Persistent B-Cell Stimulation or B-Cell Repertoire Anomalies? The Dilemma of the Origin of Chronic Lymphocytic Leukemia (CLL)



{kind=link}
1. Introduction
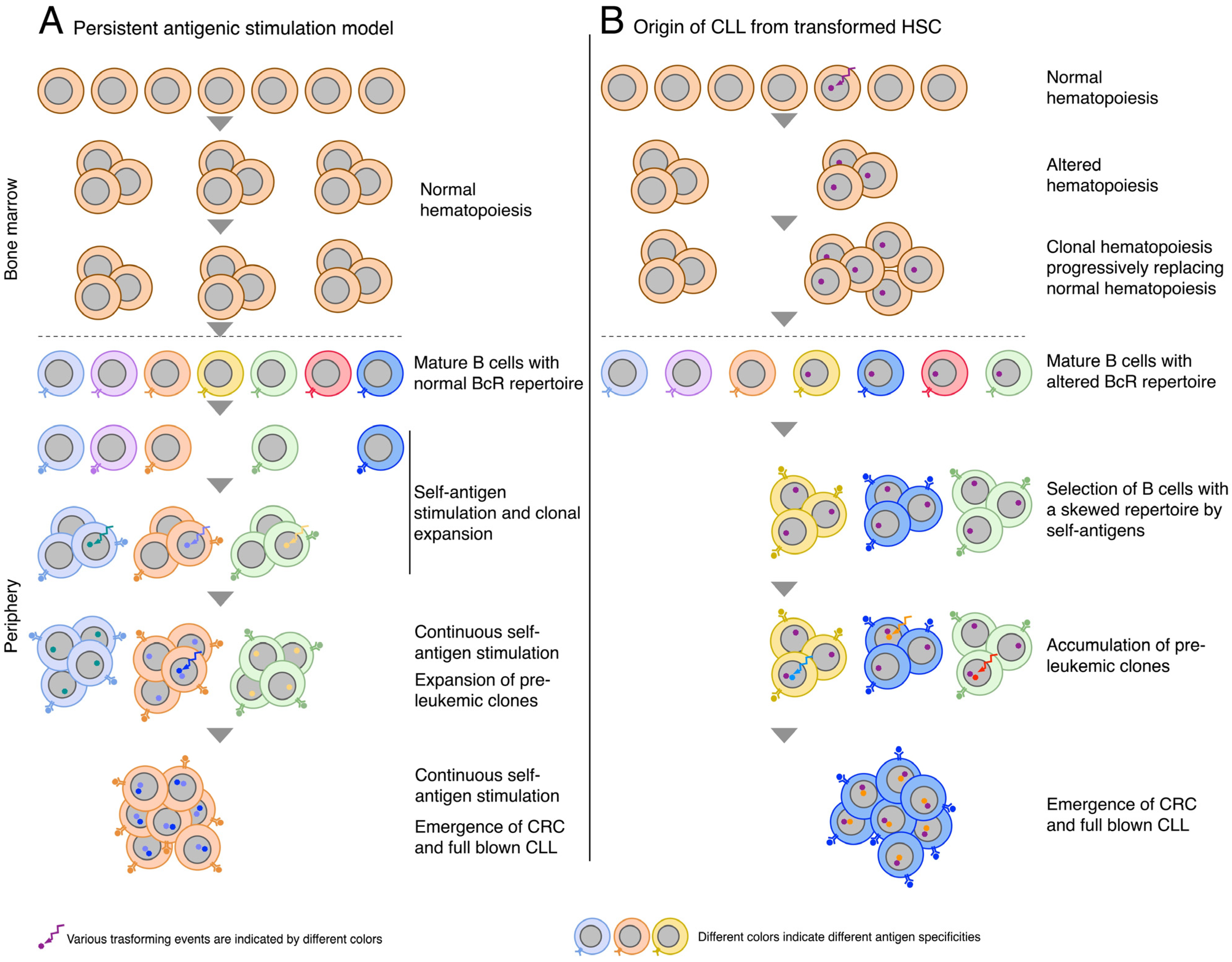
2. Origin of CLL from Mature B-Cells: The Role of Persistent Antigenic Stimulation
3. Concerns Regarding the Persistent Antigenic Stimulation Model
4. Origin of CLL Involving HSCs
5. Supporting Evidence and Concerns Regarding the Origin from HSCs
6. Concluding Remarks
7. Origin of the Article
Funding
Conflicts of Interest
References
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, F.K.; Forconi, F.; Kipps, T.J. Exploring the pathways to chronic lymphocytic leukemia. Blood 2021, 138, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.R.; Jain, P. Chronic lymphocytic leukemia (CLL)—Then and now. Am. J. Hematol. 2016, 91, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Chiorazzi, N.; Ferrarini, M. Cellular origin(s) of chronic lymphocytic leukemia: Cautionary notes and additional considerations and possibilities. Blood 2011, 117, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Dameshek, W. Chronic lymphocytic leukemia—An accumulative disease of immunolgically incompetent lymphocytes. Blood 1967, 29, 566–584. [Google Scholar] [CrossRef]
- Messmer, B.T.; Messmer, D.; Allen, S.L.; Kolitz, J.E.; Kudalkar, P.; Cesar, D.; Murphy, E.J.; Koduru, P.; Ferrarini, M.; Zupo, S.; et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J. Clin. Investig. 2005, 115, 755–764. [Google Scholar] [CrossRef]
- Burger, J.A.; Chiorazzi, N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013, 34, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Giallourakis, C.; Mostoslavsky, R.; Alt, F.W. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu. Rev. Immunol. 2006, 24, 541–570. [Google Scholar] [CrossRef]
- Victora, G.D.; Nussenzweig, M.C. Germinal Centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef]
- Takemori, T.; Kaji, T.; Takahashi, Y.; Shimoda, M.; Rajewsky, K. Generation of memory B cells inside and outside germinal centers. Eur. J. Immunol. 2014, 44, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Bagnara, D.; Mazzarello, A.N.; Ghiotto, F.; Colombo, M.; Cutrona, G.; Fais, F.; Ferrarini, M. Old and New Facts and Speculations on the Role of the B Cell Receptor in the Origin of Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2022, 23, 14249. [Google Scholar] [CrossRef] [PubMed]
- Fais, F.; Ghiotto, F.; Hashimoto, S.; Sellars, B.; Valetto, A.; Allen, S.L.; Schulman, P.; Vinciguerra, V.P.; Rai, K.; Rassenti, L.Z.; et al. Chronic lymphocytic leukemia B cells express restricted sets of mutated and unmutated antigen receptors. J. Clin. Investig. 1998, 102, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Ghiotto, F.; Fais, F.; Valetto, A.; Albesiano, E.; Hashimoto, S.; Dono, M.; Ikematsu, H.; Allen, S.L.; Kolitz, J.; Rai, K.R.; et al. Remarkably similar antigen receptors among a subset of patients with chronic lymphocytic leukemia. J. Clin. Investig. 2004, 113, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Widhopf, G.F.; Rassenti, L.Z.; Toy, T.L.; Gribben, J.G.; Wierda, W.G.; Kipps, T.J. Chronic lymphocytic leukemia B cells of more than 1% of patients express virtually identical immunoglobulins. Blood 2004, 104, 2499–2504. [Google Scholar] [CrossRef] [PubMed]
- Tobin, G.; Thunberg, U.; Karlsson, K.; Murray, F.; Laurell, A.; Willander, K.; Enblad, G.; Merup, M.; Vilpo, J.; Juliusson, G.; et al. Subsets with restricted immunoglobulin gene rearrangement features indicate a role for antigen selection in the development of chronic lymphocytic leukemia. Blood 2004, 104, 2879–2885. [Google Scholar] [CrossRef]
- Stamatopoulos, K.; Belessi, C.; Moreno, C.; Boudjograh, M.; Guida, G.; Smilevska, T.; Belhoul, L.; Stella, S.; Stavroyianni, N.; Crespo, M.; et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood 2007, 109, 259–270. [Google Scholar] [CrossRef]
- Agathangelidis, A.; Chatzidimitriou, A.; Gemenetzi, K.; Giudicelli, V.; Karypidou, M.; Plevova, K.; Davis, Z.; Yan, X.J.; Jeromin, S.; Schneider, C.; et al. Higher-order connections between stereotyped subsets: Implications for improved patient classification in CLL. Blood 2020, 137, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Bagnara, D.; Colombo, M.; Reverberi, D.; Matis, S.; Massara, R.; Cardente, N.; Ubezio, G.; Agostini, V.; Agnelli, L.; Neri, A.; et al. Characterizing Features of Human Circulating B Cells Carrying CLL-Like Stereotyped Immunoglobulin Rearrangements. Front. Oncol. 2022, 12, 894419. [Google Scholar] [CrossRef] [PubMed]
- Hoogeboom, R.; van Kessel, K.P.; Hochstenbach, F.; Wormhoudt, T.A.; Reinten, R.J.; Wagner, K.; Kater, A.P.; Guikema, J.E.; Bende, R.J.; van Noesel, C.J. A mutated B cell chronic lymphocytic leukemia subset that recognizes and responds to fungi. J. Exp. Med. 2013, 210, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Dühren-Von Minden, M.; Übelhart, R.; Schneider, D.; Wossning, T.; Bach, M.P.; Buchner, M.; Hofmann, D.; Surova, E.; Follo, M.; Köhler, F.; et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature 2012, 489, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.; Dalla-Favera, R. Chronic lymphocytic leukaemia: From genetics to treatment. Nat. Rev. Clin. Oncol. 2019, 16, 684–701. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Hansen, S.; Rodriguez, L.; Gelb, A.B.; Warnke, R.A.; Jellum, E.; Orentreich, N.; Vogelman, J.H.; Friedman, G.D. Helicobacter pylori Infection and Gastric Lymphoma. N. Engl. J. Med. 1994, 330, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- Melenotte, C.; Mezouar, S.; Mège, J.-L.; Gorvel, J.-P.; Kroemer, G.; Raoult, D. Bacterial infection and non-Hodgkin’s lymphoma. Crit. Rev. Microbiol. 2020, 46, 270–287. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.R.; Hayakawa, K. Perspectives on fetal derived CD5+ B1 B cells. Eur. J. Immunol. 2015, 45, 2978–2984. [Google Scholar] [CrossRef] [PubMed]
- Köhler, F.; Hug, E.; Eschbach, C.; Meixlsperger, S.; Hobeika, E.; Kofer, J.; Wardemann, H.; Jumaa, H. Autoreactive B Cell Receptors Mimic Autonomous Pre-B Cell Receptor Signaling and Induce Proliferation of Early B Cells. Immunity 2008, 29, 912–921. [Google Scholar] [CrossRef]
- Hayakawa, K.; Formica, A.M.; Brill-Dashoff, J.; Shinton, S.A.; Ichikawa, D.; Zhou, Y.; Morse, H.C., III; Hardy, R.R. Early generated B1 B cells with restricted BCRs become chronic lymphocytic leukemia with continued c-Myc and low Bmf expression. J. Exp. Med. 2016, 213, 3007–3024. [Google Scholar] [CrossRef]
- Förster, I.; Rajewsky, K. Expansion and functional activity of Ly-1+ B cells upon transfer of peritoneal cells into allotype-congenic, newborn mice. Eur. J. Immunol. 1987, 17, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Formica, A.M.; Colombo, M.J.; Shinton, S.A.; Brill-Dashoff, J.; Iii, H.C.M.; Li, Y.-S.; Hardy, R.R. Loss of a chromosomal region with synteny to human 13q14 occurs in mouse chronic lymphocytic leukemia that originates from early-generated B-1 B cells. Leukemia 2016, 30, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.A.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Bojnik, E.; Ruppert, A.S.; Stefanovski, M.R.; Goettl, V.M.; Smucker, K.A.; Smith, L.L.; Dubovsky, J.A.; Towns, W.H.; MacMurray, J.; et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood 2014, 123, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Schmid, V.K.; Khadour, A.; Ahmed, N.; Brandl, C.; Nitschke, L.; Rajewsky, K.; Jumaa, H.; Hobeika, E. B cell antigen receptor expression and phosphatidylinositol 3-kinase signaling regulate genesis and maintenance of mouse chronic lymphocytic leukemia. Haematologica 2022, 107, 1796–1814. [Google Scholar] [CrossRef] [PubMed]
- Berndt, S.I.; Skibola, C.F.; Joseph, V.; Camp, N.J.; Nieters, A.; Wang, Z.; Cozen, W.; Monnereau, A.; Wang, S.S.; Kelly, R.S.; et al. Genome-wide association study identifies multiple risk loci for chronic lymphocytic leukemia. Nat. Genet. 2013, 45, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Speedy, H.E.; Di Bernardo, M.C.; Sava, G.P.; Dyer, M.J.S.; Holroyd, A.; Wang, Y.; Sunter, N.J.; Mansouri, L.; Juliusson, G.; Smedby, K.E.; et al. A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat. Genet. 2014, 46, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Roessner, P.M.; Seiffert, M. T-cells in chronic lymphocytic leukemia: Guardians or drivers of disease? Leukemia 2020, 34, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Bagnara, D.; Reverberi, D.; Matis, S.; Cardillo, M.; Massara, R.; Mastracci, L.; Ravetti, J.L.; Agnelli, L.; Neri, A.; et al. Tracing CLL-biased stereotyped immunoglobulin gene rearrangements in normal B cell subsets using a high-throughput immunogenetic approach. Mol. Med. 2020, 26, 25. [Google Scholar] [CrossRef]
- Vergani, S.; Bagnara, D.; Agathangelidis, A.; Ng, A.K.Y.; Ferrer, G.; Mazzarello, A.N.; Palacios, F.; Yancopoulos, S.; Yan, X.-J.; Barrientos, J.C.; et al. CLL stereotyped B-cell receptor immunoglobulin sequences are recurrent in the B-cell repertoire of healthy individuals: Apparent lack of central and early peripheral tolerance censoring. Front. Oncol. 2023, 13, 1112879. [Google Scholar] [CrossRef]
- Plevova, K.; Francova, H.S.; Burckova, K.; Brychtova, Y.; Doubek, M.; Pavlova, S.; Malcikova, J.; Mayer, J.; Tichy, B.; Pospisilova, S. Multiple productive immunoglobulin heavy chain gene rearrangements in chronic lymphocytic leukemia are mostly derived from independent clones. Haematologica 2014, 99, 329–338. [Google Scholar] [CrossRef]
- Brazdilova, K.; Plevova, K.; Skuhrova Francova, H.; Kockova, H.; Borsky, M.; Bikos, V.; Malcikova, J.; Oltova, A.; Kotaskova, J.; Tichy, B.; et al. Multiple productive IGH rearrangements denote oligoclonality even in immunophenotypically monoclonal CLL. Leukemia 2017, 10, 1551. [Google Scholar] [CrossRef] [PubMed]
- Stamatopoulos, B.; Timbs, A.; Bruce, D.; Smith, T.; Clifford, R.; Robbe, P.; Burns, A.; Vavoulis, D.V.; Lopez, L.; Antoniou, P.; et al. Targeted deep sequencing reveals clinically relevant subclonal IgHV rearrangements in chronic lymphocytic leukemia. Leukemia 2017, 31, 837–845. [Google Scholar] [CrossRef]
- Kolijn, P.M.M.; Hosnijeh, F.S.; Späth, F.; Hengeveld, P.J.; Agathangelidis, A.; Saleh, M.; Casabonne, D.; Benavente, Y.; Jerkeman, M.; Agudo, A.; et al. High-risk subtypes of Chronic Lymphocytic Leukemia are detectable as early as 16 years prior to diagnosis. Blood 2022, 139, 1557–1563. [Google Scholar] [CrossRef]
- Klinger, M.; Zheng, J.; Elenitoba-Johnson, K.S.J.; Perkins, S.L.; Faham, M.; Bahler, D.W. Next-generation IgVH sequencing CLL-like monoclonal B-cell lymphocytosis reveals frequent oligoclonality and ongoing hypermutation. Leukemia 2016, 30, 1055–1061. [Google Scholar] [CrossRef]
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, 586. [Google Scholar] [CrossRef]
- Staudt, L.M. A Closer Look at Follicular Lymphoma. N. Engl. J. Med. 2007, 356, 741–742. [Google Scholar] [CrossRef] [PubMed]
- Pekarsky, Y.; Balatti, V.; Croce, C.M. BCL2 and miR-15/16: From gene discovery to treatment. Cell Death Differ. 2018, 25, 21–26. [Google Scholar] [CrossRef]
- López, C.; Burkhardt, B.; Chan, J.K.; Leoncini, L.; Mbulaiteye, S.M.; Ogwang, M.D.; Orem, J.; Rochford, R.; Roschewski, M.; Siebert, R. Burkitt lymphoma. Nat. Rev. Dis. Prim. 2022, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Ishikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yoshimoto, G.; Mori, Y.; Iino, T.; Yamauchi, T.; Eto, T.; et al. Self-Renewing Hematopoietic Stem Cell Is the Primary Target in Pathogenesis of Human Chronic Lymphocytic Leukemia. Cancer Cell 2011, 20, 246–259. [Google Scholar] [CrossRef]
- Bagnara, D.; Kaufman, M.S.; Calissano, C.; Marsilio, S.; Patten, P.E.M.; Simone, R.; Chum, P.; Yan, X.-J.; Allen, S.L.; Kolitz, J.E.; et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood 2011, 117, 5463–5472. [Google Scholar] [CrossRef] [PubMed]
- Damm, F.; Mylonas, E.; Cosson, A.; Yoshida, K.; Della Valle, V.; Mouly, E.; Diop, M.; Scourzic, L.; Shiraishi, Y.; Chiba, K.; et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014, 4, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Marsilio, S.; Khiabanian, H.; Fabbri, G.; Vergani, S.; Scuoppo, C.; Montserrat, E.; Shpall, E.J.; Hadigol, M.; Marin, P.; Rai, K.R.; et al. Somatic CLL mutations occur at multiple distinct hematopoietic maturation stages: Documentation and cautionary note regarding cell fraction purity. Leukemia 2018, 32, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrarini, M.; Bagnara, D.; Ghiotto, F.; Fais, F. Persistent B-Cell Stimulation or B-Cell Repertoire Anomalies? The Dilemma of the Origin of Chronic Lymphocytic Leukemia (CLL). Lymphatics 2024, 2, 147-156. https://doi.org/10.3390/lymphatics2030012
Ferrarini M, Bagnara D, Ghiotto F, Fais F. Persistent B-Cell Stimulation or B-Cell Repertoire Anomalies? The Dilemma of the Origin of Chronic Lymphocytic Leukemia (CLL). Lymphatics. 2024; 2(3):147-156. https://doi.org/10.3390/lymphatics2030012
Chicago/Turabian StyleFerrarini, Manlio, Davide Bagnara, Fabio Ghiotto, and Franco Fais. 2024. "Persistent B-Cell Stimulation or B-Cell Repertoire Anomalies? The Dilemma of the Origin of Chronic Lymphocytic Leukemia (CLL)" Lymphatics 2, no. 3: 147-156. https://doi.org/10.3390/lymphatics2030012
APA StyleFerrarini, M., Bagnara, D., Ghiotto, F., & Fais, F. (2024). Persistent B-Cell Stimulation or B-Cell Repertoire Anomalies? The Dilemma of the Origin of Chronic Lymphocytic Leukemia (CLL). Lymphatics, 2(3), 147-156. https://doi.org/10.3390/lymphatics2030012