Abstract
Xanthine oxidoreductase (XOR) is the only enzyme responsible for uric acid production and is essential for preventing gout. While XOR inhibitors effectively reduce serum urate levels, they also influence purine salvage and de novo pathways, as well as energy metabolism, raising concerns about metabolic adaptation and rebound effects upon treatment discontinuation. In this review, we outline the fundamental regulatory mechanisms of purine metabolism and summarize the mechanisms of action of XOR inhibitors and their associated metabolic effects with reference to XOR deficiency, type I xanthinuria. Furthermore, we discuss the impact of discontinuing XOR inhibitors and examine their potential for rebound.
1. Introduction
Adenine and guanine, which are major components of both DNA and RNA, are purine bases that are major components of ATP and GTP, respectively. Substances with purine skeletons are collectively referred to as purines, which include nucleic acids, purine nucleotides, purine nucleosides, and purine bases. In humans, these compounds are ultimately metabolized to uric acid. The only enzyme responsible for uric acid production is xanthine oxidoreductase (XOR), which catalyzes the two-step oxidation of hypoxanthine to xanthine and xanthine to uric acid during purine metabolism (Figure 1) [1]. The produced urate is transported across the cell membrane by transporters [2].
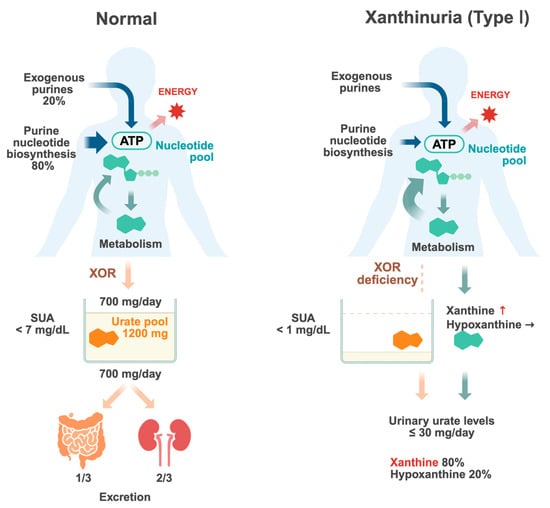
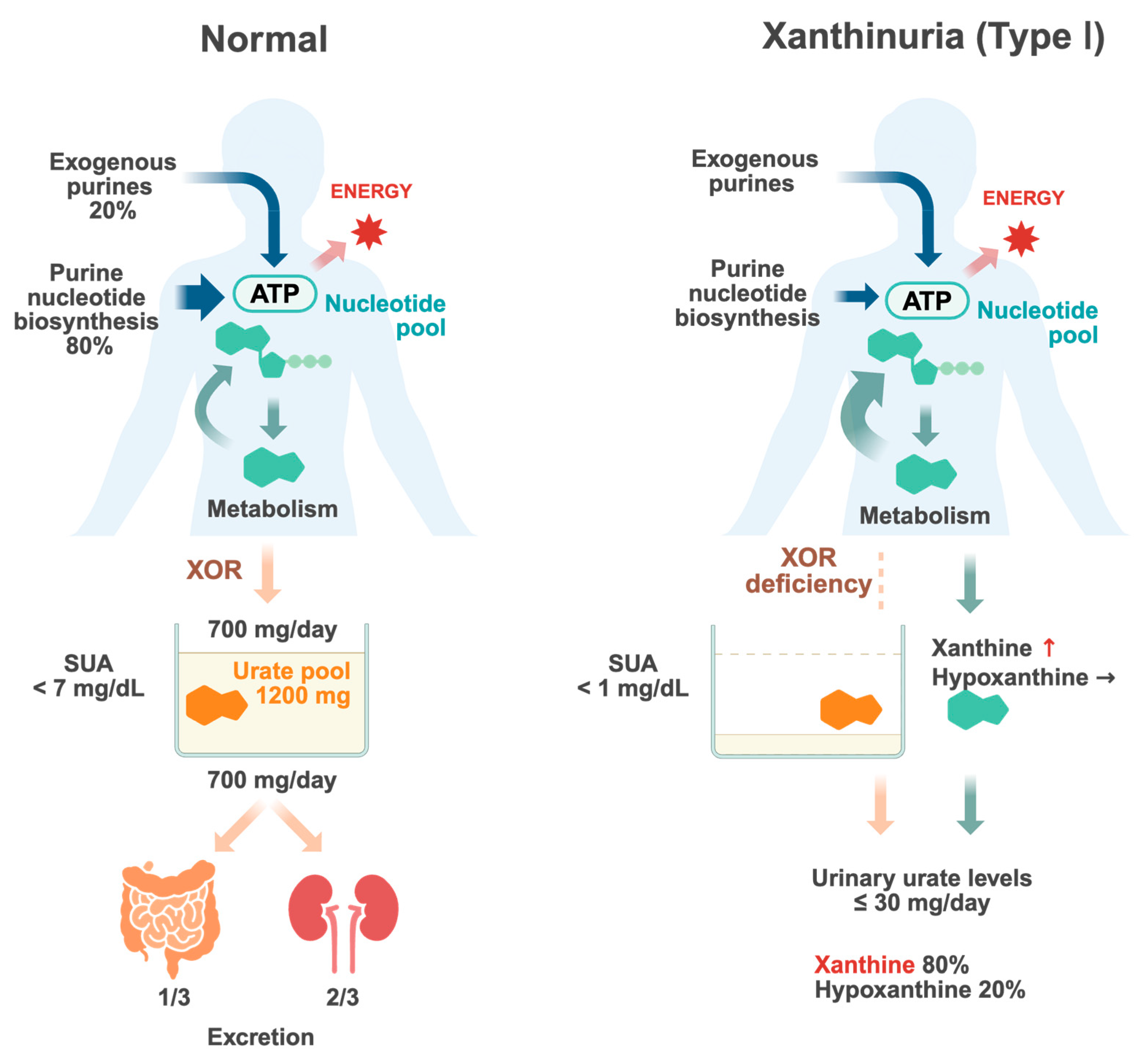
Figure 1.
Urate metabolism in normal individuals and patients with type I xanthinuria. Purines in the body are supplied through both exogenous pathways, such as dietary intake, and endogenous pathways, which involve metabolic processes within the body. Purines serve as essential components of nucleic acid and ATP, playing a crucial role in cellular turnover and energy metabolism. Purines are shown in green. Upon degradation, purines are ultimately converted into uric acid (orange). Urate is filtered by the kidneys, where a portion undergoes reabsorption in the renal tubules while the remainder is excreted. Renal excretion accounts for approximately 2/3 of urate elimination, whereas intestinal elimination constitutes the remaining approximately 1/3. The balance between urate production and excretion regulates serum urate (SUA) levels. Disruptions in this equilibrium increase the risk of hyperuricemia and gout. In type I xanthinuria, a deficiency of xanthine oxidoreductase (XOR) prevents the conversion of hypoxanthine and xanthine into uric acid, leading to elevated serum concentrations of these intermediates. Consequently, the urinary excretion of xanthine increases, resulting in xanthinuria. Since hypoxanthine can be partially salvaged via the purine salvage pathway, its accumulation is less pronounced compared to xanthine (key points in red text). Created with Biorender.com.
The total amount of urate accumulated in the body is referred to as the urate pool and is determined by the balance between urate production and excretion (Figure 1). The urate pool in healthy adults was reported to be approximately 1200 mg (ranging from 1000 to 1700 mg) in males and about 600 mg in females [3,4]. In individuals with normal renal function, approximately two-thirds of urate excretion occurs via the kidneys, while approximately one-third is eliminated through the gastrointestinal tract [5]. In the kidneys, urate is first filtered by the glomeruli and then undergoes reabsorption and secretion, primarily in the proximal tubules. Ultimately, only 6–10% of the filtered urate is excreted in urine. Approximately 700–800 mg of urate enters the urate pool daily and an equivalent amount is excreted to maintain a steady state. The serum urate levels reflect the urate pool, and hyperuricemia occurs when the balance between production and excretion shifts towards an increase in the urate pool. Gout is caused by hyperuricemia, which results in the deposition of monosodium urate crystals in and around the joints.
Uric acid is produced through two main pathways: the exogenous pathway, derived from dietary purine intake and gut microbiota, which accounts for approximately 20%, and the endogenous pathway, generated through metabolic processes within the body, which constitutes approximately 80%. While dietary purine restrictions can control serum urate levels to a certain extent, their effect on total urate production is limited; therefore, in cases of hyperuricemia, it is also necessary to control endogenous urate production.
The degradation of adenine and guanine nucleotides is an ongoing process driven by cellular energy metabolism and turnover that continuously generates a constant amount of uric acid (Figure 2). However, under conditions in which ATP breakdown is markedly accelerated, such as during intense physical exertion or excessive alcohol consumption [6], cellular hypoxanthine levels increase sharply, leading to an increase in urate production. This phenomenon is closely associated with the regulation of the adenylate pool and is quantified by the energy charge equation (([ATP] + 0.5[ADP])/([ATP] + [ADP] + [AMP])), which reflects the intracellular state of the adenylate pool [7]. AMP degradation is enhanced to maintain the energy balance, ultimately resulting in increased urate synthesis. The de novo purine synthesis pathway synthesizes phosphoribosyl pyrophosphate (PRPP) from ribose-5-phosphate, an intermediate in the oxidative branch of the pentose phosphate pathway, and subsequently produces IMP through a multi-step process involving several amino acids and formates. IMP, AMP and GMP are generated, and their accumulation leads to the feedback inhibition of rate-limiting enzymes in the de novo purine synthesis pathway. Additionally, purine nucleotide biosynthesis involves the purine salvage pathway, which recycles existing purine bases, hypoxanthine, guanine, and adenine into their corresponding nucleotides (IMP, GMP, and AMP). To suppress urate production via these pathways, the significant inhibition of XOR is required, because XOR is not a conventional regulatory enzyme in purine metabolism. This review provides an overview of the changes in purine metabolism resulting from extensive XOR inhibition, focusing on XOR deficiency as an illustrative case. Additionally, clinical studies have reported an association between the discontinuation of XOR inhibitors and an increased risk of cardiovascular-related mortality [8,9,10,11]. We examined the effects of the discontinuation of individual XOR inhibitors in this context.
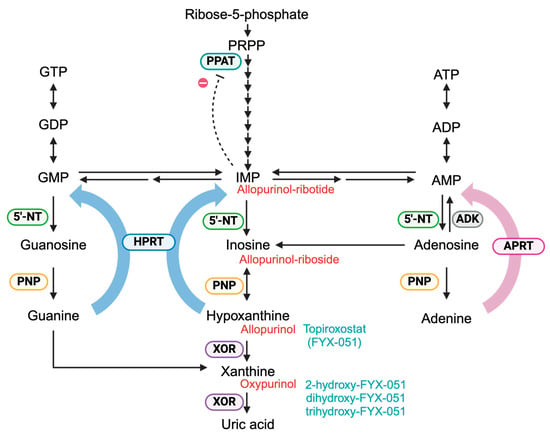
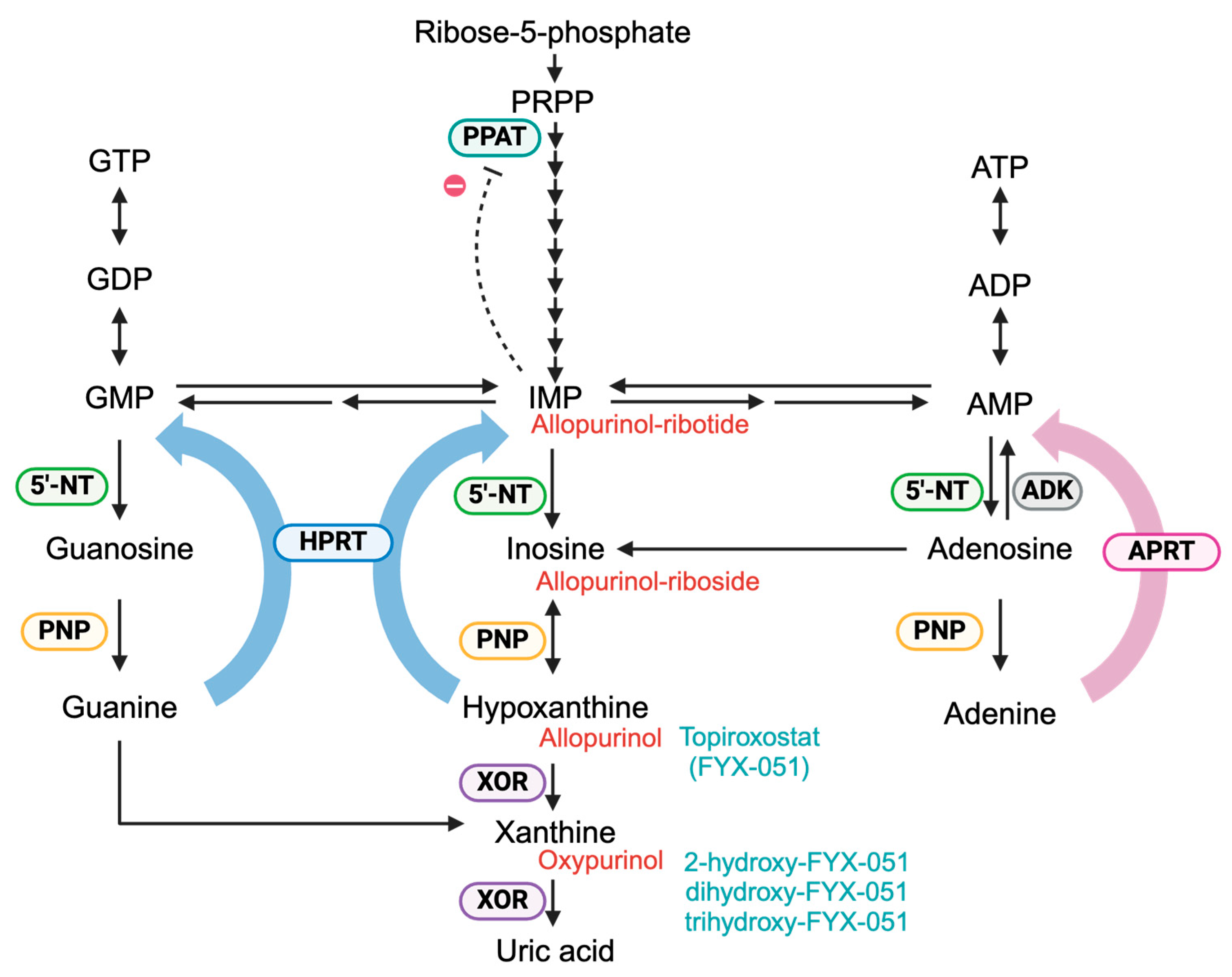
Figure 2.
Purine metabolism. Purine nucleotides are synthesized via two pathways: the de novo synthesis and the salvage pathway. In the de novo synthesis pathway, IMP is generated through multiple steps starting from phosphoribosyl pyrophosphate (PRPP). In the salvage pathway, hypoxanthine phosphoribosyltransferase (HPRT, blue) catalyzes the conversion of guanine and hypoxanthine to GMP and IMP, respectively, while adenine phosphoribosyltransferase (APRT, pink) catalyzes the conversion of adenine to AMP. The resulting purine nucleotides exert feedback inhibition on phosphoribosyl pyrophosphate amidotransferase (PPAT, light blue), thereby regulating de novo synthesis. Hypoxanthine and xanthine, generated during purine degradation, are metabolized to uric acid by xanthine oxidoreductase (XOR, purple). Allopurinol, a hypoxanthine analog, is primarily metabolized to oxypurinol by XOR. Additionally, allopurinol is converted to allopurinol-riboside and allopurinol-ribotide via purine nucleoside phosphorylase (PNP, yellow), HPRT, and 5′-nucleotidase (5′-NT, green). Allopurinol derivatives are shown in red. Topiroxostat, a non-purine XOR inhibitor, is converted into 2-hydroxy-FYX-051 by the XOR. In vitro, further hydroxylation to dihydroxy-FYX-051 and trihydroxy-FYX-051 has been observed. Topiroxostat derivatives are shown in pale blue.
2. Brief Overview of XOR
The human XOR is a large protein composed of two polypeptides, each consisting of 1333 amino acids [12,13,14]. Each monomer contains a molybdenum cofactor (Moco), two iron–sulfur clusters, and a FAD coenzyme. Structural alterations around FAD trigger a shift in the catalytic mode of XOR, converting it from its dehydrogenase form (XDH), which primarily donates electrons to NAD+, to its oxidase form (XO), which transfers electrons to O2, leading to the production of reactive oxygen species [1,15,16,17]. The conversion of XDH to XO only occurs in mammals and plays a crucial role in various physiological and pathological processes [15,18,19,20,21,22,23]. The tissue localization of XOR has been reported in various animals. In humans, the liver, kidneys, intestines and mammary glands are the primary sites of XOR expression, as demonstrated by protein expression and activity assays [12,24,25,26,27,28]. XOR is not expressed in red blood cells, and its activity in blood is normally low. However, increased plasma XOR activity has been observed in various disease states and is considered to result from its release from major organs such as the liver under stress conditions. XOR also attaches to the outer surface of endothelial cell plasma membranes. In contrast, there is no evidence that XOR is present in the human brain, as they display no detectable XOR protein expression, enzymatic activity, or urate as a product of its catalytic reaction [29,30]. The absence of XOR in such tissues is thought to result in hypoxanthine accumulation, subsequently leading to the activation of the purine salvage pathway. In mammals, XOR has been proposed to have physiological functions beyond uric acid biosynthesis, including its involvement in immune responses and intracellular signaling pathways through the production of superoxide and hydrogen peroxide [31,32,33].
3. Metabolic Effects of XOR Deficiency
Metabolic changes caused by the absence of XOR in brain and red blood cells can also be observed in the metabolism of patients with XOR deficiency. Human XOR deficiency is a rare metabolic disorder classified into three types: Type I, Type II, and Type III [34,35,36,37]. Type I results from the genetic deletion of the XOR protein, which causes a complete loss of XOR activity alone (isolated deficiency). Type II is characterized by the loss of both XOR and aldehyde oxidase (AO) activities and is known as dual enzyme deficiency. Type III involves a deficiency in XOR, AO, and sulfite oxidase (SO), resulting in triple enzyme deficiency. The metabolic effects of XOR inhibitors are similar to those of isolated XOR deficiency, which is useful in understanding their effect on metabolism.
Type I and Type II XOR deficiencies are associated with xanthinuria and typically, XOR deficiency presents with a marked reduction in urinary urate levels (often ≤30 mg/24 h), along with increased xanthine excretion (Figure 1). Under purine-restricted conditions, urate excretion is nearly or completely absent. Therefore, XOR serves as the primary pathway for uric acid production in humans. The source of the small amounts of uric acid that are sometimes detected remains unclear, but possible explanations include residual XOR activity, dietary intake, and the metabolism of endogenous hypoxanthine and xanthine by the gut microbiota [38]. Xanthinuria is well tolerated in most individuals and often diagnosed incidentally. Most patients are asymptomatic, and severe symptoms are uncommon. However, some may experience hematuria, renal colic, recurrent urinary tract infections, renal dysfunction, and muscle pain or cramps due to xanthine stone formation in the urinary tract or crystal accumulation in the muscle.
Xanthine accounts for approximately 80% of total urinary oxypurine (hypoxanthine + xanthine) excretion, a characteristic consistently observed in patients with XOR deficiency [38,39]. Serum urate levels exhibit profound hypouricemia, typically below 1 mg/dL, and xanthine concentrations are elevated in both the blood and urine. In contrast, the increase in circulating hypoxanthine levels is relatively modest, despite its minimal excretion into urine. This is likely due to the recycling of hypoxanthine via hypoxanthine-guanine phosphoribosyltransferase (HPRT) [40,41]. A report on the relationship between purine utilization and urinary oxypurine excretion in patients with xanthinuria has shown that radiolabeled adenine, hypoxanthine, and guanine were taken up to varying degrees, with adenine being the most efficiently incorporated, followed by hypoxanthine. Guanine uptake is minimal, with the majority undergoing catabolism to xanthine followed by subsequent excretion [42]. This finding implies that the incorporation of purine bases into the nucleotide pool is not uniform, with preferential conversion to adenine nucleotides, which tend to be retained in a purine storage form [29].
Furthermore, the available xanthine and hypoxanthine pools in patients with xanthinuria were smaller than those in the urate storage pools of healthy individuals [39]. This phenomenon was also observed in patients with gout treated with allopurinol, an XOR inhibitor [43,44]. Previous data indicated that although allopurinol treatment significantly increases the urinary excretion of oxypurines, it does not fully compensate for the reduced urinary excretion of urate, with the mean difference being approximately 35% [39,45], suggesting additional regulatory mechanisms. The main factors contributing to this shortfall are the increased activity of the salvage pathway and suppression of the de novo pathway by feedback inhibition [46,47,48]. Since PRPP generally exists in limited amounts, the presence of hypoxanthine preferentially drives the salvage pathway, thereby suppressing de novo purine synthesis. The metabolic alterations observed in XOR deficiency suggest that XOR inhibition not only reduces uric acid production, but also indirectly suppresses the de novo pathway, leading to an overall decrease in purine nucleotide synthesis. Allopurinol treatment increases urinary xanthine excretion, raising concerns about tubular crystal deposition and stone formation. However, to date, the reported cases have been limited only to patients with Lesch–Nyhan syndrome, a disorder caused by HPRT deficiency, or to patients with malignant tumors receiving allopurinol in combination with anticancer agents [49,50,51,52].
4. XOR Inhibitors
Cases of genetic enzyme deficiencies strongly indicate that enzyme inhibition does not lead to severe adverse effects [34,37,53]. However, the long-term administration of inhibitors may induce physiological and metabolic adaptations, potentially altering the body’s regulatory mechanisms. Therefore, the use of enzyme inhibitors requires careful assessment. The current clinically approved XOR inhibitors include allopurinol, febuxostat, and topiroxostat, which are categorized as purine-based and non-purine-based inhibitors. Given the metabolic alterations in xanthinuria, the main target during the clinical inhibition of urate production is to inhibit the conversion of hypoxanthine to xanthine rather than the conversion of xanthine to uric acid [54,55]. However, each of the inhibitors has a different mechanism of inhibition and varying levels of potency. Their duration of action also differs, and some are metabolized in the body to form active metabolites. The accumulation of these metabolites could influence the potency and duration of the urate-lowering effect. Therefore, understanding the metabolic properties of these inhibitors is essential for selecting appropriate treatments and developing effective dosing strategies. This section focuses on the metabolism of each inhibitor and its impact on purine metabolism. Furthermore, we discuss the metabolic shifts induced by treatment discontinuation and propose key considerations for the safe and effective clinical use of these inhibitors.
4.1. Allopurinol
Allopurinol, a purine-based compound, is the first clinically approved XOR inhibitor [56]. Because of its structural similarity to hypoxanthine, allopurinol is recognized by XOR as a substrate and is metabolized through enzymatic catalysis in the same manner as hypoxanthine [57]. The main metabolites of allopurinol in humans are oxypurinol (approximately 80%) and allopurinol-riboside (approximately 10%) (Figure 2) [57]. Allopurinol has a short elimination half-life of 1–2 h, whereas oxypurinol, which is reabsorbed in the renal tubules, has a much longer half-life of 18–30 h. Its clearance is primarily dependent on renal function, and impaired kidney function leads to its accumulation in the body.
Regarding the inhibitory mechanism of XOR, during the hydroxylation of allopurinol to oxypurinol by XOR, the active Mo center of XOR is reduced. The transiently reduced Mo(IV) forms a coordination bond with oxypurinol, resulting in the strong inhibition of XOR activity [58,59]. The half-life of the XOR-oxypurinol complex was approximately 1 h under physiological conditions [54]. When Mo is re-oxidized through intramolecular electron transfer, oxypurinol dissociates from the enzyme. Because free oxypurinol is not a substrate of XOR, it cannot form a coordination bond on its own and requires the presence of a substrate such as hypoxanthine, xanthine, or allopurinol. Following dissociation, allopurinol preferentially binds to the enzyme. Administration experiments in hyperuricemic mice models demonstrated that free oxypurinol required a three-fold higher dose to achieve the same urate-lowering effect as allopurinol [54]. However, oxypurinol was less effective at increasing hypoxanthine levels than allopurinol, indicating that a higher concentration of oxypurinol may be necessary for the effective feedback inhibition of the de novo pathway.
Metabolic analysis in PNP deficiency suggests that allopurinol-riboside is generated from allopurinol-ribotide, which is produced by HPRT and subsequently converted by 5′-nucleotidase [60,61,62]. The conversion rate ratio of hypoxanthine to IMP versus allopurinol to allopurinol-ribotide (IMP/allopurinol-ribotide) is 36.7 times (our data: mean 47.5 ± 11.1), indicating that the metabolic conversion of allopurinol by HPRT is relatively inefficient. Nevertheless, this metabolic pathway consumes PRPP, potentially reducing the efficiency of the salvage pathway. Furthermore, allopurinol derivatives are known to inhibit both HPRT and PNP, and their effects should also be taken into consideration. However, based on previously reported Ki values [54,60,61,63], strong direct inhibition at normal plasma concentrations is considered unlikely.
Oxypurinol is a relatively weak inhibitor of XOR and is therefore not expected to enhance the salvage pathway significantly. Consequently, the metabolic changes upon the initiation of administration are considered to be relatively gradual, although they are influenced by the dosage. When allopurinol is administered at doses ranging from 50 to 400 mg/day, plasma oxypurinol levels exhibit a broad range and may reach approximately 500 µM [64]. Since oxypurinol takes several days to be excreted entirely, its inhibitory effect continues for some time after discontinuation. Consequently, serum urate levels are expected to rise gradually rather than abruptly. Furthermore, because oxypurinol has a limited impact on the salvage pathway, it is unlikely to cause rapid metabolic shifts.
4.2. Febuxostat
Febuxostat, a non-purine XOR inhibitor, exhibits stronger inhibition than allopurinol, with high-affinity binding to the enzyme (Kd in the nanomolar range) [53,65]. Febuxostat markedly enhances the salvage pathway compared to allopurinol or oxypurinol, and does not significantly affect HPRT or PNP even at high concentrations. Febuxostat forms a stable complex with XOR and remains bound until it is degraded by proteolysis. This property enables effective inhibition with a once-daily, low-dose regimen. In healthy male subjects, the primary metabolic products of febuxostat in plasma and urine are glucuronide conjugates, with additional oxidative metabolites detected. However, these metabolites are not active and do not inhibit XOR. As febuxostat is metabolized in the liver and excreted equally via urine and feces, its pharmacokinetics are less affected by renal function. However, if liver function is impaired, the clearance rate may be reduced, thereby increasing the blood levels.
The distinctive mechanism of febuxostat has significant implications for post-discontinuation metabolic alterations. Upon cessation, XOR inhibition persists until new enzyme synthesis occurs. However, enzymatic activity is rapidly restored by newly synthesized XOR, leading to a shift in purine metabolism toward degradation. As a result, a marked increase in urate production and a pronounced rebound effect may occur, which could lead to ATP depletion and cellular dysfunction in tissues dependent on the salvage pathway for maintaining ATP levels. Although there are increasing reports on the improvement of energy metabolism via the salvage pathway in the nervous system and kidneys [29,66,67], the metabolic effects of febuxostat discontinuation have not been fully investigated.
In recent years, large-scale clinical trials comparing the cardiovascular safety of febuxostat and allopurinol have been conducted, and drug discontinuation has emerged as a critical factor in interpreting the results [8,9,10,11,68,69]. The CARES trial, which enrolled gout patients with a history of cardiovascular disease, showed that febuxostat was non-inferior to allopurinol with respect to major cardiovascular events. However, the risk of all-cause mortality and cardiovascular death were significantly higher in the febuxostat group. In contrast, the FAST trial, which enrolled gout patients with cardiovascular risk factors, found no increase in mortality in the febuxostat group. Although the CARES trial has been criticized for its design, with a high dropout rate and lack of a placebo control, further analysis revealed that a substantial proportion of deaths occurred after treatment discontinuation.
Rebound effects have been observed following the discontinuation of both allopurinol and febuxostat [70]. However, in the case of allopurinol, the onset of major adverse cardiovascular events (MACE) appears to be delayed relative to febuxostat, owing to the sustained inhibitory effect of oxypurinol. Consequently, although the timing of MACE may differ, the overall incidence seems to be comparable between the two drugs. In fact, in the CARES trial, a difference in mortality between groups was observed when events occurring more than 30 days after drug discontinuation were excluded. However, this difference was no longer statistically significant, when deaths occurring beyond that period were included. Additionally, the CARES trial included patients with more severe disease compared to the FAST trial, suggesting that these patients may have been at a higher risk of rapid clinical deterioration or increased mortality after discontinuing treatment. This observation may be partially explained by the sick stopper effect, where patients in poorer health are more likely to discontinue medication, and the healthy adherer effect, where healthier individuals are more likely to continue treatment. In conclusion, it remains uncertain whether a difference in early post-discontinuation mortality risk is present, since no evaluation was carried out using the same time frame as that employed in the CARES trial.
Both inhibitors require gradual dose titration at the initiation of treatment, whereas their discontinuation necessitates even more careful management. The primary cause of withdrawal effects is the loss of metabolic support provided by XOR inhibitors. Therefore, a gradual discontinuation does not necessarily ensure safety. Since the cessation of either drug involves risks, proper management is essential.
The effects described here are mainly based on enzymatic findings, and the direct mechanism by which XOR contributes to cardiovascular events remains unclear. XOR is thought to be involved not only in altering urate levels and energy metabolism but also in the formation of monosodium urate crystals in the blood vessels, vascular endothelial injury, inflammation, and atherosclerosis through the production of reactive oxygen species [22,71,72]. Considering these complex effects, further clinical and basic research is required to elucidate both the short- and long-term metabolic consequences of discontinuing allopurinol and febuxostat, as well as their potential association with cardiovascular events.
4.3. Topiroxostat
Topiroxostat, a non-purine XOR inhibitor, functions as a hybrid-type inhibitor that exhibits both structure-based and mechanism-based inhibition [53,73,74]. It directly interacts with XOR and undergoes hydroxylation, while simultaneously forming a reaction intermediate. This intermediate is stabilized by the formation of a strong complex with XOR via a covalent Mo-O-C bond. It has been confirmed that topiroxostat does not exhibit inhibitory effects on other purine and pyrimidine metabolic enzymes, including AO, PNP, guanine deaminase, HPRT, orotate phosphoribosyltransferase, and OMP decarboxylase.
Topiroxostat is rapidly absorbed after administration, reaching its peak plasma concentration within approximately 0.67 to 0.92 h, with a reported elimination half-life of approximately 4.56 to 7.49 h. Following a single oral dose of 80 mg, the peak plasma concentration reaches approximately 2 µM. The dosing regimen was designed for twice-daily administration to maintain stable plasma levels. The XOR–topiroxostat complex exhibits a dissociation half-life of 20.4 h at 25 °C. During this process, topiroxostat underwent hydroxylation, leading to the formation of 2-hydroxy-FYX-051. The dissociation of 2-hydroxy-FYX-051 leads to a gradual recovery of enzymatic activity; however, some inhibition persists, preventing the full restoration of activity [74]. This suggests that inhibition by both hydroxylated metabolites and additional hydroxylation reactions may be involved. XOR catalyzes the conversion of topiroxostat to 2-hydroxy-FYX-051. In addition, under artificial conditions, extended incubation for up to 72 h resulted in the formation of dihydroxy- and trihydroxy-FYX-051. Studies have demonstrated that the XOR-trihydroxy-FYX-051 complex is formed via a Mo-N-C bond between the Mo center and the nitrile group of trihydroxy-FYX-051. However, in vivo studies detected only trace amounts of 2-hydroxy-FYX-051 and no other hydroxylated metabolites were observed [75]. The likelihood that these hydroxylated derivatives exert sequential inhibitory effects appears to be low. Nevertheless, further metabolic analysis may provide a clearer understanding of their roles, overall impact, and duration of inhibition. Topiroxostat exhibited little accumulation even with repeated dosing. It is prescribed less frequently than the other two inhibitors and reports on its adverse effects are relatively rare. Upon discontinuation, XOR synthesis leads to the resumption of urate production, potentially triggering a rebound effect similar to febuxostat. However, the role of topiroxostat in energy metabolism and its contribution to ATP maintenance remains insufficiently explored, necessitating further research to assess its potential benefits.
5. Conclusions
To safely and effectively use XOR inhibitors, it is essential to understand the metabolic effects of xanthinuria. Allopurinol, febuxostat, and topiroxostat exhibit different mechanisms of action and metabolic profiles. Although these inhibitors effectively lower serum urate levels, they also affect purine and energy metabolism, potentially leading to long-term metabolic adaptation. The discontinuation of XOR inhibitors may disrupt this adaptation, resulting in a sharp increase in serum urate levels. Stronger XOR inhibitors are more likely to induce significant metabolic fluctuations, necessitating more careful dose planning and management during drug withdrawal. In the case of allopurinol, the accumulation of its active metabolite, oxypurinol, contributed to a relatively gradual increase in serum urate levels after discontinuation. However, owing to its strong dependence on renal function, individualized dose adjustments are crucial. Additionally, because XOR inhibitors affect intracellular ATP levels, it is important to consider whether metabolic fluctuations in energy levels might pose a problem to the patient. Future studies should focus on the long-term metabolic adaptations associated with continued treatment with XOR inhibitors, as well as the strategies used to mitigate metabolic risks following their discontinuation. The appropriate use and management of XOR inhibitors are critical to ensure the safety and efficacy of treatment in clinical practice.
Author Contributions
Conceptualization, M.S.; writing—original draft preparation, M.S.; writing—review and editing, M.S. and K.I.; supervision, M.S. and K.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by JSPS KAKENHI (Grant Numbers [21KK0173, 24K19311 to M.S.] and [22KK0152 to K.I.]), and in part by the Gout and Uric Acid Foundation of Japan (M.S.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| XOR | Xanthine oxidoreductase |
| IMP | Inosine monophosphate |
| XDH | Xanthine dehydrogenase |
| XO | Xanthine oxidase |
| AO | Aldehyde oxidase |
| SO | Sulfite oxidase |
| HPRT | Hypoxanthine-guanine phosphoribosyltransferase |
| PRPP | Phosphoribosyl pyrophosphate |
| PPAT | Phosphoribosyl pyrophosphate amidotransferase |
| PNP | Purine nucleoside phosphorylase |
| 5′-NT | 5′-nucleotidase |
| APRT | Adenine phosphoribosyltransferase |
| ADK | Adenosine kinase |
References
- Hille, R.; Nishino, T. Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. Faseb J. 1995, 9, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Bhatnagar, V. The systems biology of uric acid transporters: The role of remote sensing and signaling. Curr. Opin. Nephrol. Hypertens. 2018, 27, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.T.; Holloway, V.P.; Glass, H.I.; Arnot, R.N. Studies of uric acid pool size and turnover rate. Ann. Rheum. Dis. 1969, 28, 366–373. [Google Scholar] [CrossRef]
- Sloan, R.W. Hyperuricemia and gout. J. Fam. Pract. 1982, 14, 923–934. [Google Scholar]
- Sorensen, L.B. Role of the intestinal tract in the elimination of uric acid. Arthritis Rheum. 1965, 8, 694–706. [Google Scholar] [CrossRef]
- Faller, J.; Fox, I.H. Ethanol-induced hyperuricemia: Evidence for increased urate production by activation of adenine nucleotide turnover. N. Engl. J. Med. 1982, 307, 1598–1602. [Google Scholar] [CrossRef]
- Atkinson, D.E. The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 1968, 7, 4030–4034. [Google Scholar] [CrossRef]
- Kuwabara, M.; Nakai, M.; Sumita, Y.; Iwanaga, Y.; Ae, R.; Kodama, T.; Hisatome, I.; Kamatani, N. Xanthine oxidase inhibitors treatment or discontinuation effects on mortality: Evidence of xanthine oxidase inhibitors withdrawal syndrome. Front. Pharmacol. 2023, 14, 1289386. [Google Scholar] [CrossRef]
- Ghang, B.; Ahn, S.M.; Kim, J.; Kim, Y.G.; Lee, C.K.; Yoo, B. Discontinuing febuxostat might cause more deaths than continuing febuxostat: The untold story from the CARES trial. Rheumatology 2020, 59, 1439–1440. [Google Scholar] [CrossRef]
- Bubb, M.R. Excess deaths upon cessation of xanthine oxidase inhibitor treatment-data from the cardiovascular safety of febuxostat and allopurinol in patients with gout and cardiovascular morbidities trial: Comment on the article by Choi et al. Arthritis Rheumatol. 2019, 71, 1391–1392. [Google Scholar] [CrossRef]
- Choi, H.; Neogi, T.; Stamp, L.; Dalbeth, N.; Terkeltaub, R. New perspectives in rheumatology: Implications of the cardiovascular safety of febuxostat and allopurinol in patients with gout and cardiovascular morbidities trial and the associated food and drug administration public safety alert. Arthritis Rheumatol. 2018, 70, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Krenitsky, T.A.; Spector, T.; Hall, W.W. Xanthine oxidase from human liver: Purification and characterization. Arch. Biochem. Biophys. 1986, 247, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Enroth, C.; Eger, B.T.; Okamoto, K.; Nishino, T.; Nishino, T.; Pai, E.F. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proc. Natl. Acad. Sci. USA 2000, 97, 10723–10728. [Google Scholar] [CrossRef] [PubMed]
- Ichida, K.; Amaya, Y.; Noda, K.; Minoshima, S.; Hosoya, T.; Sakai, O.; Shimizu, N.; Nishino, T. Cloning of the cDNA encoding human xanthine dehydrogenase (oxidase): Structural analysis of the protein and chromosomal location of the gene. Gene 1993, 133, 279–284. [Google Scholar] [CrossRef]
- Nishino, T. The conversion of xanthine dehydrogenase to xanthine oxidase and the role of the enzyme in reperfusion injury. J. Biochem. 1994, 116, 1–6. [Google Scholar] [CrossRef]
- Corte, E.D.; Stirpe, F. The regulation of rat liver xanthine oxidase. Involvement of thiol groups in the conversion of the enzyme activity from dehydrogenase (type D) into oxidase (type O) and purification of the enzyme. Biochem. J. 1972, 126, 739–745. [Google Scholar] [CrossRef]
- Waud, W.R.; Rajagopalan, K.V. The mechanism of conversion of rat liver xanthine dehydrogenase from an NAD+-dependent form (type D) to an O2-dependent form (type O). Arch. Biochem. Biophys. 1976, 172, 365–379. [Google Scholar] [CrossRef]
- Kusano, T.; Nishino, T.; Okamoto, K.; Hille, R.; Nishino, T. The mechanism and significance of the conversion of xanthine dehydrogenase to xanthine oxidase in mammalian secretory gland cells. Redox Biol. 2023, 59, 102573. [Google Scholar] [CrossRef]
- Stevens, C.R.; Millar, T.M.; Clinch, J.G.; Kanczler, J.M.; Bodamyali, T.; Blake, D.R. Antibacterial properties of xanthine oxidase in human milk. Lancet 2000, 356, 829–830. [Google Scholar] [CrossRef]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef]
- McCord, J.M. Oxygen-derived free radicals in postischemic tissue injury. N. Engl. J. Med. 1985, 312, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Polito, L.; Bolognesi, A. Xanthine oxidoreductase in atherosclerosis pathogenesis: Not only oxidative stress. Atherosclerosis 2014, 237, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine oxidoreductase-catalyzed reactive species generation: A process in critical need of reevaluation. Redox Biol. 2013, 1, 353–358. [Google Scholar] [CrossRef]
- Jarasch, E.D.; Grund, C.; Bruder, G.; Heid, H.W.; Keenan, T.W.; Franke, W.W. Localization of xanthine oxidase in mammary-gland epithelium and capillary endothelium. Cell 1981, 25, 67–82. [Google Scholar] [CrossRef]
- Pritsos, C.A. Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chem. Biol. Interact. 2000, 129, 195–208. [Google Scholar] [CrossRef]
- Moriwaki, Y.; Yamamoto, T.; Suda, M.; Nasako, Y.; Takahashi, S.; Agbedana, O.E.; Hada, T.; Higashino, K. Purification and immunohistochemical tissue localization of human xanthine oxidase. Biochim. Biophys. Acta 1993, 1164, 327–330. [Google Scholar] [CrossRef]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef]
- Vickers, S.; Schiller, H.J.; Hildreth, J.E.; Bulkley, G.B. Immunoaffinity localization of the enzyme xanthine oxidase on the outside surface of the endothelial cell plasma membrane. Surgery 1998, 124, 551–560. [Google Scholar] [CrossRef]
- Sekine, M.; Fujiwara, M.; Okamoto, K.; Ichida, K.; Nagata, K.; Hille, R.; Nishino, T. Significance and amplification methods of the purine salvage pathway in human brain cells. J. Biol. Chem. 2024, 300, 107524. [Google Scholar] [CrossRef]
- Suzuki, G.; Okamoto, K.; Kusano, T.; Matsuda, Y.; Fuse, A.; Yokota, H. Evaluation of neuronal protective effects of xanthine oxidoreductase inhibitors on severe whole-brain ischemia in mouse model and analysis of xanthine oxidoreductase activity in the mouse brain. Neurol. Med.-Chir. 2015, 55, 77–85. [Google Scholar] [CrossRef]
- McManaman, J.L.; Palmer, C.A.; Wright, R.M.; Neville, M.C. Functional regulation of xanthine oxidoreductase expression and localization in the mouse mammary gland: Evidence of a role in lipid secretion. J. Physiol. 2002, 545, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Vorbach, C.; Capecchi, M.R.; Penninger, J.M. Evolution of the mammary gland from the innate immune system? Bioessays 2006, 28, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R. Physiological roles of xanthine oxidoreductase. Drug Metab. Rev. 2004, 36, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Ichida, K.; Amaya, Y.; Okamoto, K.; Nishino, T. Mutations associated with functional disorder of xanthine oxidoreductase and hereditary xanthinuria in humans. Int. J. Mol. Sci. 2012, 13, 15475–15495. [Google Scholar] [CrossRef]
- Dent, C.E.; Philpot, G.R. Xanthinuria, an inborn error (or deviation) of metabolism. Lancet 1954, 266, 182–185. [Google Scholar] [CrossRef]
- Simmonds, H.A.; Reiter, S.; Nishino, T. Hereditary Xanthinuria, 7th ed.; McGraw-Hill: New York, NY, USA, 1995; pp. 1781–1797. [Google Scholar]
- Sekine, M.; Okamoto, K.; Ichida, K. Association of mutations identified in xanthinuria with the function and inhibition mechanism of xanthine oxidoreductase. Biomedicines 2021, 9, 1723. [Google Scholar] [CrossRef]
- Ayvazian, J.H.; Skupp, S. The study of purine utilization and excretion in a xanthinuric man. J. Clin. Investig. 1965, 44, 1248–1260. [Google Scholar] [CrossRef]
- Bradford, M.J.; Krakoff, I.H.; Leeper, R.; Balis, M.E. Study of purine metabolism in a xanthinuric female. J. Clin. Investig. 1968, 47, 1325–1332. [Google Scholar] [CrossRef]
- Mateos, F.A.; Puig, J.G.; Jiménez, M.L.; Fox, I.H. Hereditary xanthinuria. Evidence for enhanced hypoxanthine salvage. J. Clin. Investig. 1987, 79, 847–852. [Google Scholar] [CrossRef]
- Kojima, T.; Nishina, T.; Kitamura, M.; Hosoya, T.; Nishioka, K. Biochemical studies on the purine metabolism of four cases with hereditary xanthinuria. Clin. Chim. Acta 1984, 137, 189–198. [Google Scholar] [CrossRef]
- Ayvazian, J.H.; Skupp, S. Study of the utilization and excretion of dietary purines in a xanthinuric man. J. Clin. Investig. 1966, 45, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Rundles, R.W.; Metz, E.N.; Silberman, H.R. Allopurinol in the treatment of gout. Ann. Intern. Med. 1966, 64, 229–258. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.F.; Gutman, A.B. Effect of allopurinol (4-hydroxypyrazolo-(3,4-d)pyrimidine) on serum and urinary uric acid in primary and secondary gout. Am. J. Med. 1964, 37, 885–898. [Google Scholar] [CrossRef]
- Wyngaarden, J.B.; Rundles, R.W.; Metz, E.N. Allopurinol in the treatment of gout. Ann. Intern. Med. 1965, 62, 842–847. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kondo, M.; Honda, S.; Iwahana, H.; Moritani, M.; Ii, S.; Yoshimoto, K.; Itakura, M. Amidophosphoribosyltransferase limits the rate of cell growth-linked de novo purine biosynthesis in the presence of constant capacity of salvage purine biosynthesis. J. Biol. Chem. 1997, 272, 17719–17725. [Google Scholar] [CrossRef]
- Henderson, J.F.; Khoo, K.Y. On the mechanism of feedback inhibition of purine biosynthesis de novo in ehrlich ascites tumor cells in vitro. J. Biol. Chem. 1965, 240, 3104–3109. [Google Scholar] [CrossRef]
- Yamaoka, T.; Yano, M.; Kondo, M.; Sasaki, H.; Hino, S.; Katashima, R.; Moritani, M.; Itakura, M. Feedback inhibition of amidophosphoribosyltransferase regulates the rate of cell growth via purine nucleotide, DNA, and protein syntheses. J. Biol. Chem. 2001, 276, 21285–21291. [Google Scholar] [CrossRef]
- Sikora, P.; Pijanowska, M.; Majewski, M.; Bieniaś, B.; Borzecka, H.; Zajczkowska, M. Acute renal failure due to bilateral xanthine urolithiasis in a boy with Lesch-Nyhan syndrome. Pediatr. Nephrol. 2006, 21, 1045–1047. [Google Scholar] [CrossRef]
- Greene, M.L.; Fujimoto, W.Y.; Seegmiller, J.E. Urinary xanthine stones--a rare complications of allopurinol therapy. N. Engl. J. Med. 1969, 280, 426–427. [Google Scholar] [CrossRef]
- Band, P.R.; Silverberg, D.S.; Henderson, J.F.; Ulan, R.A.; Wensel, R.H.; Banerjee, T.K.; Little, A.S. Xanthine nephropathy in a patient with lymphosarcoma treated with allopurinol. N. Engl. J. Med. 1970, 283, 354–357. [Google Scholar] [CrossRef]
- Ablin, A.; Stephens, B.G.; Hirata, T.; Wilson, K.; Williams, H.E. Nephropathy, xanthinuria, and orotic aciduria complicating Burkitt’s lymphoma treated with chemotherapy and allopurinol. Metabolism 1972, 21, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Okamoto, K. Mechanistic insights into xanthine oxidoreductase from development studies of candidate drugs to treat hyperuricemia and gout. J. Biol. Inorg. Chem. 2015, 20, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Sekine, M.; Okamoto, K.; Pai, E.F.; Nagata, K.; Ichida, K.; Hille, R.; Nishino, T. Allopurinol and oxypurinol differ in their strength and mechanisms of inhibition of xanthine oxidoreductase. J. Biol. Chem. 2023, 299, 105189. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T. XDH and XO research and drug discovery-personal history. Molecules 2023, 28, 4440. [Google Scholar] [CrossRef]
- Elion, G.B. Enzymatic and metabolic studies with allopurinol. Ann. Rheum. Dis. 1966, 25, 608–614. [Google Scholar] [CrossRef]
- Elion, G.B. Uric Acid. In Hand Book of Experimental Pharmacology; Kelley, W.N., Weiner, I.M., Eds.; Springer: Berlin, Germany, 1978; Volume 51, pp. 485–514. [Google Scholar]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Pai, E.F.; Nishino, T. Mechanism of inhibition of xanthine oxidoreductase by allopurinol: Crystal structure of reduced bovine milk xanthine oxidoreductase bound with oxipurinol. Nucleosides Nucleotides Nucleic Acids 2008, 27, 888–893. [Google Scholar] [CrossRef]
- Massey, V.; Komai, H.; Palmer, G.; Elion, G.B. On the mechanism of inactivation of xanthine oxidase by allopurinol and other pyrazolo[3,4-d]pyrimidines. J. Biol. Chem. 1970, 245, 2837–2844. [Google Scholar] [CrossRef]
- Krenitsky, T.A.; Papaioannou, R.; Elion, G.B. Human hypoxanthine phosphoribosyltransferase. I. Purification, properties, and specificity. J. Biol. Chem. 1969, 244, 1263–1270. [Google Scholar] [CrossRef]
- Nishida, Y.; Kamatani, N.; Tanimoto, K.; Akaoka, I. Inhibition of purine nucleoside phosphorylase activity and of T-cell function with allopurinol-riboside. Agents Actions 1979, 9, 549–552. [Google Scholar] [CrossRef]
- Reiter, S.; Simmonds, H.A.; Webster, D.R.; Watson, A.R. On the metabolism of allopurinol. Formation of allopurinol-1-riboside in purine nucleoside phosphorylase deficiency. Biochem. Pharmacol. 1983, 32, 2167–2174. [Google Scholar] [CrossRef]
- Krenitsky, T.A.; Elion, G.B.; Henderson, A.M.; Hitchings, G.H. Inhibition of human purine nucleoside phosphorylase. Studies with intact erythrocytes and the purified enzyme. J. Biol. Chem. 1968, 243, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Stamp, L.K.; Barclay, M.L.; O’Donnell, J.L.; Zhang, M.; Drake, J.; Frampton, C.; Chapman, P.T. Relationship between serum urate and plasma oxypurinol in the management of gout: Determination of minimum plasma oxypurinol concentration to achieve a target serum urate level. Clin. Pharmacol. Ther. 2011, 90, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Kondo, S.; Pai, E.F.; Nishino, T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J. Biol. Chem. 2003, 278, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kato, M.; Kusano, T.; Nishino, T. New strategy that delays progression of amyotrophic lateral sclerosis in G1H-G93A transgenic mice: Oral administration of xanthine oxidoreductase inhibitors that are not substrates for the purine salvage pathway. J. Neuropathol. Exp. Neurol. 2016, 75, 1124–1144. [Google Scholar] [CrossRef]
- Fujii, K.; Kubo, A.; Miyashita, K.; Sato, M.; Hagiwara, A.; Inoue, H.; Ryuzaki, M.; Tamaki, M.; Hishiki, T.; Hayakawa, N.; et al. Xanthine oxidase inhibitor ameliorates postischemic renal injury in mice by promoting resynthesis of adenine nucleotides. JCI Insight 2019, 4, e124816. [Google Scholar] [CrossRef]
- White, W.B.; Saag, K.G.; Becker, M.A.; Borer, J.S.; Gorelick, P.B.; Whelton, A.; Hunt, B.; Castillo, M.; Gunawardhana, L. Cardiovascular safety of febuxostat or allopurinol in patients with gout. N. Engl. J. Med. 2018, 378, 1200–1210. [Google Scholar] [CrossRef]
- Mackenzie, I.S.; Ford, I.; Nuki, G.; Hallas, J.; Hawkey, C.J.; Webster, J.; Ralston, S.H.; Walters, M.; Robertson, M.; De Caterina, R.; et al. Long-term cardiovascular safety of febuxostat compared with allopurinol in patients with gout (FAST): A multicentre, prospective, randomised, open-label, non-inferiority trial. Lancet 2020, 396, 1745–1757. [Google Scholar] [CrossRef]
- Ghang, B.Z.; Lee, J.S.; Choi, J.; Kim, J.; Yoo, B. Increased risk of cardiovascular events and death in the initial phase after discontinuation of febuxostat or allopurinol: Another story of the CARES trial. RMD Open 2022, 8, e001944. [Google Scholar] [CrossRef]
- Kelley, E.E. A new paradigm for XOR-catalyzed reactive species generation in the endothelium. Pharmacol. Rep. 2015, 67, 669–674. [Google Scholar] [CrossRef]
- Ryu, H.M.; Kim, Y.J.; Oh, E.J.; Oh, S.H.; Choi, J.Y.; Cho, J.H.; Kim, C.D.; Park, S.H.; Kim, Y.L. Hypoxanthine induces cholesterol accumulation and incites atherosclerosis in apolipoprotein E-deficient mice and cells. J. Cell. Mol. Med. 2016, 20, 2160–2172. [Google Scholar] [CrossRef]
- Okamoto, K.; Matsumoto, K.; Hille, R.; Eger, B.T.; Pai, E.F.; Nishino, T. The crystal structure of xanthine oxidoreductase during catalysis: Implications for reaction mechanism and enzyme inhibition. Proc. Natl. Acad. Sci. USA 2004, 101, 7931–7936. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Okamoto, K.; Ashizawa, N.; Nishino, T. FYX-051: A novel and potent hybrid-type inhibitor of xanthine oxidoreductase. J. Pharmacol. Exp. Ther. 2011, 336, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Miyata, K.; Omura, K.; Iwanaga, T.; Nagata, O. Metabolic profile of FYX-051 (4-(5-pyridin-4-yl-1h-[1,2,4]triazol-3-yl)pyridine-2-carbonitrile) in the rat, dog, monkey, and human: Identification of N-glucuronides and N-glucosides. Drug Metab. Dispos. 2006, 34, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Gout, Hyperuricemia and Crystal Associated Disease Network. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).