
Gout Basic Research: 2023 in Review


Abstract
1. Introduction
2. Fundamental Studies on the Pathophysiology of Gouty Inflammation
2.1. New Insights into Proinflammatory Mechanisms in Gout
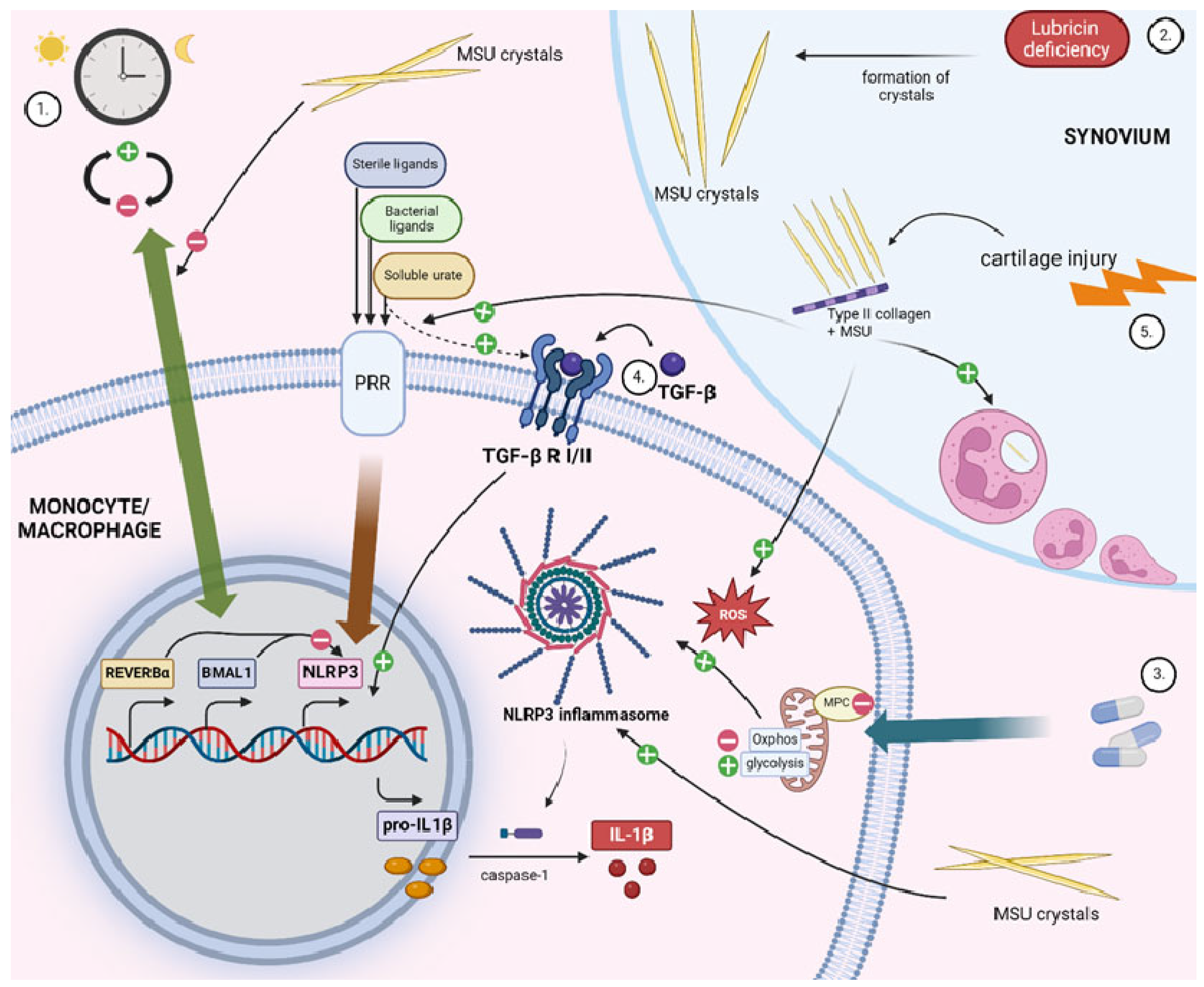
2.1.1. Circadian Clock
2.1.2. Src Family Kinases
2.1.3. Immunometabolic Reprogramming through Mitochondrial Pyruvate Carrier (MPC)
2.1.4. Transforming Growth Factor β (TGF-β)
2.1.5. Lubricin Deficiency
2.1.6. Type II Collagen (CII)
2.2. New Insights into Anti-Inflammatory Mechanisms in Gout
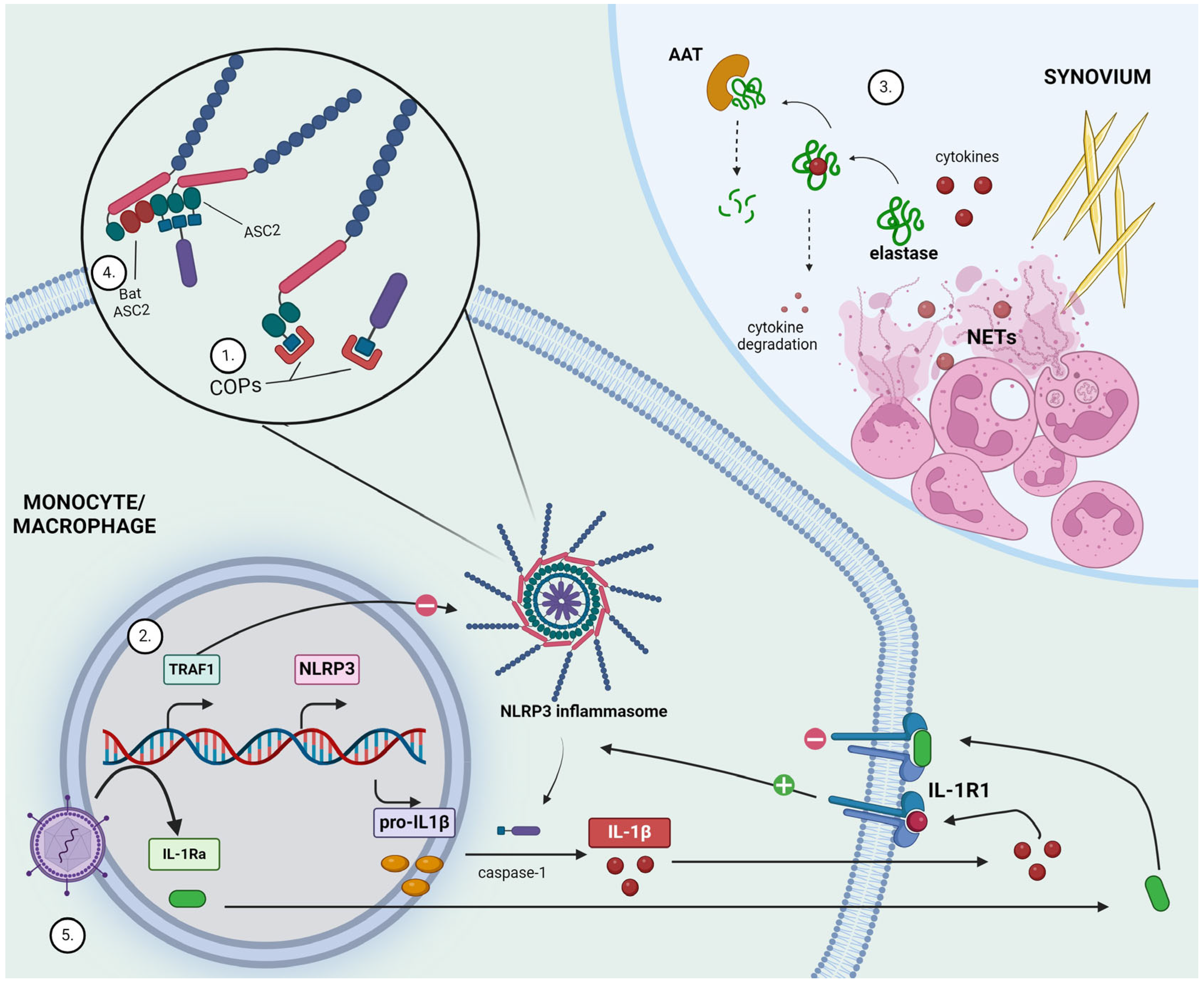
2.2.1. CARD-Only Proteins (COPs)
2.2.2. TNFR-Associated Factor 1 (TRAF1)
2.2.3. Neutrophil Extracellular Trap (NET)-Derived Elastase
2.2.4. ABCG2
2.3. Potential New Therapeutic Development in Gout Inflammation
2.3.1. Bat ASC2
2.3.2. Constitutive Interleukin-1 Receptor Antagonist (IL-1Ra)
3. Omics Studies in Gout
3.1. Single-Cell Transcriptomics and Immune Cell Phenotyping
3.1.1. Single-Cell Transcriptomics in Peripheral Blood and Synovial Fluid in Acute Gout
3.1.2. Single-Cell Transcriptomics in Intercritical and Advanced Gout
3.1.3. Single-Cell Mass Cytometry in Acute Gout
3.2. Proteome
3.3. Metabolome
3.4. Microbiome
3.4.1. Gut Anaerobic Bacteria Are Implicated in Urate Metabolism and Risk of Gout
3.4.2. Atherosclerosis Burden Depends on Gut Microbiome and Is Associated with Urate Levels
4. Closing Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dalbeth, N.; Choi, H.K.; Joosten, L.A.; Khanna, P.P.; Matsuo, H.; Perez-Ruiz, F.; Stamp, L.K. Gout (primer). Nat. Rev. Dis. Primers 2019, 5, 69. [Google Scholar] [CrossRef]
- Choi, H.K.; Mount, D.B.; Reginato, A.M. Pathogenesis of Gout. 2005. Available online: www.annals.org (accessed on 20 March 2024).
- Szekanecz, Z.; Szamosi, S.; Kovács, G.E.; Kocsis, E.; Benkő, S. The NLRP3 inflammasome—Interleukin 1 pathway as a therapeutic target in gout. Arch. Biochem. Biophys. 2019, 670, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Klück, V.; Liu, R.; Joosten, L.A.B. The role of interleukin-1 family members in hyperuricemia and gout. Jt. Bone Spine 2020, 88, 105092. [Google Scholar] [CrossRef]
- Joosten, L.A.B.; Crişan, T.O.; Bjornstad, P.; Johnson, R.J. Asymptomatic hyperuricaemia: A silent activator of the innate immune system. Nat. Rev. Rheumatol. 2020, 16, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Popov, D.; Jain, L.; Alhilali, M.; Dalbeth, N.; Poulsen, R.C. Monosodium urate crystals alter the circadian clock in macrophages leading to loss of NLRP3 inflammasome repression: Implications for timing of the gout flare. FASEB J. 2023, 37, e22940. [Google Scholar] [CrossRef]
- Futosi, K.; Németh, T.; Horváth, Á.I.; Abram, C.L.; Tusnády, S.; Lowell, C.A.; Helyes, Z.; Mócsai, A. Myeloid Src-family kinases are critical for neutrophil-mediated autoinflammation in gout and motheaten models. J. Exp. Med. 2023, 220, e20221010. [Google Scholar] [CrossRef]
- Corcoran, S.E.; O’Neill, L.A.J. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Cobo, I.; Murillo-Saich, J.; Alishala, M.; Guma, M. Epigenetic and Metabolic Regulation of Macrophages during Gout. Gout Urate Cryst. Depos. Dis. 2023, 1, 137–151. [Google Scholar] [CrossRef]
- Chen, L.; Chen, Y.; Chien, W.; Tsai, K.; Chung, C.; Wang, J.; Chen, C.; Liao, N.; Shih, C.; Lin, Y.; et al. Inactivation of mitochondrial pyruvate carrier promotes NLRP3 inflammasome activation and gout development via metabolic reprogramming. Immunology 2023, 169, 271–291. [Google Scholar] [CrossRef]
- Ye, Z.; Zhang, J.; Xu, Z.; Li, Z.; Huang, G.; Tong, B.; Xia, P.; Shen, Y.; Hu, H.; Yu, P.; et al. Pioglitazone ameliorates ischemia/reperfusion-induced acute kidney injury via oxidative stress attenuation and NLRP3 inflammasome. Hum. Cell 2024, 37, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, B.; Wang, L.; Yang, M.; Xia, Z.; Wei, W.; Zhang, F.; Yuan, X. Pioglitazone ameliorates glomerular NLRP3 inflammasome activation in apolipoprotein E knockout mice with diabetes mellitus. PLoS ONE 2017, 12, e0181248. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, C.; Zhang, D.; Zhu, Y. Pioglitazone Protects Against Hypoxia-Induced Cardiomyocyte Apoptosis Through Inhibiting NLRP3/Caspase-1 Pathway in vivo and in vitro. Int. Heart J. 2022, 63, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Klück, V.; Cabău, G.; Mies, L.; Bukkems, F.; Van Emst, L.; Bakker, R.; Van Caam, A.; Pop, I.V.; Popp, R.A.; Rednic, S.; et al. TGF-β is elevated in hyperuricemic individuals and mediates urate-induced hyperinflammatory phenotype in human mononuclear cells. Arthritis Res. Ther. 2023, 25, 30. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, K.; Merriman, T.R.; Rossitto, L.; Liu-Bryan, R.; Karsh, J.; Phipps-Green, A.; Jay, G.D.; Elsayed, S.; Qadri, M.; Miner, M.; et al. Amplification of Inflammation by Lubricin Deficiency Implicated in Incident, Erosive Gout Independent of Hyperuricemia. Arthritis Rheumatol. 2023, 75, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Waller, K.A.; Zhang, L.X.; Elsaid, K.A.; Fleming, B.C.; Warman, M.L.; Jay, G.D. Role of lubricin and boundary lubrication in the prevention of chondrocyte apoptosis. Proc. Natl. Acad. Sci. USA 2013, 110, 5852–5857. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, B.; Chen, Y.; Zeng, F.; Wang, W.; Chen, Z.; Cao, L.; Shi, J.; Chen, J.; Zhu, X.; et al. Type II collagen facilitates gouty arthritis by regulating MSU crystallisation and inflammatory cell recruitment. Ann. Rheum. Dis. 2023, 82, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Indramohan, M.; Jäger, E.; Carriere, J.; Chu, L.H.; De Almeida, L.; Greaves, D.R.; Stehlik, C.; Dorfleutner, A. CARD-only proteins regulate in vivo inflammasome responses and ameliorate gout. Cell Rep. 2023, 42, 112265. [Google Scholar] [CrossRef] [PubMed]
- Mirzaesmaeili, A.; Zangiabadi, S.; Raspanti, J.; Akram, A.; Inman, R.D.; Abdul-Sater, A.A. Cutting Edge: Negative Regulation of Inflammasome Activation by TRAF1 Can Limit Gout. J. Immunol. 2023, 210, 531–535. [Google Scholar] [CrossRef]
- Richette, P.; Doherty, M.; Pascual, E.; Barskova, V.; Becce, F.; Castañeda-Sanabria, J.; Coyfish, M.; Guillo, S.; Jansen, T.L.; Janssens, H.; et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann. Rheum. Dis. 2017, 76, 29–42. [Google Scholar] [CrossRef]
- FitzGerald, J.D.; Dalbeth, N.; Mikuls, T.; Brignardello-Petersen, R.; Guyatt, G.; Abeles, A.M.; Gelber, A.C.; Harrold, L.R.; Khanna, D.; King, C.; et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis Care Res. 2020, 72, 744–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shan, L.; Wang, H.; Schauer, C.; Schoen, J.; Zhu, L.; Lu, C.; Wang, Z.; Xue, Y.; Wu, H.; et al. Neutrophil Extracellular Trap–Borne Elastase Prevents Inflammatory Relapse in Intercritical Gout. Arthritis Rheumatol. 2023, 75, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Leask, M.P.; Merriman, T.R. The genetic basis of urate control and gout: Insights into molecular pathogenesis from follow-up study of genome-wide association study loci. Best Pract. Res. Clin. Rheumatol. 2021, 35, 101721. [Google Scholar] [CrossRef]
- Wrigley, R.; Phipps-Green, A.J.; Topless, R.K.; Major, T.J.; Cadzow, M.; Riches, P.; Tausche, A.K.; Janssen, M.; Joosten LA, B.; Jansen, T.L.; et al. Pleiotropic effect of the ABCG2 gene in gout: Involvement in serum urate levels and progression from hyperuricemia to gout. Arthritis Res. Ther. 2020, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Wrigley, R.; Phipps-Green, A.J.; Topless, R.K.; Major, T.J.; Cadzow, M.; Riches, P.; Tausche, A.-K.; Janssen, M.; Joosten, L.A.B.; Jansen, T.L.; et al. Intestinal uric acid excretion contributes to serum uric acid decrease during acute gout attack. Rheumatology 2023, 62, 3984–3992. [Google Scholar]
- Zhang, J.; Sun, W.; Gao, F.; Lu, J.; Li, K.; Xu, Y.; Li, Y.; Li, C.; Chen, Y. Changes of serum uric acid level during acute gout flare and related factors. Front. Endocrinol. 2023, 14, 1077059. [Google Scholar] [CrossRef] [PubMed]
- Notsu, T.; Kurata, Y.; Ninomiya, H.; Taufiq, F.; Komatsu, K.; Miake, J.; Sawano, T.; Tsuneto, M.; Shirayoshi, Y.; Hisatome, I. Inhibition of the uric acid efflux transporter ABCG2 enhances stimulating effect of soluble uric acid on IL-1β production in murine macrophage-like J774.1 cells. Hypertens. Res. 2023, 46, 2368–2377. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Chen, V.C.-W.; Rozario, P.; Ng, W.L.; Kong, P.S.; Sia, W.R.; Kang, A.E.Z.; Su, Q.; Nguyen, L.H.; Zhu, F.; et al. Bat ASC2 suppresses inflammasomes and ameliorates inflammatory diseases. Cell 2023, 186, 2144–2159.e22. [Google Scholar] [CrossRef]
- Colantuoni, M.; Jofra Hernandez, R.; Pettinato, E.; Basso-Ricci, L.; Magnani, L.; Andolfi, G.; Rigamonti, C.; Finardi, A.; Romeo, V.; Soldi, M.; et al. Constitutive IL-1RA Production by Modified Immune Cells Protects against IL-1-Mediated Inflammatory Disorders. 2023. Available online: https://www.science.org (accessed on 4 April 2024).
- Chang, J.-G.; Tu, S.-J.; Huang, C.-M.; Chen, Y.-C.; Chiang, H.-S.; Lee, Y.-T.; Yen, J.-C.; Lin, C.-L.; Chung, C.-C.; Liu, T.-C.; et al. Single-cell RNA sequencing of immune cells in patients with acute gout. Sci. Rep. 2022, 12, 22130. [Google Scholar] [CrossRef]
- Gu, H.; Yu, H.; Qin, L.; Yu, H.; Song, Y.; Chen, G.; Zhao, D.; Wang, S.; Xue, W.; Wang, L.; et al. MSU crystal deposition contributes to inflammation and immune responses in gout remission. Cell Rep. 2023, 42, 113139. [Google Scholar] [CrossRef]
- Wang, M.; Chen, W.; Zhang, X.; Mei, L.; Wu, X.; Chen, X.; Yang, Z.; Gao, K.; Huang, H.; Huang, R. Single-Cell Analysis in Blood Reveals Distinct Immune Cell Profiles in Gouty Arthritis. J. Immunol. 2023, 210, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Cabău, G.; Gaal, O.; Badii, M.; Nica, V.; Mirea, A.-M.; Hotea, I.; Pamfil, C.; Popp, R.A.; Netea, M.G.; Rednic, S.; et al. Hyperuricemia remodels the serum proteome toward a higher inflammatory state. iScience 2023, 26, 107909. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, R.; Qi, H.; Pang, L.; Cui, L.; Liu, Z.; Lu, J.; Wang, R.; Hu, S.; Liang, N.; et al. Metabolomics and Machine Learning Identify Metabolic Differences and Potential Biomarkers for Frequent Versus Infrequent Gout Flares. Arthritis Rheumatol. 2023, 75, 2252–2264. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jarman, J.B.; Low, Y.S.; Augustijn, H.E.; Huang, S.; Chen, H.; DeFeo, M.E.; Sekiba, K.; Hou, B.-H.; Meng, X.; et al. A widely distributed gene cluster compensates for uricase loss in hominids. Cell 2023, 186, 3400–3413.e20. [Google Scholar] [CrossRef]
- Kasahara, K.; Kerby, R.L.; Zhang, Q.; Pradhan, M.; Mehrabian, M.; Lusis, A.J.; Bergström, G.; Bäckhed, F.; Rey, F.E. Gut bacterial metabolism contributes to host global purine homeostasis. Cell Host Microbe 2023, 31, 1038–1053.e10. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Data | Study | Model | Findings |
|---|---|---|---|
| Single-cell studies | Chang et al. [31] | Gout vs. control (blood) | ↑ Levels of CD4 cells and monocytes in gout |
| Gu et al. [32] | Synovial fluid of gout patients Advanced gout vs. intercritical phase of gout vs. healthy controls | ↑ Levels of monocytes, myelocytic dendritic cells, Th1/Th17, CD8 cells, and MAIT HLA-DQA1high monocytes: advanced gout > intercritical phase > healthy controls | |
| Wang et al. [33] | Acute phase of gouty arthritis vs. intercritical phase vs. healthy controls | ↑ Levels of CCR4-expressing cells in gout (C03 monocytes, C08 NK, C13, and CD41) Distinct T-cell profile in gout ↑ Expression of CCL17 and CCL20 in acute gout | |
| Proteomics | Cabău et al. [34] | Gout vs. non-gout (asymptomatic hyperuricemia and healthy controls) | No significant differentially expressed proteins |
| Asymptomatic hyperuricemia vs. healthy controls Gout + urate-lowering therapy | 58 differentially expressed proteins (most upregulated: 4E-BP1, IL6, IL-18R1, CD40, CXCL9, PD-L1, HGF, CX3CL1, IL-10, IL-17C, and TNF) ↓ 8 proteins in subjects who reached target serum urate: LAP-TGF-beta, S100A12, HGF, CXCL1, CXCL11, MCP-3, IL-17A, and stem cell factor | ||
| Metabolomics | Wang et al. [35] | Frequent vs. infrequent gout flares | Organic acids, lipids, steroids, hormones, and transmitters downregulated in frequent gout flare group |
| Carbohydrates upregulated in frequent gout flare group | |||
| Cross-talk between purine metabolism and caffeine metabolism: most altered sub-network | |||
| Metabolite-based prediction model (4-trimethyl-ammioniobutanoic acid, 5′-methylthioadenosine, arachidic acid, taurine, uridine, and xanthine) can potentially differentiate between groups |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muntiu, M.; Joosten, L.A.B.; Crişan, T.O. Gout Basic Research: 2023 in Review. Gout Urate Cryst. Depos. Dis. 2024, 2, 220-235. https://doi.org/10.3390/gucdd2030017
Muntiu M, Joosten LAB, Crişan TO. Gout Basic Research: 2023 in Review. Gout, Urate, and Crystal Deposition Disease. 2024; 2(3):220-235. https://doi.org/10.3390/gucdd2030017
Chicago/Turabian StyleMuntiu, Maria, Leo A. B. Joosten, and Tania O. Crişan. 2024. "Gout Basic Research: 2023 in Review" Gout, Urate, and Crystal Deposition Disease 2, no. 3: 220-235. https://doi.org/10.3390/gucdd2030017
APA StyleMuntiu, M., Joosten, L. A. B., & Crişan, T. O. (2024). Gout Basic Research: 2023 in Review. Gout, Urate, and Crystal Deposition Disease, 2(3), 220-235. https://doi.org/10.3390/gucdd2030017