Cognitive and neuropsychiatric performance (N-BACE, MMSE, NPI-Q, CDR, GDS, Blessed scale), neurological status, and blood markers of inflammation and oxidative stress | Completed | [187] |
| NCT03630419 | Mito-Food Plan, Cellular Repair Therapy | Inflammation, Mini-Mental State Exam, Montreal Cognitive Assessment | Completed | [188] |
| NCT04740580 | Glycine plus N-acetylcysteine | Cognition, brain glucose uptake, inflammation, mitochondrial function, glutathione, key amino acids, oxidative stress, and BDNF | Recruiting | [189] |
| NCT00596024 | Lutein/zeaxanthin | Oxidative damage markers | Terminated | [190] |
| NCT04430517 | Nicotinamide Riboside | Cognition (RBANS), daily function (ADCS-ADL), mood (PHQ-9), neuropsychiatric symptoms (NPI-Q), brain NAD+/NADH ratio, CK/ATPase activity, and GSH levels | Recruiting | [191] |
| NCT05323812 | Edaravone | Oxidative stress markers (8-OHdG/8-OHG), plasma uric acid, amyloid (Aβ42, Aβ40), phosphorylated tau, GFAP, NfL, t-tau, YKL-40, and NRGN in plasma and CSF | Not Recruiting | [192] |
| NCT05977088 | Transcranial Magnetic Stimulation | Cognition, mood, neuropsychiatric symptoms, neurotrophic factors (BDNF, GDNF), inflammation, oxidative stress, lipid and amino acid profiles, exosomes, and brain activity (EEG) | Recruiting | [193] |
| NCT05929924 | Extra-Virgin Olive Oil | Blood metabolites and MRNA transcripts | Not Yet Recruiting | [194] |
| NCT06019117 | Probiotic K10 | Motor function (MDS-UPDRS), quality of life (PDQ-39, QOL), neuropsychiatric symptoms (NPI-Q), cognition (MMSE), and cortisol levels | Completed | [195] |
| NCT00090402 | Fish Oil and Alpha Lipoic Acid | F2-isoprostane Level Urine F2-Isoprostanes, Mini-Mental State Exam (MMSE), ADL/IADL | Completed | [196] |
| NCT04213391 | Sulforaphane | Cognition (ADAS-Cog, MMSE, MoCA, CIBIC-plus), daily function (ADCS-ADL), neuropsychiatric symptoms (NPI), oxidative stress, epigenetics, inflammation (cytokines/chemokines), metabolites, RNA expression, and gut microbiota | Unknown Status | [197] |
| NCT04559828 | Pomace Olive Oil, High-Oleic Sunflower Oil | Release of proinflammatory markers TNF-α, IL-6 and IL-1β by BV2 cells, redox markers | Completed | [198] |
| NCT04939792 | Vitamin D3, L-cysteine | Blood levels of 25(OH)VD, TNF-α, HOMA-IR | Completed | [199] |
9. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full Term |
| 3-NT | 3-nitrotyrosine |
| 4-HNE | 4-hydroxynonenal |
| 53BP1 | P53-binding protein 1 |
| 8-OHG | 7,8-dihydro-8-oxoguanine |
| 8-OHdG | 8-hydroxy-deoxyguanosine |
| ABAD | Aβ-binding alcohol dehydrogenase |
| AD | Alzheimer’s disease |
| AMP | Adenosine Monophosphate |
| APP | Amyloid precursor protein |
| ASK-1 | Apoptosis Signal-regulating Kinase 1 |
| ATP | Adenosine triphosphate |
| Akt | Akt kinase |
| Aβ | Amyloid-β |
| BBB | Blood–brain barrier |
| BER | Base excision repair |
| Bax | Bcl-2-associated X protein |
| Ca2+ | Calcium ions |
| DPL1 | Dihydrosphingosine phosphate lyase |
| DSB | Double-strand break |
| sSBs | Single-strand breaks |
| EGCG | Epigallocatechin gallate |
| EPR | Electron paramagnetic resonance |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| ETC | Electron transport chain |
| F2-IsoPs | F2-isoprostanes |
| FDA | Food and Drug Administration |
| Fe2+ | Ferrous ions |
| GLAST | Glutamate aspartate transporter |
| GLT-1 | Glutamate transporter 1 |
| GRX-1 | Glutaredoxin-1 |
| GS | Glutamine synthetase |
| GSK3-β | Glycogen synthase kinase 3β |
| GSSG | Glutathione |
| HO-1 | Heme oxygenase-1 |
| HSP60 | Heat shock protein 60 |
| H2O2 | Hydrogen peroxide |
| IL-1β | Interleukin-1 beta |
| Keap1 | Kelch-like ECH-Associated Protein 1 |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| MAPK | Mitogen-activated protein kinase |
| MnSOD | Mitochondrial superoxide dismutase |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NFTs | Neurofibrillary tangles |
| NFκ-β | Nuclear Factor kappa B |
| NMDAR | N-methyl-D-aspartate receptors |
| NOS | Nitric oxide synthase |
| NO• | Nitric oxide |
| NQO-1 | NAD(P)H quinone oxidoreductase-1 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| Nrf2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| ONOO− | Peroxynitrite |
| OS | Oxidative stress |
| NS | Nitrosative stress |
| PCAD | Preclinical Alzheimer’s disease |
| PCs | Protein carbonyls |
| PI3K | Phosphoinositide 3-kinase |
| PI3K/AKT | Phosphoinositide 3-kinase/protein kinase b |
| PI3K | Phosphoinositide 3-Kinase |
| PP2A | Protein phosphatase 2A |
| PS-1 | Presenilin-1 |
| Pin1 | Peptidyl-prolyl cis-trans isomerase 1 |
| QH2 | Ubiquinol |
| RIRR | ROS-induced ROS release |
| ROS | Reactive oxygen species |
| ROS/RNS | Reactive oxygen/nitrogen species |
| SOD | Superoxide dismutase |
| SOD1 | Copper-zinc SOD |
| SOD2 | Mitochondrial SOD |
| SSBs | Single-strand breaks |
| TNF-α | Tumor Necrosis Factor-alpha |
| Trx-1 | Thioredoxin-1 |
| aMCI | Amnestic mild cognitive impairment |
| cAMP | Adenosine monophosphate |
| mPTP | Mitochondrial permeability transition pore |
| mtDNA | Mitochondrial DNA |
References
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, J.; James, B.; Johnson, T.; Marin, A.; Weuve, J. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar]
- Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef]
- Hong, C.; Sun, L.; Liu, G.; Guan, B.; Li, C.; Luo, Y. Response of Global Health Towards the Challenges Presented by Population Aging. China CDC Wkly. 2023, 5, 884–887. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial cell-mediated neuroinflammation in Alzheimer’s disease. Int. J. Mol. Sci. 2022, 23, 10572. [Google Scholar] [CrossRef]
- Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Odum, J.; Shunnarah, J.G.; Austin, N.; Kaddoumi, A. Blood–Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies. Int. J. Mol. Sci. 2023, 24, 16288. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef]
- Mehta, R.I.; Mehta, R.I. The Vascular-Immune Hypothesis of Alzheimer’s Disease. Biomedicines 2023, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Guo, J.; Ye, X.-Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. Oxidative damage in neurodegeneration: Roles in the pathogenesis and progression of Alzheimer disease. Physiol. Rev. 2024, 104, 103–197. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease. Front. Aging Neurosci. 2021, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Wang, H.; Patterson, C. Atherosclerosis: Risks, Mechanisms, and Therapies; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Alqahtani, T.; Deore, S.L.; Kide, A.A.; Shende, B.A.; Sharma, R.; Chakole, R.D.; Nemade, L.S.; Kale, N.K.; Borah, S.; Deokar, S.S. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis-an updated review. Mitochondrion 2023, 71, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-g.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- FDA. Grants Accelerated Approval for Alzheimer’s Drug; Food and Drug Administration News Release: Silver Spring, MD, USA, 2021. [Google Scholar]
- Wang, Y. An insider’s perspective on FDA approval of aducanumab. Alzheimer’s Dement. 2023, 9, e12382. [Google Scholar] [CrossRef]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative stress in Alzheimer’s disease: Why did antioxidant therapy fail? Oxidative Med. Cell. Longev. 2014, 2014, 427318. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Nijtmans, L.G.; Dieteren, C.E.; Roestenberg, P.; Valsecchi, F.; Smeitink, J.A.; Willems, P.H. Mammalian mitochondrial complex I: Biogenesis, regulation, and reactive oxygen species generation. Antioxid. Redox Signal. 2010, 12, 1431–1470. [Google Scholar] [CrossRef]
- Forster, M.J.; Dubey, A.; Dawson, K.M.; Stutts, W.A.; Lal, H.; Sohal, R.S. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc. Natl. Acad. Sci. USA 1996, 93, 4765–4769. [Google Scholar] [CrossRef]
- Levine, R.L.; Williams, J.A.; Stadtman, E.P.; Shacter, E. Carbonyl Assays for Determination of Oxidatively Modified Proteins. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1994; Volume 233, pp. 346–357. [Google Scholar]
- Mustafa, S.A.; Karieb, S.S.; Davies, S.J.; Jha, A.N. Assessment of oxidative damage to DNA, transcriptional expression of key genes, lipid peroxidation and histopathological changes in carp Cyprinus carpio L. following exposure to chronic hypoxic and subsequent recovery in normoxic conditions. Mutagenesis 2015, 30, 107–116. [Google Scholar] [CrossRef]
- Headlam, H.A.; Davies, M.J. Markers of protein oxidation: Different oxidants give rise to variable yields of bound and released carbonyl products. Free. Radic. Biol. Med. 2004, 36, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.; Gibson, S. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef]
- Nadeau, P.J.; Charette, S.J.; Toledano, M.B.; Landry, J. Disulfide bond-mediated multimerization of Ask1 and its reduction by thioredoxin-1 regulate H2O2-induced c-Jun NH2-terminal kinase activation and apoptosis. Mol. Biol. Cell 2007, 18, 3903–3913. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Ozaki, T.; Nakanishi, M.; Kikuchi, H.; Yoshida, K.; Horie, H.; Kuwano, H.; Nakagawara, A. Oxidative stress induces p53-dependent apoptosis in hepatoblastoma cell through its nuclear translocation. Genes Cells 2007, 12, 461–471. [Google Scholar] [CrossRef]
- Cheignon, C.m.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Sheldon, R. Metal-Catalyzed Oxidations of Organic Compounds: Mechanistic Principles and Synthetic Methodology Including Biochemical Processes; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Doorn, J.A.; Petersen, D.R. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chem.Biol. Interact. 2003, 143, 93–100. [Google Scholar] [CrossRef]
- Therade-Matharan, S.; Laemmel, E.; Duranteau, J.; Vicaut, E. Reoxygenation after hypoxia and glucose depletion causes reactive oxygen species production by mitochondria in HUVEC. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1037–R1043. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Wolberg, A.; Kahwaji, C.I. Biochemistry, electron transport chain. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Kusano, T.; Nishino, T.; Okamoto, K.; Hille, R.; Nishino, T. The mechanism and significance of the conversion of xanthine dehydrogenase to xanthine oxidase in mammalian secretory gland cells. Redox Biol. 2023, 59, 102573. [Google Scholar] [CrossRef]
- Corvo, M.L.; Marinho, H.S.; Marcelino, P.; Lopes, R.M.; Vale, C.A.; Marques, C.R.; Martins, L.C.; Laverman, P.; Storm, G.; Martins, M.B.A. Superoxide dismutase enzymosomes: Carrier capacity optimization, in vivo behaviour and therapeutic activity. Pharm. Res. 2015, 32, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide radical and superoxide dismutases. Oxyg. Living Process. Interdiscip. Approach 1981, 250–272. [Google Scholar]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef]
- Butler, J.; Koppenol, W.H.; Margoliash, E. Kinetics and mechanism of the reduction of ferricytochrome c by the superoxide anion. J. Biol. Chem. 1982, 257, 10747–10750. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Andrés Juan, C.; Plou, F.J.; Pérez-Lebeña, E. Superoxide Anion Chemistry-its Role at the Core of the Innate Immunity. Int. J. Mol. Sci. 2023, 24, 1841. [Google Scholar] [CrossRef]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Bradley-Whitman, M.A.; Lovell, M.A. Biomarkers of lipid peroxidation in Alzheimer disease (AD): An update. Arch. Toxicol. 2015, 89, 1035–1044. [Google Scholar] [CrossRef]
- Schaur, R.J.; Siems, W.; Bresgen, N.; Eckl, P.M. 4-Hydroxy-nonenal—A bioactive lipid peroxidation product. Biomolecules 2015, 5, 2247–2337. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Bernard, L.P.; Sinha, C.; Ehrhart, J.; Nicklas, W.J. Excitotoxicity and oxidative stress during inhibition of energy metabolism. Dev. Neurosci. 1998, 20, 444–453. [Google Scholar] [CrossRef]
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress-A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Afzal, S.; Abdul Manap, A.S.; Attiq, A.; Albokhadaim, I.; Kandeel, M.; Alhojaily, S.M. From imbalance to impairment: The central role of reactive oxygen species in oxidative stress-induced disorders and therapeutic exploration. Front. Pharmacol. 2023, 14, 1269581. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-deMiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef]
- Eckert, A.; Schmitt, K.; Götz, J. Mitochondrial dysfunction—The beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-β toxicity. Alzheimer’s Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef]
- Khan, S.M.; Cassarino, D.S.; Abramova, N.N.; Keeney, P.M.; Borland, M.K.; Trimmer, P.A.; Krebs, C.T.; Bennett, J.C.; Parks, J.K.; Swerdlow, R.H.; et al. Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann. Neurol. 2000, 48, 148–155. [Google Scholar] [CrossRef]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1994, 36, 747–751. [Google Scholar] [CrossRef]
- Gao, R.; Ma, S.L. Is Mitochondria DNA Variation a Biomarker for AD? Genes 2022, 13, 1789. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, Y.; Wei, H. Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. J. Alzheimer’s Dis. Park. 2017, 7, 374. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Van der Paal, J.; Neyts, E.C.; Verlackt, C.C.W.; Bogaerts, A. Effect of lipid peroxidation on membrane permeability of cancer and normal cells subjected to oxidative stress. Chem. Sci. 2016, 7, 489–498. [Google Scholar] [CrossRef]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Höhn, A. Protein oxidation—Formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol. 2021, 42, 101901. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta BBA Proteins Proteom. 2005, 1703, 93–109. [Google Scholar] [CrossRef]
- Gonos, E.S.; Kapetanou, M.; Sereikaite, J.; Bartosz, G.; Naparło, K.; Grzesik, M.; Sadowska-Bartosz, I. Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging 2018, 10, 868–901. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Singh, M.; Dang, T.N.; Arseneault, M.; Ramassamy, C. Role of by-products of lipid oxidation in Alzheimer’s disease brain: A focus on acrolein. J. Alzheimer’s Dis. 2010, 21, 741–756. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef]
- Montine, T.J.; Peskind, E.R.; Quinn, J.F.; Wilson, A.M.; Montine, K.S.; Galasko, D. Increased cerebrospinal fluid F2-isoprostanes are associated with aging and latent Alzheimer’s disease as identified by biomarkers. Neuromolecular Med. 2011, 13, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.N.; Arseneault, M.; Murthy, V.; Ramassamy, C. Potential role of acrolein in neurodegeneration and in Alzheimer’s disease. Curr. Mol. Pharmacol. 2010, 3, 66–78. [Google Scholar] [PubMed]
- Ali, J.; Aziz, M.A.; Rashid, M.M.O.; Basher, M.A.; Islam, M.S. Propagation of age-related diseases due to the changes of lipid peroxide and antioxidant levels in elderly people: A narrative review. Health Sci. Rep. 2022, 5, e650. [Google Scholar] [CrossRef] [PubMed]
- Mark, R.J.; Lovell, M.A.; Markesbery, W.R.; Uchida, K.; Mattson, M.P. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J. Neurochem. 1997, 68, 255–264. [Google Scholar] [CrossRef]
- Selley, M.; Close, D.; Stern, S. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 383–388. [Google Scholar] [CrossRef]
- Tamagno, E.; Robino, G.; Obbili, A.; Bardini, P.; Aragno, M.; Parola, M.; Danni, O. H2O2 and 4-hydroxynonenal mediate amyloid β-induced neuronal apoptosis by activating JNKs and p38MAPK. Exp. Neurol. 2003, 180, 144–155. [Google Scholar] [CrossRef]
- Butterfield, D.A. Brain lipid peroxidation and alzheimer disease: Synergy between the Butterfield and Mattson laboratories. Ageing Res. Rev. 2020, 64, 101049. [Google Scholar] [CrossRef]
- Abdul, H.M.; Sultana, R.; St Clair, D.K.; Markesbery, W.R.; Butterfield, D.A. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic. Biol. Med. 2008, 45, 1420–1425. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Castegna, A.; Lauderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 2002, 23, 655–664. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid β-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Dusinska, M.; Gedik, C.M.; Stĕtina, R. Oxidative damage to DNA: Do we have a reliable biomarker? Environ. Health Perspect. 1996, 104, 465–469. [Google Scholar] [CrossRef]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef]
- Mattson, M.P.; Chan, S.L. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 2003, 34, 385–397. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999, 19, 1959–1964. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Gabbita, S.P.; Markesbery, W.R. Increased DNA oxidation and decreased levels of repair products in Alzheimer’s disease ventricular CSF. J. Neurochem. 1999, 72, 771–776. [Google Scholar] [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef]
- Chen, J.; Potlapalli, R.; Quan, H.; Chen, L.; Xie, Y.; Pouriyeh, S.; Sakib, N.; Liu, L.; Xie, Y. Exploring DNA Damage and Repair Mechanisms: A Review with Computational Insights. BioTech 2024, 13, 3. [Google Scholar] [CrossRef]
- Alexandrov, N.; Alexandrov, V. Computational science research methods for science education at PG level. Procedia Comput. Sci. 2015, 51, 1685–1693. [Google Scholar] [CrossRef]
- Milanowska, K.; Rother, K.; Bujnicki, J.M. Databases and bioinformatics tools for the study of DNA repair. Mol. Biol. Int. 2011, 2011, 475718. [Google Scholar] [CrossRef]
- Lei, T.; Du, S.; Peng, Z.; Chen, L. Multifaceted regulation and functions of 53BP1 in NHEJ-mediated DSB repair (Review). Int. J. Mol. Med. 2022, 50, 90. [Google Scholar] [CrossRef]
- Zentout, S.; Smith, R.; Jacquier, M.; Huet, S. New methodologies to study DNA repair processes in space and time within living cells. Front. Cell Dev. Biol. 2021, 9, 730998. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer’s disease. Indian J. Psychiatry 2009, 51, 55–61. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A. The 2013 discovery award from the society for free radical biology and medicine: Selected discoveries from the Butterfield Laboratory of oxidative stress and its sequelae in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment. Free. Radic. Biol. Med. 2014, 74, 157. [Google Scholar]
- Boutte, A.M.; Woltjer, R.L.; Zimmerman, L.J.; Stamer, S.L.; Montine, K.S.; Manno, M.V.; Cimino, P.J.; Liebler, D.C.; Montine, T.J. Selectively increased oxidative modifications mapped to detergent-insoluble forms of Aβ and β-III tubulin in Alzheimer’s disease. FASEB J. 2006, 20, 1473–1483. [Google Scholar] [CrossRef]
- Allan Butterfield, D. Amyloid β-peptide (1–42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer’s disease brain. A review. Free. Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef]
- Dean, R.T.; FU, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997, 324, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Takuma, K.; Yao, J.; Huang, J.; Xu, H.; Chen, X.; Luddy, J.; Trillat, A.-C.; Stern, D.M.; Arancio, O.; Yan, S.S. ABAD enhances Aβ-induced cell stress via mitochondrial dysfunction. FASEB J. 2005, 19, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Akterin, S.; Cowburn, R.F.; Miranda-Vizuete, A.; Jiménez, A.; Bogdanovic, N.; Winblad, B.; Cedazo-Minguez, A. Involvement of glutaredoxin-1 and thioredoxin-1 in β-amyloid toxicity and Alzheimer’s disease. Cell Death Differ. 2006, 13, 1454–1465. [Google Scholar] [CrossRef]
- Cenini, G.; Sultana, R.; Memo, M.; Butterfield, D.A. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 987–994. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, R.; Chaudhary, P.; Vatsyayan, R.; Pearce, V.; Jeyabal, P.V.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Arch. Biochem. Biophys. 2008, 480, 85–94. [Google Scholar] [CrossRef]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxidative Med. Cell. Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef]
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The Ambiguous Relationship of Oxidative Stress, Tau Hyperphosphorylation, and Autophagy Dysfunction in Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2015, 2015, 352723. [Google Scholar] [CrossRef]
- Dhapola, R.; Beura, S.K.; Sharma, P.; Singh, S.K.; HariKrishnaReddy, D. Oxidative stress in Alzheimer’s disease: Current knowledge of signaling pathways and therapeutics. Mol. Biol. Rep. 2024, 51, 48. [Google Scholar] [CrossRef]
- Lee, K.Y.; Koh, S.H.; Noh, M.Y.; Park, K.W.; Lee, Y.J.; Kim, S.H. Glycogen synthase kinase-3beta activity plays very important roles in determining the fate of oxidative stress-inflicted neuronal cells. Brain Res. 2007, 1129, 89–99. [Google Scholar] [CrossRef]
- Morel, M.; Authelet, M.; Dedecker, R.; Brion, J.P. Glycogen synthase kinase-3beta and the p25 activator of cyclin dependent kinase 5 increase pausing of mitochondria in neurons. Neuroscience 2010, 167, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Mudher, A.; Shepherd, D.; Newman, T.A.; Mildren, P.; Jukes, J.P.; Squire, A.; Mears, A.; Drummond, J.A.; Berg, S.; MacKay, D.; et al. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry 2004, 9, 522–530. [Google Scholar] [CrossRef]
- Ibáñez-Salazar, A.; Bañuelos-Hernández, B.; Rodríguez-Leyva, I.; Chi-Ahumada, E.; Monreal-Escalante, E.; Jiménez-Capdeville, M.E.; Rosales-Mendoza, S. Oxidative Stress Modifies the Levels and Phosphorylation State of Tau Protein in Human Fibroblasts. Front. Neurosci. 2017, 11, 495. [Google Scholar] [CrossRef] [PubMed]
- Sanacora, G.; Rothman, D.L.; Mason, G.; Krystal, J.H. Clinical studies implementing glutamate neurotransmission in mood disorders. Ann. NY Acad. Sci. 2003, 1003, 292–308. [Google Scholar] [CrossRef]
- Krystal, J.H.; Tolin, D.F.; Sanacora, G.; Castner, S.A.; Williams, G.V.; Aikins, D.E.; Hoffman, R.E.; D’Souza, D.C. Neuroplasticity as a target for the pharmacotherapy of anxiety disorders, mood disorders, and schizophrenia. Drug Discov. Today 2009, 14, 690–697. [Google Scholar] [CrossRef]
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [PubMed]
- Meldrum, B.S. The role of glutamate in epilepsy and other CNS disorders. Neurology 1994, 44, S14–S23. [Google Scholar] [PubMed]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689. [Google Scholar] [CrossRef]
- Kew, J.N.; Kemp, J.A. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology 2005, 179, 4–29. [Google Scholar] [CrossRef]
- Gjessing, L.; Gjesdahl, P.; Sjaastad, O. The free amino acids in human cerebrospinal fluid. J. Neurochem. 1972, 19, 1807–1808. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar]
- Girouard, H.; Wang, G.; Gallo, E.F.; Anrather, J.; Zhou, P.; Pickel, V.M.; Iadecola, C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J. Neurosci. 2009, 29, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef]
- Szule, J.A.; Jung, J.H.; McMahan, U.J. The structure and function of ‘active zone material’at synapses. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140189. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Jang, S.-S.; Chung, H.J. Emerging link between Alzheimer’s disease and homeostatic synaptic plasticity. Neural Plast. 2016, 2016, 7969272. [Google Scholar] [CrossRef]
- Chen, T.S.; Huang, T.H.; Lai, M.C.; Huang, C.W. The Role of Glutamate Receptors in Epilepsy. Biomedicines 2023, 11, 783. [Google Scholar] [CrossRef]
- Bliss, T.V.; Cooke, S.F. Long-term potentiation and long-term depression: A clinical perspective. Clinics 2011, 66 (Suppl. 1), 3–17. [Google Scholar] [CrossRef]
- Srivastava, A.; Das, B.; Yao, A.Y.; Yan, R. Metabotropic Glutamate Receptors in Alzheimer’s Disease Synaptic Dysfunction: Therapeutic Opportunities and Hope for the Future. J. Alzheimer’s Dis. 2020, 78, 1345–1361. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory, and brain aging. Dialogues Clin. Neurosci. 2000, 2, 219–232. [Google Scholar] [CrossRef]
- Frankland, P.W.; Bontempi, B. The organization of recent and remote memories. Nat. Rev. Neurosci. 2005, 6, 119–130. [Google Scholar] [CrossRef]
- Trushina, E.; McMurray, C. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M. The role of lipid peroxidation in neurological disorders. J. Clin. Biochem. Nutr. 2014, 54, 151–160. [Google Scholar] [CrossRef]
- Westra, M.; Gutierrez, Y.; MacGillavry, H.D. Contribution of Membrane Lipids to Postsynaptic Protein Organization. Front. Synaptic Neurosci. 2021, 13, 790773. [Google Scholar] [CrossRef]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef] [PubMed]
- McDaid, J.; Mustaly-Kalimi, S.; Stutzmann, G.E. Ca2+ Dyshomeostasis Disrupts Neuronal and Synaptic Function in Alzheimer’s Disease. Cells 2020, 9, 2655. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Cohen, G.; Kesler, N. Monoamine oxidase and mitochondrial respiration. J. Neurochem. 1999, 73, 2310–2315. [Google Scholar] [CrossRef]
- Cai, Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer’s disease. Mol. Med. Rep. 2014, 9, 1533–1541. [Google Scholar] [CrossRef]
- Thomas, T. Monoamine oxidase-B inhibitors in the treatment of Alzheimers disease. Neurobiol. Aging 2000, 21, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, L.; Saetre, P.; Balciuniene, J.; Castensson, A.; Cairns, N.; Jazin, E.E. Increased monoamine oxidase messenger RNA expression levels in frontal cortex of Alzheimer’s disease patients. Neurosci. Lett. 2002, 326, 56–60. [Google Scholar] [CrossRef]
- Gulyás, B.; Pavlova, E.; Kása, P.; Gulya, K.; Bakota, L.; Várszegi, S.; Keller, E.; Horváth, M.C.; Nag, S.; Hermecz, I. Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-L-deprenyl using whole hemisphere autoradiography. Neurochem. Int. 2011, 58, 60–68. [Google Scholar] [CrossRef]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Magni, G.; Meibach, R. Lazabemide for the long-term treatment of Alzheimer’s disease. Eur. Neuropsychopharmacol. 1999, 9, 142. [Google Scholar] [CrossRef]
- Schneier, F.R. Pharmacotherapy of social anxiety disorder. Expert Opin. Pharmacother. 2011, 12, 615–625. [Google Scholar] [CrossRef]
- Dhull, D.K.; Jindal, A.; Dhull, R.K.; Aggarwal, S.; Bhateja, D.; Padi, S.S. Neuroprotective effect of cyclooxygenase inhibitors in ICV-STZ induced sporadic Alzheimer’s disease in rats. J. Mol. Neurosci. 2012, 46, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Muck-Seler, D.; Presecki, P.; Mimica, N.; Mustapic, M.; Pivac, N.; Babic, A.; Nedic, G.; Folnegovic-Smalc, V. Platelet serotonin concentration and monoamine oxidase type B activity in female patients in early, middle and late phase of Alzheimer’s disease. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2009, 33, 1226–1231. [Google Scholar] [CrossRef] [PubMed]
- Joe, E.; Ringman, J.M. Cognitive symptoms of Alzheimer’s disease: Clinical management and prevention. Br. Med. J. 2019, 367, l6217. [Google Scholar] [CrossRef]
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, Aducanumab, and Gantenerumab—Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Barthold, D.; Joyce, G.; Ferido, P.; Drabo, E.F.; Marcum, Z.A.; Gray, S.L.; Zissimopoulos, J. Pharmaceutical Treatment for Alzheimer’s Disease and Related Dementias: Utilization and Disparities. J. Alzheimer’s Dis. 2020, 76, 579–589. [Google Scholar] [CrossRef]
- Arias-Sánchez, R.A.; Torner, L.; Fenton Navarro, B. Polyphenols and Neurodegenerative Diseases: Potential Effects and Mechanisms of Neuroprotection. Molecules 2023, 28, 5415. [Google Scholar] [CrossRef]
- Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Kaddoumi, A. Extra-Virgin Olive Oil in Alzheimer’s Disease: A Comprehensive Review of Cellular, Animal, and Clinical Studies. Int. J. Mol. Sci. 2024, 25, 1914. [Google Scholar] [CrossRef]
- Rathod, N.B.; Elabed, N.; Punia, S.; Ozogul, F.; Kim, S.K.; Rocha, J.M. Recent Developments in Polyphenol Applications on Human Health: A Review with Current Knowledge. Plants 2023, 12, 1217. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Leri, M.; Nardiello, P.; Casamenti, F.; Stefani, M. Olive Polyphenols: Antioxidant and Anti-Inflammatory Properties. Antioxidants 2021, 10, 1044. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Wang, D.W.; Xu, S.F.; Zhang, S.; Fan, Y.G.; Yang, Y.Y.; Guo, S.Q.; Wang, S.; Guo, T.; Wang, Z.Y.; et al. α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018, 14, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Afzal, O.; Dalhat, M.H.; Altamimi, A.S.A.; Rasool, R.; Alzarea, S.I.; Almalki, W.H.; Murtaza, B.N.; Iftikhar, S.; Nadeem, S.; Nadeem, M.S.; et al. Green Tea Catechins Attenuate Neurodegenerative Diseases and Cognitive Deficits. Molecules 2022, 27, 7604. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.; Fernandez, F.; Johnson, J.B.; Naiker, M.; Owoola, A.G.; Broszczak, D.A. Oxidative stress in alzheimer’s disease: A review on emergent natural polyphenolic therapeutics. Complement. Ther. Med. 2020, 49, 102294. [Google Scholar] [CrossRef]
- Kim, J.H.; Ju, I.G.; Kim, N.; Huh, E.; Son, S.R.; Hong, J.P.; Choi, Y.; Jang, D.S.; Oh, M.S. Yomogin, Isolated from Artemisia iwayomogi, Inhibits Neuroinflammation Stimulated by Lipopolysaccharide via Regulating MAPK Pathway. Antioxidants 2022, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Y.; Shi, X.; Ma, C. An overview on therapeutics attenuating amyloid β level in Alzheimer’s disease: Targeting neurotransmission, inflammation, oxidative stress and enhanced cholesterol levels. Am. J. Transl. Res. 2016, 8, 246–269. [Google Scholar] [PubMed]
- Ekert, J.O.; Gould, R.L.; Reynolds, G.; Howard, R.J. TNF alpha inhibitors in Alzheimer’s disease: A systematic review. Int. J. Geriatr. Psychiatry 2018, 33, 688–694. [Google Scholar] [CrossRef]
- López, P.R. Tributyrin Treatment in Mild Alzheimer Disease: Assessment of Butyric Acid Effects Via the Gut-Brain (TRIM–GUT). Available online: https://clinicaltrials.gov/study/NCT06797817?term=targeting%20oxidative%20stress%20in%20Alzheimer%0A%20disease&rank=1#study-overview (accessed on 25 March 2025).
- Alltech Life Sciences INC. Safety and Tolerability of Antioxidant (AT-001) for Reducing Brain Oxidative Stress. Available online: https://clinicaltrials.gov/study/NCT01731093?term=NCT01731093&rank=1 (accessed on 28 March 2025).
- Virginia Commonwealth University. Safety Study of R(+)Pramipexole to Treat Early Alzheimer’s Disease. Available online: https://clinicaltrials.gov/study/NCT01388478?term=Oxidative%20Stress%20and%20Alzheimer%20disease&rank=14#study-record-dates (accessed on 28 March 2025).
- Fundació ACE Institut Català de Neurociències Aplicades. Effect of Treatment With EGb 761(r) on Blood Markers of Inflammation and Oxidative Stress in Patients With MCI. Available online: https://clinicaltrials.gov/study/NCT01731093?term=NCT01731093&rank=1#study-record-dates (accessed on 28 March 2025).
- Perseverance Research Center. Specialized Food Plan Based on Individual Physiological Comprehensive Body Assessments Accompanied With Cellular Repair Therapy to Decrease Inflammation Cognitively Impaired Patients. Available online: https://clinicaltrials.gov/study/NCT03630419 (accessed on 28 March 2025).
- Sekhar, R.V. Glutathione, Brain Metabolism and Inflammation in Alzheimer’s Disease. Available online: https://clinicaltrials.gov/study/NCT04740580 (accessed on 28 March 2025).
- Oregon Health and Science University. Lutein and Alzheimer’s Disease Study (LAD). Available online: https://clinicaltrials.gov/study/NCT00596024 (accessed on 28 March 2025).
- Du, F. Effects of Nicotinamide Riboside on Bioenergetics and Oxidative Stress in Mild Cognitive Impairment/Alzheimer’s Dementia. Available online: https://clinicaltrials.gov/study/NCT04430517 (accessed on 28 March 2025).
- Treeway, B.V. ASURE: Alzheimer Study Using ORal Edaravone. Available online: https://clinicaltrials.gov/study/NCT05323812 (accessed on 28 March 2024).
- Hanoglu, L. Prediction of Effectiveness of rTMS Application in Alzheimer’s Patients. Available online: https://clinicaltrials.gov/study/NCT05977088 (accessed on 28 March 2025).
- Kaddoumi, A. Does EVOO Induce Gene and Metabolic Changes in Healthy Subjects. Available online: https://clinicaltrials.gov/study/NCT05929924 (accessed on 28 March 2025).
- Guimaraes, D.d.O. Effectiveness of Probiotic K10 in Managing Health Outcomes in Parkinson and Alzheimer Disease (Probiótico). Available online: https://clinicaltrials.gov/study/NCT06019117 (accessed on 28 March 2025).
- Shinto, L. Fish Oil and Alpha Lipoic Acid in Treating Alzheimer’s Disease. Available online: https://clinicaltrials.gov/study/NCT00090402 (accessed on 28 March 2025).
- Second Affiliated Hospital, School of Medicine, Zhejiang University. Effects of Sulforaphane in Patients with Prodromal to Mild Alzheimer’s Disease. Available online: https://clinicaltrials.gov/study/NCT04213391 (accessed on 28 March 2025).
- Sánchez Perona, J. Attenuation of Inflammatory Processes Associated with Alzheimer’s Disease After Consumption of Pomace Olive Oil. (ORIVA2). Available online: https://clinicaltrials.gov/study/NCT04559828 (accessed on 28 March 2025).
- Jain, S. Optimization of Blood Levels of 25(OH)-Vitamin D in African Americans. Available online: https://clinicaltrials.gov/study/NCT04939792 (accessed on 28 March 2025).
- Lương, K.; Nguyen, L.T. The role of Beta-adrenergic receptor blockers in Alzheimer’s disease: Potential genetic and cellular signaling mechanisms. Am. J. Alzheimer’s Dis. Other Demen. 2013, 28, 427–439. [Google Scholar] [CrossRef]
- Qin, J.; Ma, Z.; Chen, X.; Shu, S. Microglia activation in central nervous system disorders: A review of recent mechanistic investigations and development efforts. Front. Neurol. 2023, 14, 1103416. [Google Scholar] [CrossRef]
- Kisby, B.; Jarrell, J.T.; Agar, M.E.; Cohen, D.S.; Rosin, E.R.; Cahill, C.M.; Rogers, J.T.; Huang, X. Alzheimer’s Disease and Its Potential Alternative Therapeutics. J. Alzheimer’s Dis. Park. 2019, 9, 477. [Google Scholar] [CrossRef]
- Ouyang, Y.; Chen, Z.; Tan, M.; Liu, A.; Chen, M.; Liu, J.; Pi, R.; Fang, J. Carvedilol, a third-generation β-blocker prevents oxidative stress-induced neuronal death and activates Nrf2/ARE pathway in HT22 cells. Biochem. Biophys. Res. Commun. 2013, 441, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, S.S.; Hafez, M.M.; Mehanna, E.T.; Mesbah, N.M.; Abo-Elmatty, D.M. Combined vildagliptin and memantine treatment downregulates expression of amyloid precursor protein, and total and phosphorylated tau in a rat model of combined Alzheimer’s disease and type 2 diabetes. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 685–695. [Google Scholar] [CrossRef]
- Shukla, M.; Govitrapong, P.; Boontem, P.; Reiter, R.J.; Satayavivad, J. Mechanisms of Melatonin in Alleviating Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 1010–1031. [Google Scholar] [CrossRef]
- Sun, T.C.; Liu, X.C.; Yang, S.H.; Song, L.L.; Zhou, S.J.; Deng, S.L.; Tian, L.; Cheng, L.Y. Melatonin Inhibits Oxidative Stress and Apoptosis in Cryopreserved Ovarian Tissues via Nrf2/HO-1 Signaling Pathway. Front. Mol. Biosci. 2020, 7, 163. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Sortino, M.A. Early compensatory responses against neuronal injury: A new therapeutic window of opportunity for Alzheimer’s Disease? CNS Neurosci. Ther. 2019, 25, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.; Akram, M.; Shahrukh, M.; Ishrat, T.; Parvez, S. Effects of pramipexole on beta-amyloid(1–42) memory deficits and evaluation of oxidative stress and mitochondrial function markers in the hippocampus of Wistar rat. Neurotoxicology 2022, 92, 91–101. [Google Scholar] [CrossRef]
- Wang, J.; Jia, Y.; Li, G.; Wang, B.; Zhou, T.; Zhu, L.; Chen, T.; Chen, Y. The Dopamine Receptor D3 Regulates Lipopolysaccharide-Induced Depressive-Like Behavior in Mice. Int. J. Neuropsychopharmacol. 2018, 21, 448–460. [Google Scholar] [CrossRef]
- Chen, J.Y.; Zhu, Q.; Zhang, S.; OuYang, D.; Lu, J.H. Resveratrol in experimental Alzheimer’s disease models: A systematic review of preclinical studies. Pharmacol. Res. 2019, 150, 104476. [Google Scholar] [CrossRef]
- Wang, N.; He, J.; Pan, C.; Wang, J.; Ma, M.; Shi, X.; Xu, Z. Resveratrol Activates Autophagy via the AKT/mTOR Signaling Pathway to Improve Cognitive Dysfunction in Rats with Chronic Cerebral Hypoperfusion. Front. Neurosci. 2019, 13, 859. [Google Scholar] [CrossRef]
- Schweiger, S.; Matthes, F.; Posey, K.; Kickstein, E.; Weber, S.; Hettich, M.M.; Pfurtscheller, S.; Ehninger, D.; Schneider, R.; Krauß, S. Resveratrol induces dephosphorylation of Tau by interfering with the MID1-PP2A complex. Sci. Rep. 2017, 7, 13753. [Google Scholar] [CrossRef]
- Detrait, E.R.; Danis, B.; Lamberty, Y.; Foerch, P. Peripheral administration of an anti-TNF-α receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-α levels and memory deficits in mice. Neurochem. Int. 2014, 72, 10–13. [Google Scholar] [CrossRef]
- Lee, I.T.; Luo, S.F.; Lee, C.W.; Wang, S.W.; Lin, C.C.; Chang, C.C.; Chen, Y.L.; Chau, L.Y.; Yang, C.M. Overexpression of HO-1 protects against TNF-alpha-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am. J. Pathol. 2009, 175, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Ortí-Casañ, N.; Wu, Y.; Naudé, P.J.W.; De Deyn, P.P.; Zuhorn, I.S.; Eisel, U.L.M. Targeting TNFR2 as a Novel Therapeutic Strategy for Alzheimer’s Disease. Front. Neurosci. 2019, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D.H. Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation 2023, 46, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, J.; Zhao, J.; Meng, M. Etanercept attenuates myocardial ischemia/reperfusion injury by decreasing inflammation and oxidative stress. PLoS ONE 2014, 9, e108024. [Google Scholar] [CrossRef]
- Khalatbary, A.R.; Khademi, E. The green tea polyphenolic catechin epigallocatechin gallate and neuroprotection. Nutr. Neurosci. 2020, 23, 281–294. [Google Scholar] [CrossRef]
- Han, J.; Wang, M.; Jing, X.; Shi, H.; Ren, M.; Lou, H. (-)-Epigallocatechin gallate protects against cerebral ischemia-induced oxidative stress via Nrf2/ARE signaling. Neurochem. Res. 2014, 39, 1292–1299. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Podlacha, M.; Gaffke, L.; Majkutewicz, I.; Mantej, J.; Węgrzyn, A.; Osiadły, M.; Myślińska, D.; Węgrzyn, G. Autophagy-dependent mechanism of genistein-mediated elimination of behavioral and biochemical defects in the rat model of sporadic Alzheimer’s disease. Neuropharmacology 2019, 148, 332–346. [Google Scholar] [CrossRef]
- Devi, K.P.; Shanmuganathan, B.; Manayi, A.; Nabavi, S.F.; Nabavi, S.M. Molecular and Therapeutic Targets of Genistein in Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 7028–7041. [Google Scholar] [CrossRef]
- Guo, J.; Yang, G.; He, Y.; Xu, H.; Fan, H.; An, J.; Zhang, L.; Zhang, R.; Cao, G.; Hao, D.; et al. Involvement of α7nAChR in the Protective Effects of Genistein Against β-Amyloid-Induced Oxidative Stress in Neurons via a PI3K/Akt/Nrf2 Pathway-Related Mechanism. Cell. Mol. Neurobiol. 2021, 41, 377–393. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhalifa, A.E.; Alkhalifa, O.; Durdanovic, I.; Ibrahim, D.R.; Maragkou, S. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease: Insights into Pathophysiology and Treatment. J. Dement. Alzheimer's Dis. 2025, 2, 17. https://doi.org/10.3390/jdad2020017
Alkhalifa AE, Alkhalifa O, Durdanovic I, Ibrahim DR, Maragkou S. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease: Insights into Pathophysiology and Treatment. Journal of Dementia and Alzheimer's Disease. 2025; 2(2):17. https://doi.org/10.3390/jdad2020017
Chicago/Turabian StyleAlkhalifa, Amer E., Oula Alkhalifa, Iva Durdanovic, Dalia R. Ibrahim, and Sofia Maragkou. 2025. "Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease: Insights into Pathophysiology and Treatment" Journal of Dementia and Alzheimer's Disease 2, no. 2: 17. https://doi.org/10.3390/jdad2020017
APA StyleAlkhalifa, A. E., Alkhalifa, O., Durdanovic, I., Ibrahim, D. R., & Maragkou, S. (2025). Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease: Insights into Pathophysiology and Treatment. Journal of Dementia and Alzheimer's Disease, 2(2), 17. https://doi.org/10.3390/jdad2020017