

Harnessing AI and Quantum Computing for Accelerated Drug Discovery: Regulatory Frameworks for In Silico to In Vivo Validation



Abstract
1. Introduction
1.1. Background
1.2. The Role of AI and Quantum Computing in New Drug Discovery
1.2.1. AI in Drug Discovery (Machine Learning, Deep Learning, and Generative Models)
1.2.2. Quantum Computing’s Potential in Molecular Simulations and Optimization
1.3. Purpose of the Study
1.3.1. Understanding the Integration of AI and Quantum Computing in Drug Discovery
1.3.2. Identifying Regulatory Challenges in the In Silico to In Vivo Transition
1.3.3. Proposing a Regulatory Framework to Facilitate AI-Driven Drug Validation
2. State of the Art: AI and Quantum Computing in Drug Discovery
2.1. AI-Driven Approaches to Drug Discovery
2.1.1. Data-Driven Techniques: Molecular Docking, Virtual Screening, and De Novo Drug Design
2.1.2. Predictive Modeling of Toxicity, Efficacy, and Pharmacokinetics
2.2. Quantum Computing Applications
- (1)
- Cryptography: Quantum computing breaks Rivest-Shamir-Adleman (RSA) encryption using Shor’s algorithm, posing a significant threat to current public-key systems. In post-quantum cryptography development to resist quantum attacks [12].
- (2)
- Drug Discovery and Chemistry: Simulating quantum systems at the molecular level to model complex molecules and reactions, discover new drugs and materials, and understand protein folding [13].
- (3)
- Optimization Problems: These include logistics (e.g., route optimization, supply chain management), financial portfolio optimization, scheduling problems, and the application of quantum algorithms, such as the Quantum Approximation Optimization Algorithm (QAOA) [14].
- (4)
- Machine Learning and AI: Speeding up specific tasks like pattern recognition, clustering and classification, and feature selection. Quantum-enhanced machine learning models could outperform classical ones in particular domains [15].
- (5)
- Financial Modeling: Risk analysis and fraud detection [16]. Option pricing using quantum Monte Carlo simulations for faster convergence.
- (6)
- Search and Database: Grover’s algorithms can search unsorted databases [17].
- (7)
- Cybersecurity: Development of new encryption methods based on quantum principles, e.g., quantum key distribution (QKD) [18].
- (8)
- Material Science: Modeling new materials at the atomic level, like superconductors and advanced alloys [19].
- (9)
- Climate and Weather Modeling: Simulating complex systems with many interacting variables more efficiently [20].
- (10)
- Energy: Modeling and optimization for chemical reactions and battery or fuel cell systems in renewable energy systems [21].
- (1)
- Hardware Limitations: Qubit decoherence gate errors (NISQ constraints).
- (2)
- (3)
- Regulatory Gaps: Standards for quantum-AI hybrids are missing, e.g., [13] UK Good Microbiological Laboratory Practice (GMLP).
- (4)
- Recommendation: Focus on hybrid quantum-classical approaches, e.g., [22] to mitigate current limitations.
2.2.1. Quantum Simulations for Protein–Ligand Interactions
2.2.2. Quantum Computing in Drug Discovery
2.2.3. Enhancing Computational Efficiency in Chemical Space Exploration
2.2.4. Hybrid AI–Quantum Approaches
2.3. Case Studies
3. Regulatory Challenges in AI- and Quantum-Driven Drug Discovery
3.1. Current Drug Approval and Validation Processes
- (1)
- Discovery and Preclinical Testing [53]
- Researchers identify and develop new drug candidates.
- Preclinical studies are conducted in labs (in vitro) and animals (in vivo) to evaluate safety, toxicity, dosage, and pharmacological effects.
- (2)
- Investigational New Drug (IND) Application
- Before clinical trials, sponsors submit an IND to the FDA, including the following:
- ○
- Preclinical data.
- ○
- Proposed clinical study protocols.
- ○
- Safety information and investigator qualifications.
- The FDA reviews the IND to determine if human testing can begin, ensuring the safety of the study.
- (3)
- Clinical TrialsClinical trials are conducted in phases:
- Phase 1: Safety
- ○
- Small group (20–100 healthy volunteers or patients).
- ○
- Evaluate safety, side effects, and optimal dosage.
- Phase 2: Effectiveness
- ○
- Larger group (100–300 patients).
- ○
- Assesses the effectiveness of drugs and further evaluates their safety and efficacy.
- Phase 3: Confirmatory Studies
- ○
- Large-scale (hundreds to thousands of patients).
- ○
- Confirms efficacy, monitors adverse reactions, and compares drugs with existing treatments or placebo.
- (4)
- New Drug Application (NDA)
- Upon successful completion of clinical trials, the sponsor submits an NDA containing the following:
- ○
- Clinical trial results.
- ○
- Manufacturing processes.
- ○
- Proposed labeling information.
- The FDA thoroughly reviews efficacy, safety data, and manufacturing practices.
- (5)
- FDA Review and Approval
- FDA experts review the data submitted with the New Drug Application (NDA).
- Advisory committees may provide independent recommendations.
- The FDA decides whether to approve, request additional studies, or deny approval based on the following:
- ○
- The drug’s demonstrated safety and efficacy.
- ○
- Risks vs. benefits profile.
- ○
- Manufacturing quality and controls.
- (6)
- Post-Marketing Surveillance (Phase 4)
- After approval, drugs continue to be monitored:
- ○
- Identify rare or long-term side effects.
- ○
- Ensure ongoing safety, efficacy, and quality standards.
- ○
- FDA may require additional studies or modifications based on surveillance data.
- (1)
- Pre-Submission (Preparation)
- Pharmaceutical companies conduct extensive preclinical and clinical studies to gather data on the quality, safety, and efficacy of medicines.
- Companies consult with EMA (Scientific Advice procedure) to ensure their data and development plans meet regulatory standards.
- (2)
- Application Submission
- The company submits a Marketing Authorisation Application (MAA) through a centralized procedure, which is mandatory for certain medicines, e.g., biotechnology products, cancer treatments, rare diseases.
- (3)
- Validation
- EMA validates the application to confirm that it meets regulatory and technical requirements, including all required documentation.
- (4)
- Scientific Evaluation
- Assessment by the Committee for Medicinal Products for Human Use (CHMP): A Rapporteur and Co-Rapporteur from CHMP lead the evaluation, assessing quality, safety, and efficacy data.
- CHMP can request additional information or clarification from the applicant. The clock stops and restarts to allow the company time to respond.
- (5)
- Opinion
- CHMP issues a recommendation based on their evaluation:
- ○
- Positive opinion: recommending authorization.
- ○
- Negative opinion: refusing approval.
- The CHMP’s recommendation is typically provided within 210 days (excluding clock stops).
- (6)
- European Commission Decision
- The CHMP opinion is sent to the European Commission (EC).
- EC makes a legally binding decision (usually within 67 days):
- ○
- Approval of marketing authorization is valid throughout the entire EU.
- ○
- Refusal of authorization.
- (7)
- Post-Approval Monitoring
- Pharmacovigilance: Continuous monitoring for safety after approval, including reporting adverse effects.
- Risk Management Plans (RMPs): Companies must implement strategies to monitor and minimize risks.
- Periodic Safety Update Reports (PSURs): Regular submission of safety updates and ongoing evaluation.
- (8)
- Renewal and Variations
- Initial marketing authorization is valid for five years; after this period, it may be renewed based on reassessment.
- EMA must also submit and review variations to authorization, such as label updates or new indications.
3.1.1. Regulatory Pathways (FDA, EMA, ICH Guidelines)
- Clinical trials (human testing) in three main phases [53]:
- Phase I:
- ○
- Tests a small number (20–100) of healthy volunteers or patients.
- ○
- Determines safety, dosage range, side effects, and pharmacokinetics.
- Phase II:
- ○
- Includes a larger group (100–300 patients).
- ○
- Evaluate effectiveness, optimal dosing, and side effects.
- Phase III:
- ○
- Tests an even larger group (hundreds to thousands of patients).
- ○
- Confirms efficacy, monitors adverse reactions, and compares them to standard treatments or placebo.
3.1.2. Preclinical and Clinical Validation Requirements
3.2. Challenges of In Silico Validation
3.2.1. Trustworthiness and Interpretability of AI Models
3.2.2. Reproducibility and Reliability of Quantum-Enhanced Simulations
3.2.3. Data Biases and Ethical Considerations
3.2.4. Integration of Computational and Experimental Validation
3.2.5. Standardizing AI and Quantum Model Validation
4. Proposed Regulatory Framework for AI and Quantum Computing in Drug Discovery
4.1. AI Model Validation and Transparency
4.1.1. Explainable AI (XAI) Requirements for Regulatory Compliance
4.1.2. Model Auditing and Documentation and Bias Assessment
4.2. Quantum Computing Guidelines
4.3. Ethical and Data Governance Considerations
5. Future Directions and Conclusions
5.1. Technological Advancements and Industry Adoption
5.1.1. Summary of Key Takeaways
5.1.2. Recommendations for Researchers, Industry Stakeholders, and Policymakers
For Researchers
For Industry Stakeholders
For Policymakers and Regulators
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bassman, L.; Urbanek, M.; Metcalf, M.; Carter, J.; Kemper, A.F.; de Jong, W.A. Simulating quantum materials with digital quantum computers. Quantum Sci. Technol. 2021, 6, 043002. [Google Scholar] [CrossRef]
- Bordukova, M.; Makarov, N.; Rodriguez-Esteban, R.; Schmich, F.; Menden, M.P. Generative artificial intelligence empowers digital twins in drug discovery and clinical trials. Expert Opin. Drug Discov. 2024, 19, 33–42. [Google Scholar] [CrossRef]
- Golbraikh, A.; Wang, X.S.; Zhu, H.; Tropsha, A. Predictive QSAR Modeling: Methods and Applications in Drug Discovery and Chemical Risk Assessment. In Handbook of Computational Chemistry; Leszczynski, J., Kaczmarek-Kedziera, A., Puzyn, T., Papadopoulos, M.G., Reis, H., Shukla, M.K., Eds.; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Singh, N.; Vayer, P.; Tanwar, S.; Poyet, J.L.; Tsaioun, K.; Villoutreix, B.O. Drug discovery and development: Introduction to the general public and patient groups. Front. Drug Discov. 2023, 3, 1201419. [Google Scholar] [CrossRef]
- Cao, Y.; Romero, J.; Aspuru-Guzik, A. Potential of quantum computing for drug discovery. IBM J. Res. Dev. 2018, 62, 6:1–6:20. [Google Scholar] [CrossRef]
- de la Cruz Calvo, H.I.; Cuartero Gómez, F.; Paulet González, J.J.; Mezzini, M.; Pelayo, F.L. Functional Matrices on Quantum Computing Simulation. Mathematics 2023, 11, 3742. [Google Scholar] [CrossRef]
- Vora, L.K.; Gholap, A.D.; Jetha, K.; Thakur, R.R.S.; Solanki, H.K.; Chavda, V.P. Artificial Intelligence in Pharmaceutical Technology and Drug Delivery Design. Pharmaceutics 2023, 15, 1916. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Qiu, X.; Li, H.; Ver Steeg, G.; Godzik, A. Advances in AI for protein structure prediction: Implications for cancer drug discovery and development. Biomolecules 2024, 14, 339. [Google Scholar] [CrossRef]
- Niazi, S.K.; Mariam, Z. Computer-Aided Drug Design and Drug Discovery: A Prospective Analysis. Pharmaceuticals 2024, 17, 22. [Google Scholar] [CrossRef]
- Altman, R.B. A Holy Grail—The Prediction of Protein Structure. N. Engl. J. Med. 2023, 389, 1431–1434. [Google Scholar] [CrossRef]
- Kumar, G.; Yadav, S.; Mukherjee, A.; Hassija, V.; Guizani, M. Recent advances in quantum computing for drug discovery and development. IEEE Access 2024, 12, 64491–64509. [Google Scholar] [CrossRef]
- Mayers, D. Unconditional security in quantum cryptography. J. Assoc. Comput. Mach. 2001, 48, 351–406. [Google Scholar] [CrossRef]
- Breakthrough in High-Performance Computing and Quantum Chemistry Revolutionises Drug Discovery. (17 July 2024). Targeted News Service. Available online: https://library.capella.edu/login?url=https://www.proquest.com/wire-feeds/breakthrough-high-performance-computing-quantum/docview/3081756756/se-2 (accessed on 1 March 2025).
- Bozhedarov, A.A.; Usmanov, S.R.; Salakhov, G.V.; Boev, A.S.; Kiktenko, E.O.; Fedorov, A.K. Quantum and quantum-inspired optimization for solving the minimum bin packing problem. J. Phys. Conf. Ser. 2024, 2701, 012129. [Google Scholar] [CrossRef]
- Janke, E.; Zhang, M.; Ryu, S.E.; Bhattarai, J.P.; Schreck, M.R.; Moberly, A.H.; Luo, W.; Ding, L.; Wesson, D.W.; Ma, M. Machine learning-based clustering and classification of mouse behaviors via respiratory patterns. iScience 2022, 25, 105625. [Google Scholar] [CrossRef]
- Aboussalah, A.M.; Chi, C.; Lee, C.G. Quantum computing reduces systemic risk in financial networks. Sci. Rep. 2023, 13, 3990. [Google Scholar] [CrossRef]
- AbuGhanem, M. Characterizing Grover search algorithm on large-scale superconducting quantum computers. Sci. Rep. 2025, 15, 1281. [Google Scholar] [CrossRef]
- Amirkhanova, D.S.; Iavich, M.; Mamyrbayev, O. Lattice-Based Post-Quantum Public Key Encryption Scheme Using ElGamal’s Principles. Cryptography 2024, 8, 31. [Google Scholar] [CrossRef]
- Zhichao, L.; Dong, M.; Xiongjun, L.; Lu, Z. High-throughput and data-driven machine learning techniques for discovering high-entropy alloys. Commun. Mater. 2024, 5, 76. [Google Scholar] [CrossRef]
- Kupczynski, M. Mathematical Modeling of Physical Reality: From Numbers to Fractals, Quantum Mechanics, and the Standard Model. Entropy 2024, 26, 991. [Google Scholar] [CrossRef]
- Rurański, A.; Kuś, W. The Quantum-Inspired Evolutionary Algorithm in the Parametric Optimization of Lithium-Ion Battery Housing in the Multiple-Drop Test. Batter 2024, 10, 308. [Google Scholar] [CrossRef]
- Jeyaraman, N.; Jeyaraman, M.; Yadav, S.; Ramasubramanian, S.; Balaji, S. Revolutionizing Healthcare: The Emerging Role of Quantum Computing in Enhancing Medical Technology and Treatment. Cureus 2024, 16, e67486. [Google Scholar] [CrossRef] [PubMed]
- Jaya, V.P.; Bopardikar, S.; Avinash, D.; Ashwini, K.; Dasgupta, K.; Senapati, S. An Approach to Solve the Coarse-Grained Protein Folding Problem in a Quantum Computer. arXiv 2023, arXiv:2311.14141. [Google Scholar]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- Ja’afaru, S.C.; Uzairu, A.; Bayil, I.; Sallau, M.S.; Ndukwe, G.I.; Ibrahim, M.T.; Moin, A.T.; Mollah, A.K.M.M.; Absar, N. Unveiling potent inhibitors for schistosomiasis through ligand-based drug design, molecular docking, molecular dynamics simulations and pharmacokinetics predictions. PLoS ONE 2024, 19, e0302390. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.V.; Varma, M.; Rai, M.; Pratap Singh, S.; Bansod, G.; Laux, P.; Luch, A. Advancing Predictive Risk Assessment of Chemicals via Integrating Machine Learning, Computational Modeling, and Chemical/Nano-Quantitative Structure-Activity Relationship Approaches. Adv. Intell. Syst. 2024, 6, 2300366. [Google Scholar] [CrossRef]
- Flores-Hernandez, H.; Martinez-Ledesma, E. A systematic review of deep learning chemical language models in recent era. J. Cheminform. 2024, 16, 129. [Google Scholar] [CrossRef]
- Martinelli, D.D. Generative machine learning for de novo drug discovery: A systematic review. Comput. Biol. Med. 2022, 145, 105403. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhou, T.; Wu, B.; Chen, C.; Wang, Y. A Review and Experimental Evaluation on Split Learning. Future Internet 2025, 17, 87. [Google Scholar] [CrossRef]
- Ballester, P.J.; Mangold, M.; Howard, N.I.; Robinson, R.L.; Abell, C.; Blumberger, J.; Mitchell, J.B. Hierarchical virtual screening for the discovery of new molecular scaffolds in antibacterial hit identification. J. R. Soc. Interface 2012, 9, 3196–3207. [Google Scholar] [CrossRef]
- Cheng, M.; Jiang, P.; Jiexiang, H.; Leshi, S.; Zhou, Q. A multi-fidelity surrogate modeling method based on variance-weighted sum for the fusion of multiple non-hierarchical low-fidelity data. Struct. Multidiscip. Optim. 2021, 64, 3797–3818. [Google Scholar] [CrossRef]
- Retherford, J.Q.; McDonald, M. Estimation and Validation of Gaussian Process Surrogate Models for Sensitivity Analysis and Design Optimization. Transp. Res. Rec. 2011, 2, 119–126. [Google Scholar] [CrossRef]
- Guerrero, G.D.; Imbernón, B.; Pérez-Sánchez, H.; Sanz, F.; García, J.M.; Cecilia, J.M. A Performance/Cost Evaluation for a GPU-Based Drug Discovery Application on Volunteer Computing. BioMed Res. Int. 2014, 2014, 474219. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
- Huang, B.; von Lilienfeld, O.A. Quantum machine learning using atom-in-molecule-based fragments selected on the fly. Nat. Chem. 2020, 12, 945–951. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Yamamori, Y.; Tomii, K. Protein-protein interaction prediction methods: From docking-based to AI-based approaches. Biophys. Rev. 2022, 14, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Aliper, A.; Chen, J.; Zhao, H.; Rao, S.; Kuppe, C.; Ozerov, I.V.; Zhang, M.; Witte, K.; Kruse, C.; et al. A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models. Nat. Biotechnol. 2025, 43, 63–75. [Google Scholar] [CrossRef]
- The Atomwise AIMS Program. AI is a viable alternative to high throughput screening: A 318-target study. Sci. Rep. 2024, 14, 7526. [Google Scholar] [CrossRef]
- Kavitha, K.S.; Kamalakumari, J.; Krishna, G.; Najma, U.; Rama, S. AI and Quantum Computing for Accelerating Drug Discovery and Precision Medicine. In Modern SuperHyperSoft Computing Trends in Science and Technology; IGI Global Scientific Publishing: Hershey, PA, USA, 2025; pp. 33–60. [Google Scholar]
- Nature Biotechnology Study Reveals That Tempus’ xT Platform Increases Cancer Patients’ Personalized Therapeutic Opportunities. Tempus. Available online: http://tempus.com (accessed on 21 March 2025).
- de Jesus, G.F.; Teixeira, E.S.; Galvão, L.Q.; da Silva, M.H.F.; Neto, M.Q.N.; Fernandez, B.O.; de Lima, A.M.; de Aguiar Silva, E.D.R.; Cruz, C.D.S. Evaluating Variational Quantum Eigensolver Approaches for Simplified Models of Molecular Systems: A Case Study on Protocatechuic Acid. Molecules 2024, 30, 119. [Google Scholar] [CrossRef]
- Swayne, M. CQC, Roche Partner to Use Quantum Algorithms to Tackle Drug Discovery for Alzheimer’s Disease. Available online: https://thequantuminsider.com/2021/01/31/cqc-roche-partner-to-use-quantum-algorithms-to-tackle-drug-discovery-for-alzheimers-disease/ (accessed on 1 March 2025).
- Quantum Computing: Boehringer Ingelheim Pioneers with Google for Pharma R&D. Available online: https://www.boehringer-ingelheim.com/science-innovation/cooperation-google-quantum-ai (accessed on 1 March 2025).
- Merck and HQS Quantum Simulations Cooperate in Quantum Computing. Available online: https://quantumsimulations.de/news/merck-and-hqs-quantum-simulations-cooperate-in-quantum-computing (accessed on 1 March 2025).
- Quantum-Enhanced AI Boosts Drug Discovery with Zapata Computing and Insilico Medicine Collaboration. Available online: https://quantumzeitgeist.com/quantum-enhanced-ai-boosts-drug-discovery-with-zapata-computing-and-insilico-medicine-collaboration/ (accessed on 1 March 2025).
- Johnson & Johnson Homepage. Available online: www.jnj.com (accessed on 1 March 2025).
- ProteinQure Homepage. Available online: https://www.proteinqure.com/pipeline/ (accessed on 1 March 2025).
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef]
- Bayer to Accelerate Drug Discovery with Google Cloud’s High-Performance Compute Power. Available online: https://www.bayer.com/media/en-us/bayer-to-accelerate-drug-discovery-with-google-clouds-high-performance-compute-power/ (accessed on 1 March 2025).
- QC Ware Homepage. Available online: https://www.qcware.com/ (accessed on 1 March 2025).
- Emmert-Streib, F. What Is the Role of AI for Digital Twins? AI 2023, 4, 721–728. [Google Scholar] [CrossRef]
- Sarkar, C.; Das, B.; Rawat, V.S.; Wahlang, J.B.; Nongpiur, A.; Tiewsoh, I.; Lyngdoh, N.M.; Das, D.; Bidarolli, M.; Sony, H.T. Artificial Intelligence and Machine Learning Technology Driven Modern Drug Discovery and Development. Int. J. Mol. Sci. 2023, 24, 2026. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Development & Approval Process: Drugs. Available online: https://www.fda.gov/drugs/development-approval-process-drugs (accessed on 1 March 2025).
- EMA. Legal Basis & Types of Approvals. Available online: https://www.ema.europa.eu/en/documents/presentation/presentation-legal-basis-and-types-approvals-s-prilla_en.pdf (accessed on 1 March 2025).
- Greganti, C.; Demarie, T.F.; Ringbauer, M.; Jones, J.A.; Saggio, V.; Calafell, I.A.; Rozema, L.A.; Erhard, A.; Meth, M.; Postler, L.; et al. Cross-verification of independent quantum devices. Phys. Rev. X 2021, 11, 031049. [Google Scholar]
- Braga, D.M.; Rawal, B.S. Quantum and AI-Enhanced Digital Twin Systems. In Proceedings of the 2024 15th International Conference on Computing Communication and Networking Technologies (ICCCNT), Kamand, India, 24–28 June 2024; IEEE: New York, NY, USA, 2024; pp. 1–8. [Google Scholar]
- Lotaiq, N.; Dermawan, D. Advancements in Virtual Bioequivalence: A Systematic Review of Computational Methods and Regulatory Perspectives in the Pharmaceutical Industry. Pharmaceutics 2024, 16, 1414. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- U.S. Food and Drug Administration. The Use of Physiologically Based Pharmacokinetic Analyses—Biopharmaceutics Applications for Oral Drug Product Development, Manufacturing Changes, and Controls. 2020. Available online: https://www.fda.gov/media/142500/download (accessed on 1 July 2022).
- Wu, D.; Sanghavi, M.; Kollipara, S.; Ahmed, T.; Saini, A.K.; Heimbach, T. Physiologically Based Pharmacokinetics Modeling in Biopharmaceutics: Case Studies for Establishing the Bioequivalence Safe Space for Innovator and Generic Drugs. Pharm. Res. 2023, 40, 337–357. [Google Scholar] [CrossRef]
- Jha, D.; Durak, G.; Sharma, V.; Keles, E.; Cicek, V.; Zhang, Z.; Srivastava, A.; Rauniyar, A.; Hagos, D.H.; Tomar, N.K.; et al. A Conceptual Framework for Applying Ethical Principles of AI to Medical Practice. Bioengineering 2025, 12, 180. [Google Scholar] [CrossRef] [PubMed]
- Mirakhori, F.; Niazi, S.K. Harnessing the AI/ML in Drug and Biological Products Discovery and Development: The Regulatory Perspective. Pharmaceuticals 2025, 18, 47. [Google Scholar] [CrossRef]
- Vela, D.; Sharp, A.; Zhang, R.; Nguyen, T.; Hoang, A.; Pianykh, O.S. Temporal quality degradation in AI models. Sci. Rep. 2022, 12, 11654. [Google Scholar] [CrossRef]
- Mujahid, M.; Kına, E.; Rustam, F.; Villar, M.G.; Alvarado, E.S.; Isabel, D.L.T.D.; Ashraf, I. Data oversampling and imbalanced datasets: An investigation of performance for machine learning and feature engineering. J. Big Data 2024, 11, 87. [Google Scholar] [CrossRef]
- David Winster Praveenraj, D.; Melvin, V.; Vennila, C.; Ahmed, H.A.; Diyora, P.; Vasudevan, N.; Avudaiappan, T. Exploring Explainable Artificial Intelligence for Transparent Decision Making. EDP Sci. 2023, 399, 04030. [Google Scholar] [CrossRef]
- Taherdoost, H.; Madanchian, M. AI Advancements: Comparison of Innovative Techniques. AI 2024, 5, 38–54. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, T.; Xu, Y.; Li, L.; Tan, Z.; Tan, P.; Huang, P.; Zeng, G. Continuous-variable quantum key distribution robust against environmental disturbances. Opt. Express 2024, 32, 7783–7799. [Google Scholar] [CrossRef] [PubMed]
- Gardas, B.; Deffner, S. Quantum fluctuation theorem for error diagnostics in quantum annealers. Sci. Rep. 2018, 8, 17191. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Valiente, J.; Romero-Álvarez, J.; Moguel, E.; García-Alonso, J.; Murillo, J.M. Technological diversity of quantum computing providers: A comparative study and a proposal for API Gateway integration. Softw. Qual. J. 2024, 32, 53–73. [Google Scholar] [CrossRef]
- Gibbs, J.; Gili, K.; Holmes, Z.; Commeau, B.; Arrasmith, A.; Cincio, L.; Coles, P.J.; Sornborger, A. Long-time simulations for fixed input states on quantum hardware. NPJ Quantum Inf. 2022, 8, 135. [Google Scholar] [CrossRef]
- Brandão, F.G.; Ramanathan, R.; Grudka, A.; Horodecki, K.; Horodecki, M.; Horodecki, P.; Szarek, T.; Wojewódka, H. Realistic noise-tolerant randomness amplification using finite number of devices. Nat. Commun. 2016, 7, 11345. [Google Scholar] [CrossRef]
- Gu, T.; Pan, W.; Yu, J.; Ji, G.; Meng, X.; Wang, Y.; Li, M. Mitigating bias in AI mortality predictions for minority populations: A transfer learning approach. BMC Med. Inform. Decis. Mak. 2025, 25, 30. [Google Scholar] [CrossRef]
- Zafar, A. Balancing the scale: Navigating ethical and practical challenges of artificial intelligence (AI) integration in legal practices. Discov. Artif. Intell. 2024, 4, 27. [Google Scholar] [CrossRef]
- Amandeep, S.B.; Bernal Neira, D.E. Federated learning with tensor networks: A quantum AI framework for healthcare. Mach. Learn. Sci. Technol. 2024, 5, 045035. [Google Scholar] [CrossRef]
- Humr, S.; Canan, M. Intermediate Judgments and Trust in Artificial Intelligence-Supported Decision-Making. Entropy 2024, 26, 500. [Google Scholar] [CrossRef]
- Feretzakis, G.; Papaspyridis, K.; Aris Gkoulalas-Divanis Verykios, V.S. Privacy-Preserving Techniques in Generative AI and Large Language Models: A Narrative Review. Information 2024, 15, 697. [Google Scholar] [CrossRef]
- Ojha, I.; Bhattacharya, M. Addressing AI Inequality: A Path Towards Fair and Equitable Systems. Globsyn Manag. J. 2024, 18, 84–86. Available online: https://library.capella.edu/login?url=https://www.proquest.com/scholarly-journals/addressing-ai-inequality-path-towards-fair/docview/3180218654/se-2 (accessed on 1 March 2025).
- Nicolas, T.; Lukas, E.; Hessler, G.; Friedemann, S.K.; Güssregen Stefan Kast, S.M. Quantum–mechanical property prediction of solvated drug molecules: What have we learned from a decade of SAMPL blind prediction challenges? J. Comput.-Aided Mol. Des. 2021, 35, 453–472. [Google Scholar] [CrossRef]
- Kamuntavičius, G.; Prat, A.; Paquet, T.; Bastas, O.; Aty, H.A.; Sun, Q.; Andersen, C.B.; Harman, J.; Siladi, M.E.; Rines, D.R.; et al. Accelerated hit identification with target evaluation, deep learning and automated labs: Prospective validation in IRAK1. J. Cheminf. 2024, 16, 127. [Google Scholar] [CrossRef] [PubMed]
- Madushanka, A.; Moura, R.T.; Jr Verma, N.; Kraka, E. Quantum Mechanical Assessment of Protein-Ligand Hydrogen Bond Strength Patterns: Insights from Semiempirical Tight-Binding and Local Vibrational Mode Theory. Int. J. Mol. Sci. 2023, 24, 6311. [Google Scholar] [CrossRef]
- Lunghini, F.; Fava, A.; Pisapia, V.; Sacco, F.; Iaconis, D.; Beccari, A.R. ProfhEX: AI-based platform for small molecule liability profiling. J. Cheminform. 2023, 15, 60. [Google Scholar] [CrossRef]
- Odah, M. Artificial Intelligence Meets Drug Discovery: A Systematic Review on AI-Powered Target Identification and Molecular Design. Med. Pharmacol. 2025; preprint. [Google Scholar] [CrossRef]
- Maramraju, S.; Kowalczewski, A.; Kaza, A.; Liu, X.; Jathin, P.S.; Albert, M.V.; Ma, Z.; Yang, H. AI-organoid integrated systems for biomedical studies and applications. Bioeng. Transl. Med. 2024, 9, e10641. [Google Scholar] [CrossRef]
- Nene, L.; Flepisi, B.T.; Brand, S.J.; Basson, C.; Balmith, M. Evolution of Drug Development and Regulatory Affairs: The Demonstrated Power of Artificial Intelligence. Clin. Ther. 2024, 46, e6–e14. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Anand, O.; Pepin, X.J.H.; Kolhatkar, V.; Seo, P. The Use of Physiologically Based Pharmacokinetic Analyses—In Biopharmaceutics Applications -Regulatory and Industry Perspectives. Pharm. Res. 2022, 39, 1681–1700. [Google Scholar] [CrossRef]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhao, X.; Li, H.; Yu, X. A Personalized Federated Learning Method Based on Knowledge Distillation and Differential Privacy. Electronics 2024, 13, 3538. [Google Scholar] [CrossRef]
- van Outersterp, R.E.; Martens, J.; Berden, G.; Lubin, A.; Cuyckens, F.; Oomens, J. Identification of Drug Metabolites with Infrared Ion Spectroscopy-Application to Midazolam in vitro Metabolism**. Chem. Methods 2023, 3, e202200068. [Google Scholar] [CrossRef]
- Santini, D.; Botticelli, A.; Galvano, A.; Iuliani, M.; Incorvaia, L.; Gristina, V.; Taffon, C.; Foderaro, S.; Paccagnella, E.; Simonetti, S.; et al. Quantum Machine Learning—An Overview. Electronics 2023, 12, 2379. [Google Scholar] [CrossRef]
- Newsfile Corp. GNQ Insilico’s AI-Driven Digital Twin Platform Shows Promising Results in First Virtually Simulated Clinical Drug Trial; Newstex: Riviera Beach, FL, USA, 2024; Available online: https://www.biospace.com/gnq-insilico-s-ai-driven-digital-twin-platform-shows-promising-results-in-first-virtually-simulated-clinical-drug-trial (accessed on 1 March 2025).
- Bettanti, A.; Beccari, A.R.; Biccarino, M. Exploring the future of biopharmaceutical drug discovery: Can advanced AI platforms overcome current challenges? Discov. Artif. Intell. 2024, 4, 102. [Google Scholar] [CrossRef]
- Yadav, C.S.; Azad, I.; Nasibullah, M.; Ahmad, N.; Lohani, M.B.; Khan, A.R. Synthesis, characterization, quantum chemical modelling, molecular docking, in silico and in vitro assessment of 3-(2-bromo-5-fluorophenyl))-1-(thiophen-2-yl)prop-2-en-1-one. Sci. Rep. 2024, 14, 29838. [Google Scholar] [CrossRef]
- Geris, L.; Guyot, Y.; Schrooten, J.; Papantoniou, I. In silico regenerative medicine: How computational tools allow regulatory and financial challenges to be addressed in a volatile market. Interface Focus 2016, 6, 20150105. [Google Scholar] [CrossRef]
- Wenteler, A.; Cabrera, C.P.; Wei, W.; Neduva, V.; Barnes, M.R. AI approaches for the discovery and validation of drug targets. Camb. Prism. Precis. Med. 2024, 2, e7. [Google Scholar] [CrossRef]
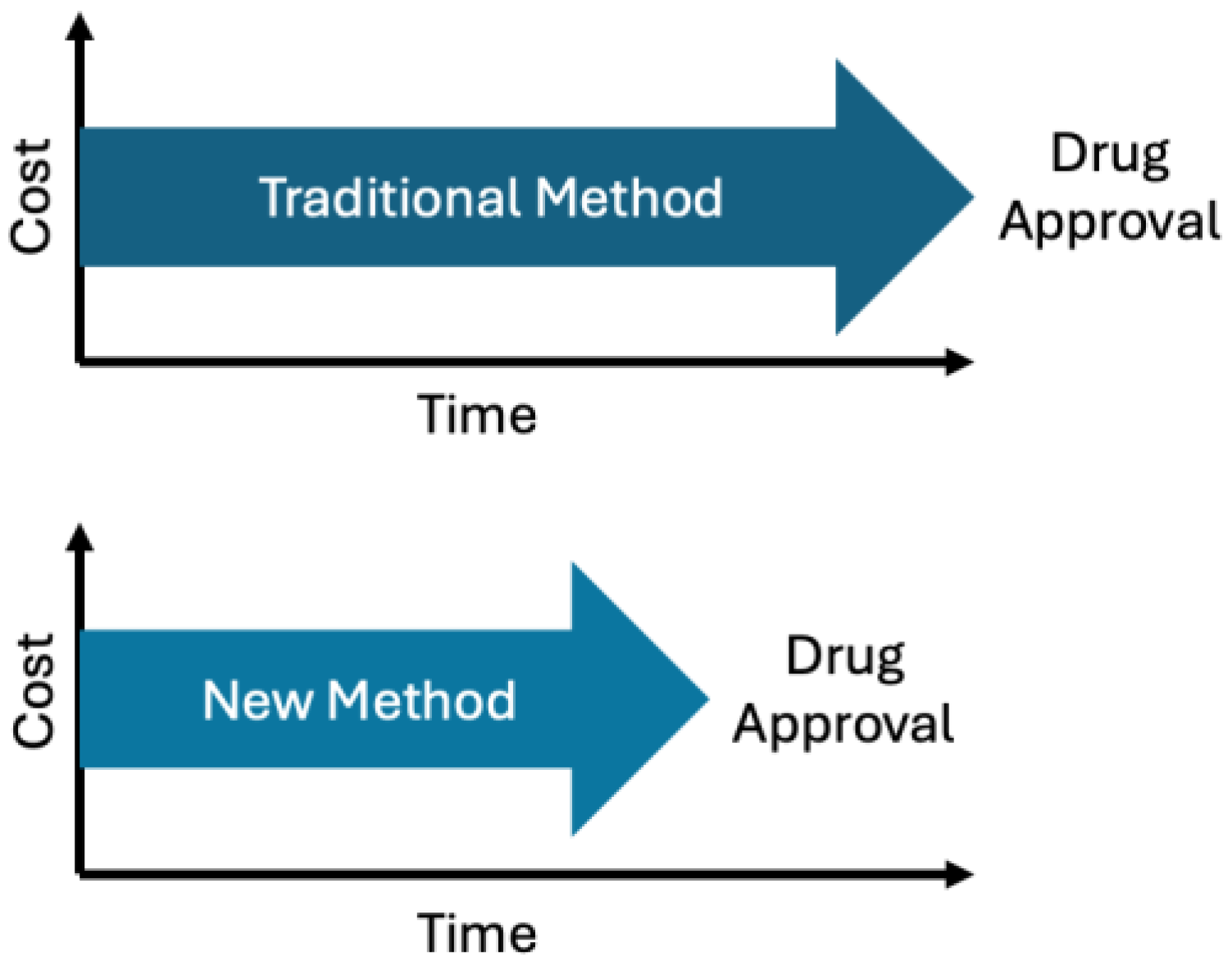
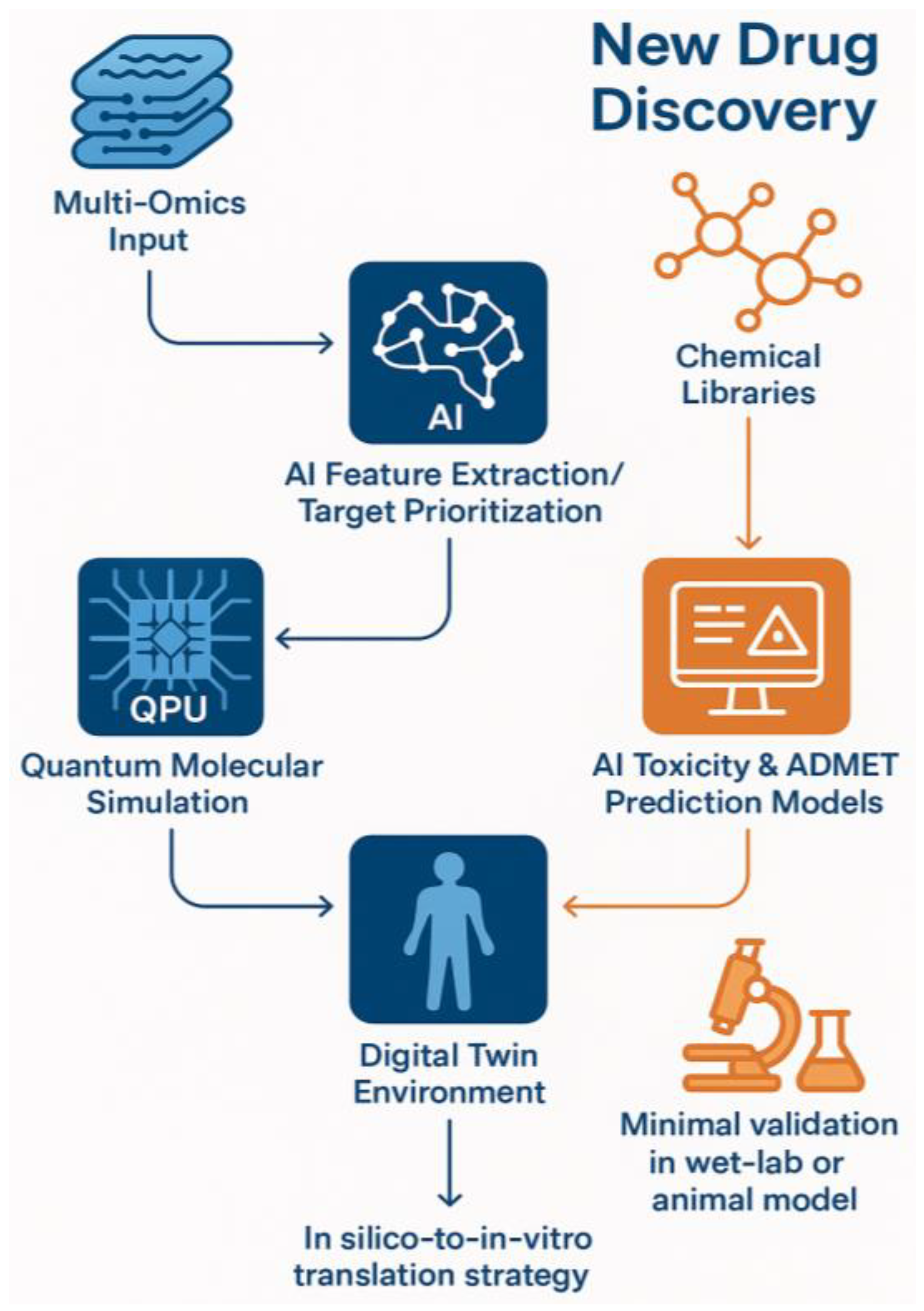
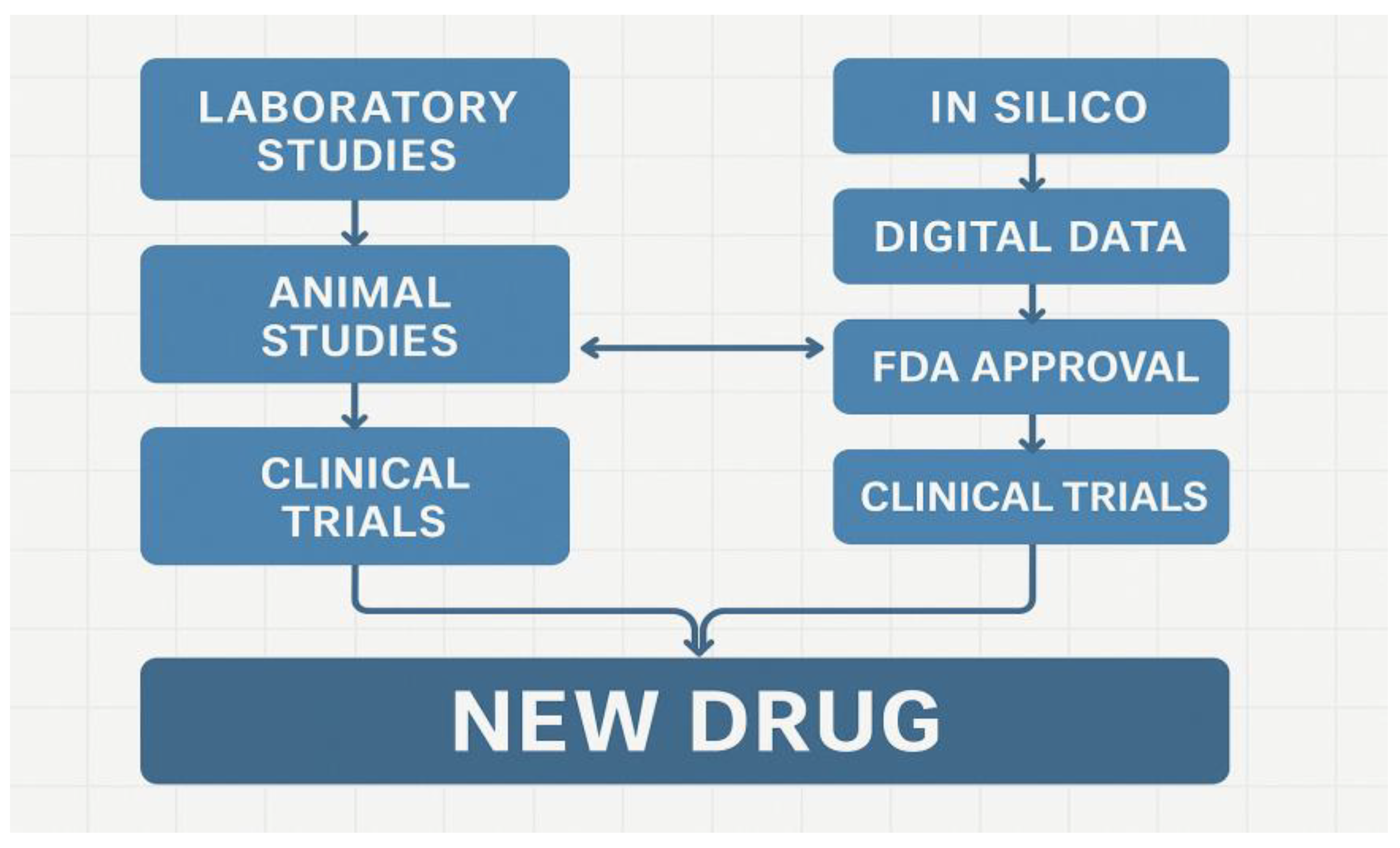

{kind=link}
{kind=link}
{kind=link}
| Component | Digital Twin (In Silico) | Traditional Laboratory |
|---|---|---|
| Input Data | Genomic data, protein structures, chemical libraries, clinical datasets | Biological specimens, assays, physical compounds |
| Core Technology | Quantum chemistry, simulators, machine learning, molecular dynamics, digital twins | Wet lab technologies (e.g., NMR, HPLC, cell assays) |
| Computation Engine | Hybrid AI–quantum system (e.g., VGG, GAN, DNN, BERT, TensorFlow) | Manual or automated lab protocols, practical experiments |
| Validation Process | Simulated vs. historical or parallel experimental datasets | N/A |
| Knowledge Process | Reinforcement learning, Bayesian inference, model selection on results and hypotheses | Traditional hypothesis testing |
| Time and Cost Efficiency | High throughput, low-cost iterations | Low (sophisticated) |
| Ethical and Regulation Scope | Ethical AI, FAIR data model, FDA/EMA submission readiness | Standard GLP/GCP (animal) trials |
| Method | Quantum Calculation | Laboratory Validation | Comparison |
|---|---|---|---|
| Molecular Structure Prediction | Uses quantum computing methods (including quantum chemistry algorithms) to simulate the molecular structure, interactions, and stability of a potential drug candidate. This involves calculating properties, including bond lengths, angles, and molecular energy states. | Experimentally determines the molecular structure using methods such as X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy. | The calculated molecular structure (e.g., bond lengths, angles) is compared to the experimentally derived structure. Discrepancies may indicate areas where the quantum model needs refinement or where experimental conditions differ from theoretical assumptions. |
| Binding Affinity to Targets | Quantum refers to the study and prediction of how strongly a molecule (e.g., a drug or ligand) binds to its target, e.g., a protein, using quantum mechanics (QM) principles. By calculating the interactions at the electronic level, QM-based methods provide highly accurate insights into binding energetics and mechanisms, surpassing traditional approaches in procession. | Measures binding affinities experimentally using techniques such as surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), or enzyme-linked assays. | Compare the predicted binding energies or disassociation constants from quantum calculations with the experimental results. A close match indicates that the quantum molecule accurately represents the molecular interactions. |
| Reaction Pathways and Mechanisms | It utilizes quantum computing to simulate the chemical reactions a drug may induce in the body, including metabolic pathways and interactions with enzymes. This provides insights into potential metabolites or degradation products. | Experimentally determines the reaction products or metabolites using techniques such as mass spectrometry (MS) or liquid chromatography (LC). | Check whether the reaction pathways and products predicted using quantum simulations align with experimental observations. This helps to validate whether the quantum model accurately predicts real biochemical reactions. |
| Thermodynamics in Kinetic Properties | Simulates thermodynamic properties, including free energy and entropy changes for reactions involving the drug, as well as kinetic parameters such as reaction rates and activation energy. | Experimentally measures thermodynamic properties using calorimetry or kinetic properties through reaction rate analysis (e.g., spectroscopic methods or chromatographic separation). | Compares the predicted thermal dynamic and kinetic values with those obtained from lab experiments. Deviations can help refine quantum models to include additional factors such as solvation effects or specific experimental conditions. |
| Solubility in Pharmacokinetics | Quantum solubility refers to the application of quantum mechanical (QM) principles to predict and understand the solubility of drug molecules in various solvents, a key parameter in pharmacokinetics (PK). Solubility impacts drug absorption, distribution, metabolism, and excretion (ADME) and influences a drug’s bioavailability in terms of therapeutic efficacy. | Tests solubility experimentally and excess pharmacokinetics in vitro (e.g., cell-based assays) or in vitro (e.g., animal models). | Compare the predicted solubility, permeability, and metabolic stability with experimental data to validate the model’s accuracy. If they match closely, the quantum predictions can be considered reliable for further development. |
| Toxicity Predictions | It utilizes quantum models to predict potential toxic interactions by simulating the interactions of the drug candidate with off-target proteins or DNA or by predicting potential toxic metabolites. | Conducts in vitro toxicity tests (e.g., using cell cultures) and in vivo toxicity studies in animal models to measure toxic effects. | Compare the predicted toxicity levels with laboratory findings. If the predictions align, this may help reduce reliance on extensive animal testing, as quantum molecules can be trusted for early-stage toxicity screenings. |
| AI-Model | Description of Model Function |
|---|---|
| DeepTox | It utilizes deep learning to predict toxicological endpoints, including mutagenicity, carcinogenicity, and organ toxicity. It analyzes molecular structures to identify potentially toxic properties. |
| Tox21 Challenge Models | Developed in collaboration with the NIH, EP, and FDA, these machine learning models predict drug toxicity by screening compounds for various toxicological effects, utilizing large datasets such as Tox21. |
| IBM Watson for Drug Discovery | Machine learning is used to predict adverse drug reactions and potential toxicity by analyzing extensive datasets, including chemical properties, biological activities, and clinical trials. |
| ADMET Predictor | From Simulations Plus, this software predicts absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, focusing on identifying any red flags in a compound’s structure. |
| ToxCast | An EPA initiative, ToxCast, uses computational models and machine learning to assess chemical toxicity. It is beneficial for screening chemicals without extensive toxicology data. |
| Multi-Instance Multi-Label (MIML) Models | These are advanced machine learning models tailored to predict multiple toxicological endpoints simultaneously and are used by some drug development companies. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braga, D.M.; Rawal, B.S. Harnessing AI and Quantum Computing for Accelerated Drug Discovery: Regulatory Frameworks for In Silico to In Vivo Validation. J. Pharm. BioTech Ind. 2025, 2, 11. https://doi.org/10.3390/jpbi2030011
Braga DM, Rawal BS. Harnessing AI and Quantum Computing for Accelerated Drug Discovery: Regulatory Frameworks for In Silico to In Vivo Validation. Journal of Pharmaceutical and BioTech Industry. 2025; 2(3):11. https://doi.org/10.3390/jpbi2030011
Chicago/Turabian StyleBraga, David Melvin, and Bharat S. Rawal. 2025. "Harnessing AI and Quantum Computing for Accelerated Drug Discovery: Regulatory Frameworks for In Silico to In Vivo Validation" Journal of Pharmaceutical and BioTech Industry 2, no. 3: 11. https://doi.org/10.3390/jpbi2030011
APA StyleBraga, D. M., & Rawal, B. S. (2025). Harnessing AI and Quantum Computing for Accelerated Drug Discovery: Regulatory Frameworks for In Silico to In Vivo Validation. Journal of Pharmaceutical and BioTech Industry, 2(3), 11. https://doi.org/10.3390/jpbi2030011