Abstract
Although traditionally contraindicated, the coadministration of tamoxifen and estradiol may hold clinical relevance in specific contexts, particularly in breast cancer survivors with premature menopause and a high risk of osteoporosis, thereby justifying the need to re-evaluate this therapeutic combination. This study presents an innovative physiologically based pharmacokinetic (PBPK) modeling approach to evaluate the coadministration of tamoxifen and estradiol in women with breast cancer and a high risk of osteoporosis. Using GastroPlus® software, PBPK models were developed and validated for both drugs, based on physicochemical and kinetic data obtained from the literature and, where necessary, supplemented by estimates generated in ADMET Predictor®. The simulations considered different hormonal profiles (pre and postmenopausal) and therapeutic regimens, evaluating potential interactions mediated by the CYP3A4 enzyme. Analysis of the pharmacokinetic parameters (F, Cmax, Tmax and AUC) revealed strong agreement between the simulated and experimental values, with prediction errors of less than twofold. The drug interaction studies, carried out in dynamic and stationary modes, indicated that estradiol does not significantly alter the pharmacokinetics of tamoxifen, even at increasing doses or in enlarged virtual populations. These results represent the first in silico evidence that, under certain conditions, the concomitant use of estradiol does not compromise the pharmacokinetic efficacy of tamoxifen. Although the study is computational, it provides a solid scientific basis for re-evaluating this therapeutic combination and proposes a pioneering model for personalized strategies in complex oncological contexts. All simulations assumed average enzyme abundance/activity without CYP polymorphism parameterization; findings are restricted to parent-tamoxifen pharmacokinetics and do not infer metabolite (e.g., endoxifen) exposure or phenotype effects.
1. Introduction
Globally, breast cancer (BC) is one of the most frequently diagnosed tumors, being responsible for the majority of cancer-related deaths in women, with a ratio of 30.7% of the cases registered [1]. According to GLOBOCAN 2020, an estimated 2.3 million new cases were reported globally, hinting at the high public health impact of the disease [2].
The classification of BC is divided based on histological and molecular criteria. Histological types divide the tumor into carcinoma in situ (ductal in situ or lobular in situ), when the tumors remain confined within the anatomical site of origin, and invasive carcinoma (invasive ductal or invasive lobular), when it spreads outside the epithelium and reaches the contiguous tissues, which is more dangerous for the formation of metastasis [3,4]. Molecular profiling, on the other hand, plays a crucial role in the personalized treatment of breast cancer. This classification distinguishes between subtypes such as Luminal A, Luminal B, HER2-enriched and Basal-like (triple-negative), based on the expression of hormone receptors (estrogen and progesterone) and the HER2 protein [3]. Such segregation has promoted more precise and efficient treatments, addressing the specific biological characteristics of each tumor. By this categorization, tumors are separated into four broad molecular subtypes: HR+/HER2−, HR+/HER2+, HR-/HER2+ and triple-negative [5] and all these subtypes have unique therapeutic implications. For BC, there are approximately 75% of tumors that are positive for hormone receptors (HR+), which are susceptible to hormone therapy, also known as endocrine therapy [6,7]. Among them, HR+/HER2- tumors, the most common, are helped by this class of therapy, either tamoxifen (TAM) or aromatase inhibitors, depending on the menopausal status of the patient [8]. TAM is a selective estrogen receptor modulator (SERM), whose primary therapeutic action consists of competitively inhibiting the binding of estrogen to its receptor. This interaction prevents the hormone’s proliferative effect on neoplastic breast cells with hormone receptor expression [9]. However, although established to be effective in tumor growth suppression, tamoxifen can weaken the bone with variable effects on the tissue, but also involves actions with a harmful impact on the bone [10].
In the case of women with early menopause, caused by surgery or ovarian suppression, using gonadotropin-releasing hormone agonists results in an acute decrease in the level of estrogen, simulating an early menopause [11]. This abrupt hormonal change disrupts bone metabolism, increases bone resorption and reduces bone mineral density (BMD), thus increasing the risk of osteoporosis and fractures [12,13,14]. Furthermore, it is estimated that up to 80% of breast cancer survivors suffer a prolonged reduction in BMD, which reinforces the importance of integrating bone preservation strategies into the follow-up of an oncology patient [15].
Similarly, natural menopause also interrupts ovarian function, accelerating the loss of bone mass and the development of osteoporosis. To combat this problem, estradiol can be administered as part of hormone replacement therapy, helping to preserve bone health and reduce the risk of fractures [16,17].
Although the concomitant use of estradiol in breast cancer treatment is not currently recommended due to its potential oncological risks, certain clinical scenarios may justify a re-evaluation of this strategy. Hormone replacement therapy (HRT), particularly with estradiol, is a cornerstone in the management of premature ovarian insufficiency (POI) and treatment-induced early menopause, conditions frequently observed in breast cancer survivors [18,19]. HRT is mainly administered to suppress the symptoms of menopause, but it is also widely used to prevent post-menopausal osteoporosis. It reduces postmenopausal bone loss and reduces the incidence of osteoporotic fractures by around 50% [20]. For women under 60 years of age or with a high risk of osteoporosis, HRT may be clinically indicated after oncological remission [18]. Studies show that HRT based on estrogens such as estradiol has a significant protective effect against osteoporosis and fragility fractures [17,19]. Therefore, it becomes essential to understand how exogenous estradiol might interfere with tamoxifen metabolism in such patients.
Although tamoxifen and estradiol exert opposite pharmacodynamic effects at the level of estrogen receptors, the former acting as a SERM and an antagonist in breast tissue [10], and the latter as a full agonist [21], promoting cell proliferation, both share common metabolic pathways, mainly involving cytochrome P450 enzymes, such as CYP3A4 [22]. Any alteration in the activity of these enzymes can affect the absorption, metabolism, distribution and elimination of both compounds. This opposition of mechanisms raises the possibility of competitive pharmacodynamic antagonism, especially in coadministration contexts, as both drugs compete for the same molecular target—the estrogen receptor. When administered simultaneously, there is a risk that estradiol may compete with tamoxifen for estrogen receptors in breast tissue, potentially reducing the therapeutic efficacy of tamoxifen [21]. On the other hand, in tissues such as bone, where both drugs promote the maintenance of bone mineral density in postmenopausal patients, this competition may not compromise the clinical benefits [23]. This analysis is essential to understand the potential implications of coadministration on therapeutic efficacy and safety, particularly in oncological patients at increased risk of osteoporosis.
Therefore, from a theoretical perspective, the possibility of pharmacokinetic interactions between tamoxifen and exogenous estradiol should be carefully considered, as these interactions may influence the plasma exposure, therapeutic efficacy, or safety of one or both drugs.
In this context, this is the first study dedicated to the drug-drug interaction (DDI) between tamoxifen and estradiol. As is well known, physiologically based pharmacokinetic (PBPK) models have proved to be promising tools for the mechanistic assessment of these drug interactions, allowing the impact of one drug on the pharmacokinetic profile of another to be accurately simulated. In this study, we used the DDI simulation module of the GastroPlus® software, using previously parameterized and validated PBPK models.
The development of effective pharmaceutical formulations requires an integrated evaluation of pharmacokinetics and pharmacodynamics (PK/PD), enabling a deeper understanding of drug mechanisms of action and the optimization of compound design. The early incorporation of PK/PD studies into drug development contributes to a faster and more efficient selection of promising candidates while reducing costs and the need for in vivo studies. In this context, computational approaches have gained increasing prominence, driven by advances in hardware, software, and algorithmic scenarios [24,25].
Although the interaction between tamoxifen and estradiol is primarily described as pharmacodynamic, due to competition at the estrogen receptor, potential pharmacokinetic drug–drug interactions (PK-DDIs) cannot be excluded, since both compounds share metabolic pathways involving cytochrome P450 enzymes. PBPK modeling is particularly well-suited to explore these PK-DDIs, as it allows for mechanistic evaluation of drug metabolism, enzyme competition, and interindividual variability. This approach has become especially relevant in oncology, where treatment regimens are complex and require careful consideration of pharmacokinetics across diverse patient populations. According to FDA data, oncology was the therapeutic area with the highest number of PBPK model submissions between 2018 and 2019. These models are used not only to assess DDIs but also to predict drug behavior in specific populations such as pediatric or organ-impaired patients, and to account for internal and external variability, including genetic polymorphisms and food effects. In breast cancer, PBPK models can support clinical decision-making by anticipating potential risks and optimizing treatment regimens in susceptible patients [26].
Among the various available platforms, GastroPlus® has emerged as a robust and widely used tool for PBPK modeling. The software integrates advanced modules, including the ACAT model for oral absorption, the ADMET Predictor® for estimating physicochemical and biopharmaceutical properties, and a dedicated module for simulating drug–drug interactions. Its application has been validated in multiple studies and recognized by regulatory authorities such as the FDA. GastroPlus® is particularly useful for translating in vitro data into in vivo contexts and for predicting complex clinical scenarios [24,25].
The analysis of pharmacokinetic parameters under different conditions, both dynamic and steady-state, associated with population simulations with several virtual subjects, aims to deepen knowledge about the coadministration of tamoxifen and estradiol. Given the scarcity of clinical trials in the literature that directly evaluate this pharmacokinetic interaction, in silico simulations are particularly relevant for anticipating potential risks and guiding therapeutic decisions. The results obtained in silico should, in the future, be complemented by clinical data, reinforcing the proposal for personalized medicine based on computational evidence, to ensure greater safety and efficacy in the treatment of vulnerable clinical populations.
This work provides a mechanistic prediction of the clinical PK interaction between tamoxifen and oral estradiol across clinically used doses and interindividual variability. The simulations consistently indicate PK-DDI neutrality on parent tamoxifen exposure, while highlighting that pharmacodynamic antagonism and metabolite-/genotype-dependent effects require separate evaluation.
Also, this work provides a mechanistic prediction of the PK interaction on parent tamoxifen with oral estradiol across clinically used doses and interindividual variability, while acknowledging that metabolite-/genotype-dependent effects (e.g., endoxifen, CYP2D6) lie outside the present PBPK setup and require separate evaluation
2. Materials and Methods
2.1. PBPK Model Flowchart
This study employed a stepwise approach, as illustrated in the flowchart in Figure 1. This schematic representation summarizes the strategy adopted to develop PBPK models to characterize the pharmacokinetic behavior of the drugs tamoxifen and estradiol in different female population profiles.
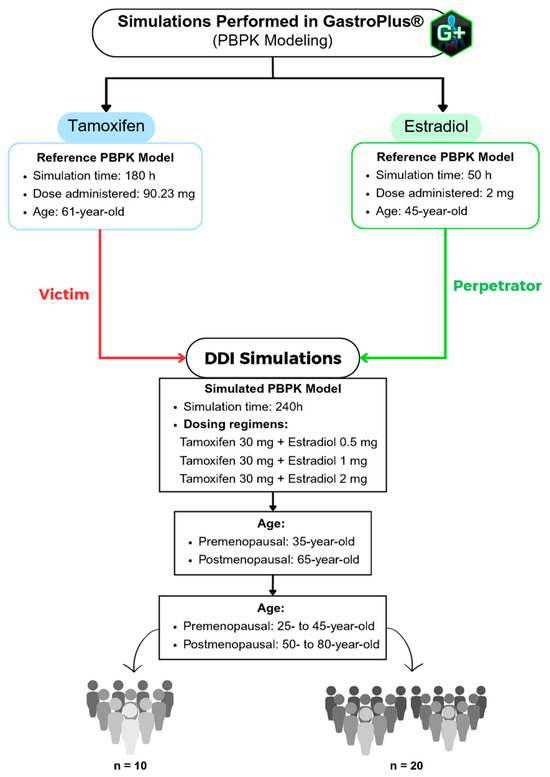
Figure 1.
Flowchart of the PBPK simulations performed using the GastroPlus® software, including the reference models for tamoxifen and estradiol, as well as the drug–drug interaction (DDI) simulation scenarios across different age groups (premenopausal and postmenopausal).
In the first phase, published PBPK models, known as Reference PBPK Models, were used to validate the parameters simulated in this study. For tamoxifen, an oral dose of 90.23 mg was considered, with a simulation time of 180 h, in 61-year-old American women. In the case of estradiol, the simulation time was 50 h, with an administered dose of 2 mg, in 45-year-old American women.
Subsequently, a PBPK model, called the Simulated PBPK Model, was developed to assess the pharmacokinetic interaction between the two drugs. This was done using the “DDI” module of the GastroPlus® software (version 9.9; Simulation Plus Inc., Lancaster, CA, USA), based on a standard population of American women with a body weight of 57 kg. The combined dosage regimens studied included tamoxifen 30 mg + estradiol 0.5 mg, 1 mg and 2 mg, with a simulation time of 240 h, with estradiol modeled as the “perpetrator” drug and tamoxifen as the “victim” compound. We selected a 30 mg dose of tamoxifen because the daily dose recommended by INFARMED ranges from 20 mg to 40 mg [27]. Thus, our study focuses on the average dose commonly administered in clinical practice, enabling a representative simulation of the typical systemic exposure to the drug. Regarding the therapeutic regimen of estradiol, the oral doses recommended by the FDA were used, namely 0.5 mg, 1 mg and 2 mg per day. These doses reflect usual clinical practice and enable a comprehensive assessment of the pharmacokinetic behavior of estradiol in different age groups [28]. For the physiological parameterization, the “Population Estimates for Age-Related” (PEAR) model was applied, which allows the age of the simulated women to be accurately adjusted, representing different age profiles throughout the study.
Next, simulations were carried out for two age groups, premenopausal women (35-year-old) and postmenopausal women (65-year-old). Finally, in order to study inter-individual variability, population simulations were carried out with 10 (n = 10) and 20 (n = 20) virtual individuals, extending the age range to 25- to 45-year-old (pre-menopausal) and 50- to 80-year-old (post-menopausal). This set of simulations made it possible to systematically assess the potential impact of coadministration of estradiol on the pharmacokinetics of tamoxifen in different physiological conditions, providing fundamental data for a personalized approach to the treatment of breast cancer and osteoporosis in women at different stages of life.
PK metrics and windows. For single-dose simulations, Tmax is defined as the earliest time at which C(t) = Cmax 0–24 h post-dose (“first tie wins”). For steady-state once-daily dosing (τ =24 h), we summarize Cmax,ss, Ctrough,ss and AUCτ (and Tmax,ss only within the [dose, dose + τ] window). Global maxima over extended horizons (e.g., 0–240 h) are not used to derive Tmax to avoid spurious late selections caused by repeated dose peaks, numerical plateaus, or terminal noise. Population summaries report C_max,ss, C_trough,ss, and AUC_τ as the primary steady-state PK metrics.
All DDI simulations assumed average enzyme abundance/activity (population defaults) and did not parameterize CYP polymorphisms (including CYP2D6 activity-score strata). Accordingly, outcomes are restricted to parent-tamoxifen PK in the presence of estradiol.
2.2. Development and Initial Parameterization of the Tamoxifen PBPK Model
The development of the tamoxifen PBPK model began with the definition of its chemical structure, designed in MedChem Designer® software (Version 5.5; Simulation Plus Inc., Lancaster, CA, USA) and then imported into ADMET Predictor® (Version 9.5; Simulation Plus Inc., Lancaster, CA, USA), in MOL format. Using the latter software, it was possible to determine various relevant physicochemical and pharmacokinetic properties, making it possible to fill gaps in the literature, especially when experimental data were unavailable [29].
ADMET Predictor® makes it possible to quickly predict more than 175 properties, including solubility, lipophilic-hydrophilic partition coefficient (logP), base pKa, molecular weight, human jejunum effective permeability (Peff), diffusion coefficient (Diff. Coeff.), metabolism and transport mediated by cytochrome P450 (CYP) enzymes, as well as permeability in the blood-brain barrier (BBB). Based on these data and the information provided by Chen et al. in the study “Predicting the Effects of Different Triazole Antifungal Agents on the Pharmacokinetics of Tamoxifen” [30], the PBPK model for tamoxifen was initially built using the GastroPlus® software. The human physiology model adopted considered a healthy American woman, 61-years-old, and with a body weight of 57 kg.
The main physicochemical parameters used were: Peff, pKa, chemical structure, molecular weight, solubility, dose administered, fraction not bound in plasma (fup), blood-plasma concentration ratio (Rbp) and logP, as described in Table 1. The parameters missing from the literature were estimated and optimized using the ADMET Predictor® module, except the variables entered in the “Gut Physiology” tab of GastroPlus®, where the individual physiological characteristics were specified, assuming “fasted” conditions.

Table 1.
Physicochemical parameters of the tamoxifen PBPK model. Adapted from [30].
The enzymatic component of the model was based on the maximum reaction speed (Vmax) and Michaelis-Menten constant (Km) described by Chen et al. [30] with values for the enzymes CYP2D6, CYP3A4 and CYP3A5. The reported values were 96 μM for the Km and 5.5 nmol/min/nmol for the Vmax; for CYP3A4, 50 μM and 10 nmol/min/nmol, respectively; and for CYP3A5, 24 μM and 6 nmol/min/nmol, respectively. Given that the GastroPlus® software does not support these units, it was necessary to convert them to mg/L in the case of the Km, and for mg/s/mg enzyme in the case of Vmax, as shown in Table 2.
Table 2.
Kinetic parameters for each tamoxifen enzyme, adjusted to units compatible with GastroPlus®.
Finally, the tissue-plasma partition coefficients (Kp) of tamoxifen were also extracted from the Chen et al. [30] study and were inserted into the “Pharmacokinetics” tab of GastroPlus®.
2.3. Development and Initial Parameterization of the Estradiol PBPK Model
Like tamoxifen, the PBPK model for estradiol was built based on data obtained from the literature and predictions generated in ADMET Predictor® (Version 9.5; Simulation Plus Inc., Lancaster, CA, USA), after modeling the chemical structure in MedChem Designer® (Version 5.5; Simulation Plus Inc., Lancaster, CA, USA). This approach made it possible to gather the essential physicochemical parameters for parameterizing the model, listed in Table 3.
Table 3.
Physicochemical parameters of estradiol PBPK model.
Enzyme kinetic parameters (Vmax and Km) were obtained from the study “Role of Human Cytochrome P450 1A1, 1A2, 1B1, and 3A4 in the 2-, 4-, and 16α-Hydroxylation of 17β-Estradiol” by Badawi et al. [33]. To study DDI drug interactions, both drugs must share at least one enzyme. That said, one of the enzymes in common is CYP3A4. Thus, the values reported for the CYP3A4 enzyme are 102 ± 30 μmol/L for the Km and 0.7 ± 0.4 nmol/min/nmol CYP3A4 enzyme for Vmax. As the GastroPlus® software does not support these units, it was necessary to convert them: Km values were expressed in mg/L and Vmax values in mg/s/mg enzyme, as presented in Table 4.
Table 4.
Kinetic parameters for each estradiol enzyme, adjusted to units compatible with GastroPlus®.
2.4. PBPK Modeling Final Development
With the physicochemical and kinetic data duly defined, we proceeded with the final implementation of the PBPK models for the two compounds, tamoxifen and estradiol, in the GastroPlus® software. To do this, the molecular structures and parameters generated in ADMET Predictor® were imported and integrated into the simulation environment.
The GastroPlus® «DDI» module was used to simulate possible drug interactions, including competitive inhibition, time-dependent inhibition and autoinduction, with simulations in dynamic mode.
Finally, the graphs generated during these analyses were exported to GraphPad Prism® (Version 8.0.1) for visual processing and graphical representation of the data obtained.
2.5. PBPK Model Validation
The PK parameter values obtained from the developed models were compared with data previously reported in the literature. In addition, a visual inspection of the plasma concentration profiles was conducted to assess the concordance between the PBPK models and similar studies described in the literature. Accordingly, the PBPK models were considered validated.
2.6. DDI Simulation Between Tamoxifen and Estradiol
As previously mentioned, the pharmacokinetic interaction between tamoxifen and estradiol was investigated using the «DDI» module of the GastroPlus® software, through simulations in dynamic and stationary mode. The stationary mode uses constant average (steady-state) plasma concentrations of the drugs as the basis for interaction assessment, without considering temporal variation in compound concentrations. This approach follows a rationale similar to mechanistic static models, often used to flag potential interactions in early stages of drug development. In contrast, the dynamic mode is based on PBPK models and allows for the simulation of time-dependent variation in the concentrations of perpetrator and victim drugs across different physiological compartments (organs, tissues, and systemic circulation). This approach is considered more robust, as it incorporates factors such as absorption, metabolism, distribution, and excretion over time, as well as inter-individual variability. Thus, the dynamic mode provides more realistic and comprehensive predictions, enabling the evaluation of, for example, the impact of active metabolites, different dosing regimens, or specific populations [34].
Initially, representative hormonal profiles were simulated: a premenopausal woman (30-year-old) and a postmenopausal woman (65-year-old), both over 240 h. Three estradiol dosage regimens were tested (0.5 mg, 1 mg and 2 mg/day), administered together with a single dose of tamoxifen (30 mg). In stationary mode, the magnitude of the interaction was assessed based on the AUC ratio (with and without the perpetrator), according to the standard classification (none, weak, moderate or strong).
Subsequently, to study inter-individual variability, additional simulations were carried out with two groups of 10 virtual women (25- to 45-year-old and 50- to 80-year-old) on a continuous regimen of tamoxifen (30 mg/day) and estradiol (0.5 mg/day). These simulations were carried out using the «Population Simulation» module of GastroPlus®, population simulations (n = 10 and n = 20) were conducted to sample inter-individual variability; they do not emulate a clinical trial. This module combines physiological parameters with the expected pharmacokinetic variability between subjects, also considering formulation characteristics and experimental data entered as input into the software.
Finally, the number of simulated subjects was increased to 20 women, while maintaining the same experimental conditions. This decision followed FDA guidelines, which recommend a minimum of 20 participants in phase I clinical trials to better represent the inter-individual variability expected in a clinical context [35].
3. Results
3.1. PBPK Model Tamoxifen
To validate the PBPK model of tamoxifen under study, a simulation was conducted in GastroPlus® with a dose of 90.23 mg, in agreement with the study by Chen et al. [30], as mentioned in Section 2.2.
Table 5 shows the comparison between the observed pharmacokinetic values and the values estimated by the PBPK model. The accuracy of the predicted pharmacokinetic parameters was assessed using the fold error, and predictions were considered reliable whenever they were less than 2. The fold error was calculated as follows: when the observed value was lower than the estimated value, the formula predicted/observed was applied; when the observed value was higher, observed/predicted was used [36]. Based on this criterion, all the parameters evaluated Cmax, Tmax, AUC0-inf and AUC0-t, showed folding errors of 1.34, 1.09, 1.33 and 1.01, respectively.
Table 5.
Observed [30] and estimated (GastroPlus®) pharmacokinetic properties of 90.23 mg tamoxifen administered to a 61-year-old American female after a 180-h simulation.
We can therefore say that the results show good agreement between the estimated and observed data, demonstrating the predictive reliability of the parameterized PBPK model.
In addition, a graphical analysis of the model’s predictive performance was carried out. Figure 2 shows the comparison between the plasma concentration profiles simulated in GastroPlus® and the experimental data observed in the study by Chen et al. [30]. Overall, the data obtained confirm the predictive capacity of the PBPK model, validating its applicability for subsequent simulations.
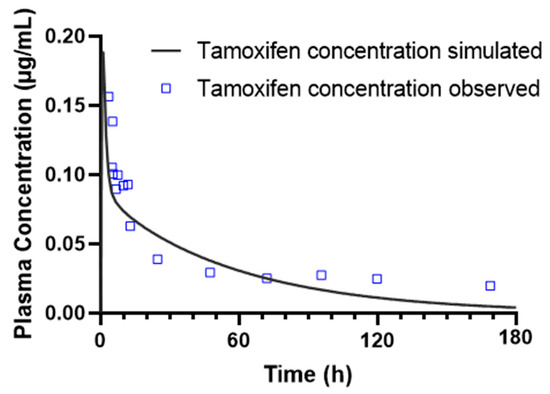
Figure 2.
Pharmacokinetics of 90.23 mg tamoxifen over a 180-h simulation in a 61-year-old American female, including simulated and observed plasma concentration-time profiles of tamoxifen after oral administration.
3.2. PBPK Model Estradiol
To validate the PBPK model developed for estradiol, a simulation was carried out in GastroPlus® with an oral dose of 2 mg, according to the protocol described by Sier et al. in the study “Linking physiologically based pharmacokinetic and genome-scale metabolic networks to understand estradiol biology” [37]. The simulation considered a population profile corresponding to healthy American women aged 45 years and 57 kg, with no variation in weight in relation to tamoxifen.
Table 6 shows the comparison between the experimental pharmacokinetic parameters described in the study by Sier et al. and the values estimated by the PBPK model after a 50-h simulation. The results obtained show an acceptable agreement between the simulated data and the observed values, reflecting the validity of the model to represent the pharmacokinetic behavior of estradiol after oral administration.
Table 6.
Comparison between pharmacokinetic parameters observed [37,38] in the literature and those estimated by the estradiol PBPK model developed in GastroPlus®, following oral administration of 2 mg to a 45-year-old female.
In addition to the numerical comparison of the pharmacokinetic parameters, a graphical analysis was also carried out to assess the predictive accuracy of the model. Figure 3 shows the plasma concentration profile of estradiol over time, simulated in GastroPlus®, superimposed on the experimental data reported by Sier et al. The simulated curve satisfactorily follows the trend of the observed values, especially during the absorption and initial distribution phases. Despite some discrepancies observed in the plasma concentration values, the overall agreement between the simulated data and the experimental values reinforces the model’s ability to adequately describe the pharmacokinetic behavior of estradiol after oral administration. It is important to note that these differences occur at extremely low plasma concentrations, in the order of µg/mL, which can accentuate numerical and visual variations that are modest. Even so, the accuracy of the predicted pharmacokinetic parameters was assessed using fold error. Based on this criterion, all the parameters evaluated, F, Cmax, Tmax, AUC0-inf and AUC0-t, showed fold errors of 1.64, 1.64, 1.09, 1.19 and 1.15, respectively, which confirms the adequate predictive capacity of the PBPK model developed for estradiol.
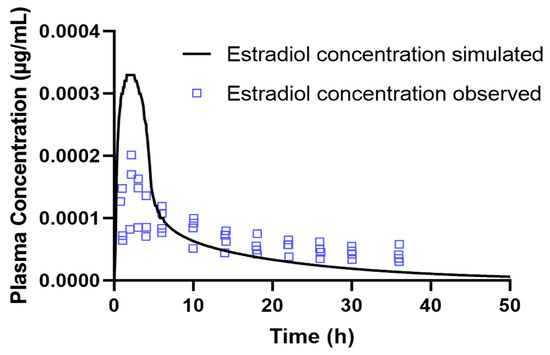
Figure 3.
Comparison between the simulated plasma concentration-time profile of estradiol obtained in GastroPlus® and the experimental data reported by [37], following oral administration of 2 mg to a 45-year-old female.
3.3. DDI Simulation Between Tamoxifen and Estradiol
To assess whether the concomitant administration of estradiol influences the pharmacokinetics of tamoxifen, the therapeutic regimen described in Section 2.6. This approach is justified by the frequent use of estradiol in the treatment of osteoporosis and tamoxifen in the treatment of breast cancer. Therefore, understanding the interaction between these two drugs is essential to ensure therapeutic efficacy and clinical safety.
Drug-drug interaction (DDI) simulations were carried out using PBPK models previously developed and validated for each compound, using representative population profiles.
3.3.1. Effect of Menopausal Status on Tamoxifen Parameters When Administered with Estradiol
To investigate whether menopausal status influenced the coadministration of tamoxifen and estradiol, two virtual female populations were simulated: one premenopausal (35-year-old) and one postmenopausal (65-year-old).
Age is a recognized factor in the variation in hepatic clearance, since drug metabolization depends on multiple physiological parameters that change with age, including hepatic perfusion, hepatocellular uptake and enzymatic activity. In the case of tamoxifen and estradiol, both are metabolized by CYP3A4, with tamoxifen also depending significantly on CYP2D6 for conversion into its active metabolites.
The interaction between the two compounds was initially simulated in steady-state mode, evaluating the AUC ratios of tamoxifen as a function of the estradiol dose. For all doses tested, the total AUC ratio values ranged between 1.027 and 1.033, reflecting a lower variation compared to tamoxifen administration alone (baseline). Based on these results, all interactions were classified by GastroPlus® as “No interaction”.
Figure 4 illustrates this relationship, showing that although there is a linear correlation between increasing the dose of estradiol and the AUC ratio of tamoxifen, the impact is minimal. Linear regression analysis confirmed that the increase in tamoxifen exposure was minimal and did not warrant significant clinical concerns.
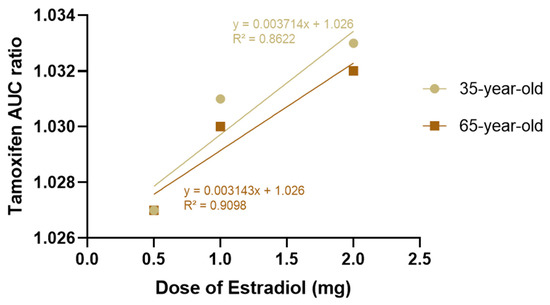
Figure 4.
Effect of increasing oral estradiol dose (0.5 mg, 1 mg, and 2 mg/day) on the tamoxifen (30 mg/day) AUC ratio in premenopausal (35 years old) and postmenopausal (65 years old) women, estimated using steady-state simulation in the GastroPlus® DDI module. AUC values were calculated with and without estradiol coadministration. Observed variations (<3%) fall within expected interindividual variability and do not indicate a clinically relevant pharmacokinetic interaction.
Subsequently, a dynamic simulation was carried out to compare the pharmacokinetic parameters of each combination (tamoxifen 30 mg + estradiol 0.5 mg, tamoxifen 30 mg + estradiol 1 mg, tamoxifen 30 mg + estradiol 2 mg) with the tamoxifen baseline (administered alone), through dynamic simulation, as represented in Table 7.
Table 7.
Effect of increasing estradiol dose on the pharmacokinetics of tamoxifen. Pharmacokinetic parameters were estimated by dynamic simulation over 240 h in virtual female subjects (35- and 65-year-old) receiving a fixed daily dose of tamoxifen (30 mg) and coadministration of estradiol (0.5, 1, or 2 mg).
In all cases, the coadministration of estradiol did not cause significant changes in the pharmacokinetics of tamoxifen, with the values being practically the same as in the baseline condition. This lack of impact was also seen in the simulations for the two age groups, indicating that age does not influence the magnitude of the interaction.
The data presented show that the coadministration of estradiol, even at increasing doses, did not cause clinically significant changes in the pharmacokinetics of tamoxifen, with the values being practically the same as in the baseline condition.
These data support the hypothesis that, despite sharing CYP3A4 as a metabolizing pathway, estradiol does not significantly affect tamoxifen clearance or systemic exposure. This may be due to a lower enzymatic affinity or to the predominance of tamoxifen metabolism by other routes, particularly CYP2D6. In any case, the results suggest that, in a clinical context, the coadministration of estradiol and tamoxifen is safe from a pharmacokinetic point of view, with no changes expected in the efficacy or toxicity of tamoxifen due to the presence of estradiol.
In short, the data obtained indicate that, at the doses and ages simulated, estradiol is not a relevant clinical perpetrator of pharmacokinetic interactions with tamoxifen, which could have positive implications for clinical practice. However, it is important to note that these results come from simulations and should be complemented with clinical data for validation.
At both simulated ages, the values of Cmax and Tmax remained constant, and AUC0-inf varied at most from 17,500 to 17,600 ng∙h/mL, a negligible difference. The same pattern was observed for AUC0-t, with a slight increase compared to baseline. These results reinforce the conclusion that estradiol does not significantly interfere with systemic exposure to tamoxifen, supporting the safety of coadministration of these drugs from a pharmacokinetic point of view.
In addition, the results suggest that age alone does not potentiate the pharmacokinetic interaction between estradiol and tamoxifen, which reinforces the safety of coadministration of these drugs in postmenopausal women with breast cancer and the need for hormonal therapy to prevent or treat osteoporosis. However, it will be important to complement these data with clinical evidence, given the high inter-individual variability in hepatic metabolism associated with aging.
To complement the quantitative data, Figure 5 shows the plasma concentration profiles of tamoxifen over time for both ages. The curves obtained show an almost total overlap between the different doses of estradiol, which visually confirms the absence of a relevant pharmacokinetic impact. These results support the hypothesis that estradiol, at the doses simulated, is not a relevant clinical perpetrator of interactions with tamoxifen, and that coadministration is safe from a pharmacokinetic point of view.
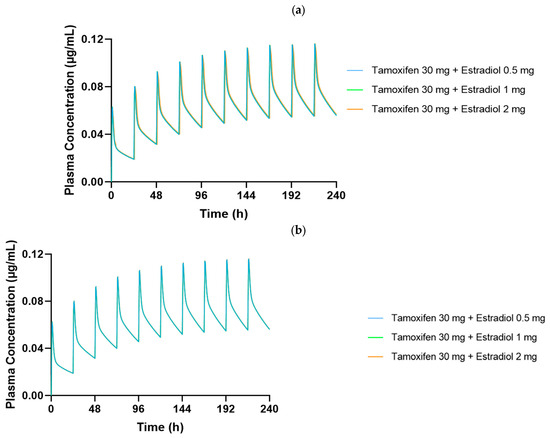
Figure 5.
Plasma concentration–time profiles of tamoxifen (30 mg/day) over 240 h in dynamic simulation for (a) a premenopausal 35-year-old woman and (b) a postmenopausal 65-year-old woman, coadministered with estradiol at doses of 0.5 mg, 1 mg, and 2 mg/day. Curves show near-complete overlap among estradiol dosing regimens, suggesting no significant alteration in tamoxifen pharmacokinetics.
3.3.2. Population-Based Simulation to Assess Interindividual Variability in the Tamoxifen–Estradiol Interaction
To further evaluate the pharmacokinetic interaction between tamoxifen and estradiol, a population simulation was carried out using the «Population Simulation» module of GastroPlus®, with the «DDI» mode activated. This approach makes it possible to incorporate inter-individual variability and observe the behavior of the drugs in a simulated population. Population simulations (n = 10 and n = 20) were conducted in the GastroPlus® Population Simulation module to sample inter-individual variability across the predefined age groups; these simulations do not emulate a clinical trial. DDI was assessed in dynamic and stationary modes with estradiol as perpetrator and tamoxifen as victim, using AUC and C_max ratios (with/without perpetrator) for interaction classification.
The simulation was conducted in groups of 10 and 20 healthy virtual women, divided into two age groups: 25- to 45-year-old (representing the premenopausal population) and 50 to 80-year-old (representing the postmenopausal population), with a fixed body weight of 57 kg. All the participants underwent the same therapeutic regimen as in the previous Section 3.3.1.
Population Simulation with 10 Virtual Subjects
We performed virtual population simulations with n = 10 and n = 20 subjects to characterize inter-individual variability; these simulations are not intended to reproduce clinical trial design. Table 8 shows the main pharmacokinetic parameters for a group of 10 people resulting from the simulation, including the mean values (Mean), the geometric mean (Geom Mean), the coefficient of variation (CV%) and the 90% confidence interval (90% CI). The parameters analyzed were F, Fa, FDp, Cmax, Tmax, AUC0-t and AUC0–inf. For each parameter, the ratio between the values obtained with estradiol coadministration (DDI) and the baseline values was calculated, allowing the impact of estradiol to be assessed. Overall, the results showed robust pharmacokinetic stability between the scenarios with and without estradiol. The Fa remained constant in all the simulated conditions, with values very close to 100% and DDI/baseline ratios fixed at 1.000 in both age groups, reflecting absolute stability in the effective absorption of the drug. In the case of FDp, the values remained between 79 and 81%, with DDI/baseline ratios also at 1.000, which means that a considerable part of the dissolution takes place in the proximal intestine, which is typical of drugs with good intestinal absorption, although with slightly greater variability (CV between 3.4% and 5.5%).
Table 8.
Summary of pharmacokinetic parameters of tamoxifen with and without estradiol (0.5 mg/day) over 10 days based on a population-based simulation (n = 10) in virtual female subjects with 25- to 45-year-olds and 50- to 80-year-olds. Tmax is reported only within the specified window (0–24 h single dose; [dose, dose + τ] at steady state). Extended-horizon maxima are not used.
As for F, the values remained stable, with DDI/baseline ratios close to 1000 in all the conditions tested. There was a slight reduction in older individuals (50 to 80-year-old), with a F of 70%, compared to younger individuals (25- to 45-year-old), who had a F of 73%. This difference could be associated with age-related physiological changes, such as a decrease in hepatic flow, the activity of metabolizing enzymes or the efficiency of absorption mechanisms. However, this is a discrete variation with no apparent clinical relevance.
Cmax values also remained practically unchanged with estradiol coadministration, with DDI/baseline ratios between 1.000 and 1.009 in both age groups. The coefficients of variation were approximately 20% in the 25–45 age group and 27% in the 50–80 age group, with variability ratios (DDI/baseline) of 0.045 and 0.28, respectively. Although there was a slight increase in inter-individual variability in the older group, these values remained within the expected limits, suggesting no relevant effect of estradiol on maximum tamoxifen levels.
With regard to Tmax, a different behavior was observed between the two age groups. In the 25–45 age group, the value of this parameter was 217.4 h, with a ratio of 1.000 and a very low CV of 0.18%, reflecting low variability between the simulated individuals. Using the revised window-restricted definitions, all Tmax values occur within the first dosing interval; no late (>24 h) Tmax values are observed. Consequently, we report Cmax,ss, Ctrough,ss and AUCτ as primary PK metrics at steady state and provide Tmax only when computed within the appropriate window.
In the 50–80 age group, there was a reduction in Tmax to 176.8 h, maintaining the ratio of 1.000, but with a considerable increase in CV to 52.2%. Although this difference may point to faster absorption in older individuals, the absolute Tmax values are disproportionately high, indicating the possible occurrence of a simulation artifact. It is plausible that the prolonged duration of the model (240 h) led to the identification of secondary peaks as Tmax, which compromises the physiological validity of these results. This suspicion is supported by data from individual simulations lasting 24 h, in which a Tmax of 1 h was observed, a value much more compatible with the expected pharmacokinetics.
Finally, the systemic exposure parameters, represented by AUC0–t and AUC0–inf, showed minimal variations with estradiol coadministration, with DDI/baseline ratios between 1.000 and 1.004 in both age groups. These differences correspond to changes of less than 0.5%, compatible with simulated inter-individual variability, and do not indicate any relevant influence of estradiol at a dose of 0.5 mg on the extent of tamoxifen exposure.
Figure 6 generated show the plasma concentration profiles of tamoxifen over time, with and without coadministration of estradiol. In these graphs, the continuous line represents the mean of the simulated concentrations, while the shaded area indicates the 90% confidence interval (90% CI), reflecting the expected dispersion of the data in a population with variable physiological characteristics. The overlap of the curves with and without estradiol, as well as the coincidence of the respective confidence intervals, indicate that the coadministration of 0.5 mg of estradiol did not significantly alter the pharmacokinetic profile of tamoxifen in any of the age groups studied.
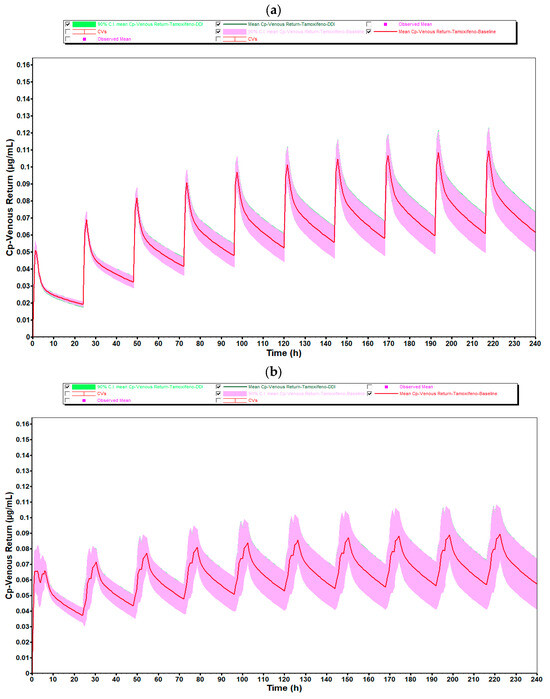
Figure 6.
Mean plasma concentration–time profiles of tamoxifen (30 mg/day) from population simulations (n = 10) for (a) women aged 25–45 years and (b) women aged 50–80 years, with (green line) and without (red line) coadministration of estradiol 0.5 mg/day. Shaded areas represent the 90% confidence interval. Overlapping curves and variability ranges indicate that estradiol does not meaningfully alter tamoxifen’s plasma profile.
In both age groups, 25- to 45-year-olds and 50- to 80-year-olds, the plasma concentration profiles of tamoxifen over 10 days of repeated administration show an overlap between the DDI (green line) and baseline (red line). The shape and amplitude of the peaks and valleys are very similar, reflecting a stable and predictable pharmacokinetic behavior with and without the presence of estradiol. This observation is in complete agreement with the numerical values obtained, which indicated DDI/baseline ratios very close to 1.000 for Cmax, F and Fa.
It should be noted, however, that in the 50- to 80-year-old group, there was a slight discrepancy in the plasma concentration profile after the first administration, with a sharper peak and a shift to subsequent days. This behavior is compatible with the initial pre-accumulation phase of the drug, before the establishment of steady-state, and is common in compounds with a long half-life and high tissue distribution, such as tamoxifen. In addition, physiological changes associated with aging, particularly in liver function, splanchnic blood flow and body composition, may contribute to a more variable initial distribution in this age group. After this stage, the profiles become visibly more consistent and overlapping, showing that the presence of estradiol does not compromise the pharmacokinetic stability of tamoxifen over time.
The greater width of the 90% confidence interval in the 50- to 80-year-olds reflects the greater inter-individual physiological variability associated with aging. Despite this greater dispersion, the mean plasma concentrations remained similar to those of the younger group, and overall systemic exposure was not affected by the coadministration of estradiol, reinforcing the robustness and reliability of the results obtained.
In addition, a graph showing the variation in AUC0-inf as a function of estradiol dose (0.5, 1 and 2 mg) was drawn for both age groups, as shown in Figure 7. This graph showed a slight upward trend in the 50 to 80-year-old group, suggesting that higher doses of estradiol may be associated with a slight reduction in tamoxifen clearance in postmenopausal women. However, this change was marginal and did not indicate a relevant clinical impact in the simulated conditions.
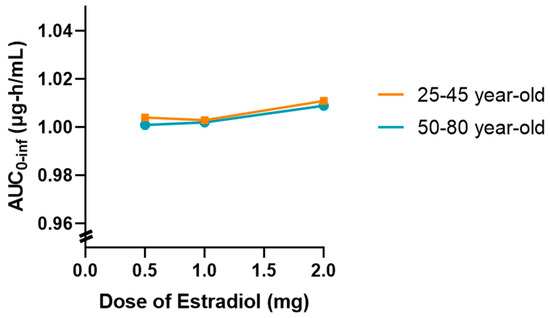
Figure 7.
Effect of increasing oral estradiol dose (0.5 mg, 1 mg, and 2 mg/day) on tamoxifen (30 mg/day) AUC0-inf in premenopausal (25–45 years) and postmenopausal (50–80 years) women, based on population simulation (n = 10). A slight upward trend in AUC is observed in older women, but the magnitude remains within expected pharmacokinetic variability and is not clinically relevant in the studied context.
Overall, the results obtained indicate that the coadministration of estradiol at a dose of 0.5 mg does not significantly affect the pharmacokinetics of tamoxifen in either young or older women. However, the tendency for exposure to increase with increasing doses of estradiol in older women may warrant further investigation with larger samples or different dosage regimens.
Population Simulation with 20 Virtual Subjects
Table 9 summarizes the main pharmacokinetic parameters obtained from the simulation with 20 virtual subjects, conducted in the Population Simulation module of GastroPlus®. As in Table 7, the mean values (arithmetic and geometric), the coefficient of variation (CV%) and the 90% confidence interval (90% CI) are shown for the absorption parameters (F, Fa, FDp), systemic exposure (AUC0–t and AUC0–inf) and plasma profile (Cmax and Tmax). Also in this case, the DDI/baseline ratio was calculated for each parameter in order to assess the impact of estradiol coadministration on the pharmacokinetics of tamoxifen in a larger population.
Table 9.
Summary of pharmacokinetic parameters of tamoxifen with and without estradiol (0.5 mg/day) over 10 days based on a population-based simulation (n = 20) in virtual female subjects with 25- to 45-year-olds and 50- to 80-year-olds.
Overall, the results obtained with 20 subjects confirmed the pharmacokinetic stability observed in the previous simulation. Fa remained constant, with values close to 100% and DDI/baseline ratios fixed at 1.000 in both age groups, reflecting full and unchanged absorption of tamoxifen, as seen in the simulation with 10 individuals. FDp was between 79% and 80%, with ratios also at 1.000 and slight variability (CV between 3.8% and 4.7%), maintaining the same pattern as previously observed.
Across both age groups, DDI/baseline ratios for AUC and C_max remained ≈1.00, indicating no clinically meaningful PK interaction of estradiol on parent tamoxifen under the simulated regimens.
F also remained stable with estradiol coadministration, with DDI/baseline ratios close to 1.000. The values ranged from 72% in the 25- to 45-year-old group to 71% in the 50- to 80-year-old group, differences which were practically identical to those recorded in the simulation with 10 individuals (73% and 70%, respectively). This consistency between simulations reinforces the interpretation that the slight reduction observed in older individuals may be due to age-related physiological changes, with no relevant pharmacokinetic or clinical impact.
With regard to Cmax, the values remained practically unchanged with the coadministration of estradiol, with DDI/baseline ratios very close to 1.000 in both age groups. Variations in the coefficients of variation between the scenarios with and without estradiol were minimal, with ratios ranging from 0.165 to 0.097, showing a pattern of variability similar to that observed in the 10 individuals. Thus, as before, the data do not point to any significant influence of estradiol on the maximum plasma levels of tamoxifen.
With regard to Tmax, the results were once again high and not very plausible from a physiological point of view. In the 25- to 45-year-old group, this parameter showed values of 217.3 h, a ratio of 1.000 and an extremely low CV of 0.16%. In the 50- to 80-year-old group, there was an apparent reduction in Tmax to 132.1 h with the ratio remaining at 1.000, but with a high CV (82.6%), indicating high variability between the simulated individuals. These values are consistent with those obtained in the population of 10 individuals and again suggest the occurrence of an artifact related to the excessive duration of the simulation (240 h), which may have led to the identification of secondary peaks as Tmax.
Finally, systemic exposure, assessed by the AUC0-t and AUC0-inf parameters, showed marginal differences with the coadministration of estradiol, with ratios between 1.000 and 1.003. These results confirm the pattern observed in the simulation with 10 individuals, where the variations also did not exceed 0.5%. In both cases, the data are within the limits of the simulated variability and do not point to any significant change in the extent of exposure to tamoxifen.
From a pharmacological point of view, the results obtained in the two simulations are consistent with the characteristics of tamoxifen, a drug with high bioavailability, effective absorption and complex metabolization through multiple cytochrome P450 isoenzymes, such as CYP3A4 and CYP2D6. Considering that estradiol is administered at a low dose (0.5 mg) and also shares similar metabolization pathways, a relevant inhibitory or inducing effect would not be expected. The absence of changes in absorption parameters (F, Fa, FDp) reinforces this interpretation.
Thus, the data obtained from 10 and 20 subjects validate the conclusion that, under the conditions tested, estradiol does not have a clinically significant impact on the pharmacokinetics of tamoxifen. The absence of relevant changes in the main pharmacokinetic parameters makes it possible to exclude, with reasonable certainty, the existence of a relevant pharmacokinetic interaction between the two drugs in this context.
Figure 8 illustrates the plasma concentration profiles of tamoxifen over 10 days of repeated administration, with and without coadministration of estradiol, obtained through population simulation with 20 virtual subjects. In these graphs, the continuous line represents the mean of the simulated concentrations in each group, while the shaded area corresponds to the 90% confidence interval (90% CI), reflecting the inter-individual pharmacokinetic variability.
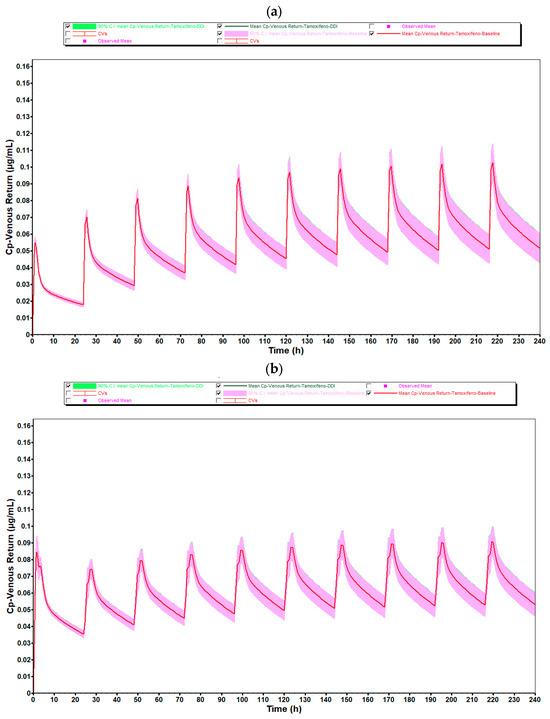
Figure 8.
Mean plasma concentration–time profiles of tamoxifen (30 mg/day) from population simulations (n = 20) for (a) women aged 25–45 years and (b) women aged 50–80 years, with (green line) and without (red line) coadministration of estradiol 0.5 mg/day. Shaded areas correspond to the 90% confidence interval. The high degree of overlap between profiles and variability ranges confirms the absence of a meaningful pharmacokinetic impact of estradiol on tamoxifen.
As observed in the simulation with 10 individuals, there is a complete overlap between the curves corresponding to the coadministration condition (green line) and the reference condition (red line), in both simulated age groups (25- to 45-year-old and 50- to 80-year-old). In the case of premenopausal women, the shape and amplitude of the peaks and valleys remain practically unchanged, indicating that the presence of estradiol (0.5 mg/day) did not significantly alter the pharmacokinetic profile of tamoxifen. This consistency is reinforced by the coincidence of the confidence intervals, which show stability in the pharmacokinetic behavior even as the sample size increases.
In postmenopausal women, there was again a slight discrepancy in the plasma concentration profile after the first administration, with a sharper and more displaced peak to subsequent cycles. As identified in the simulation with 10 individuals, this phenomenon is compatible with the pre-accumulation phase before steady-state is established, and is typical of drugs with a long half-life and high tissue distribution, such as tamoxifen. The physiological changes associated with aging, such as reduced hepatic flow, changes in body composition and decreased metabolic function, could explain the greater initial variability observed in this age group.
As for the average systemic exposure and maximum concentration values, as well as the absorption parameters (F and Fa), these showed DDI/baseline ratios very close to 1.000, quantitatively confirming the absence of any relevant pharmacokinetic interaction. This pattern remained stable when comparing the simulations with 10 and 20 individuals, which reinforces the robustness of the model and the reliability of the results obtained.
It should also be noted that, despite the increase in the number of simulated individuals, the average plasma concentrations remained the same as in the group of 10 individuals, with no changes in the overall shape of the profiles. However, as expected, the 90% confidence interval was slightly wider in the 50- to 80-year-old group, reflecting greater physiological variability among older individuals. Even so, this dispersion did not compromise the interpretation of the data or indicate any relevant impact of estradiol on the pharmacokinetics of tamoxifen.
Thus, the results of the simulations with 20 individuals validate and reinforce the conclusions previously obtained with 10 individuals, showing that the coadministration of estradiol at the tested dose does not significantly modify the pharmacokinetic profile of tamoxifen in a clinically relevant manner, regardless of the age group evaluated.
In addition, Figure 9 illustrates the variation in AUC0-inf as a function of estradiol dose (0.5, 1.0, and 2.0 mg), based on population simulations conducted with n = 20 virtual subjects in two age groups (25- to 45-year-old and 50- to 80-year-old). This graph summarizes the main results obtained from complementary simulations conducted to explore the impact of increasing estradiol doses on systemic exposure to tamoxifen.
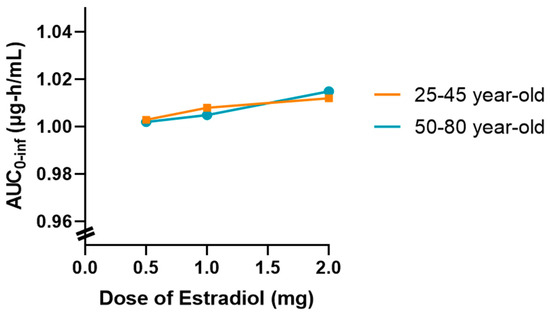
Figure 9.
Variation in tamoxifen (30 mg/day) AUC0-inf as a function of oral estradiol dose (0.5 mg, 1 mg, and 2 mg/day) in two age groups (25–45 years and 50–80 years), based on population simulations (n = 20). A slight upward trend in AUC is seen at higher estradiol doses in older women; however, the magnitude of this change (<4%) remains within normal interindividual variability and without clinical implications.
Thus, there is a slightly upward trend in AUC0-inf as the dose of estradiol increases, which is more evident in the 50–80 age group. However, it should be noted that this variation occurs within a very narrow AUC scale (0.96–1.04 μg∙h/mL), which visually amplifies differences that are marginal in practice. Despite this graphical representation, the absolute values remain very close, and the observed change remains within the limits of the expected inter-individual variability.
This behavior may be associated with a slight reduction in tamoxifen clearance as the dose of estradiol increases in postmenopausal women, possibly related to metabolic competition at the hepatic level. However, in the simulated conditions, this effect does not translate into a clinically relevant difference.
In summary, the results confirm that the coadministration of estradiol at a dose of 0.5 mg does not significantly alter the pharmacokinetics of tamoxifen in any of the age groups studied. Simulations with higher doses, although showing a slight tendency for the AUC to increase, especially in the older group, do not warrant concern from a clinical point of view. Nevertheless, this finding could be relevant in specific contexts and warrants further investigation in scenarios of prolonged use or in populations with compromised metabolism.
4. Discussion
This study introduces an innovative physiologically based pharmacokinetic (PBPK) modeling framework to investigate a previously unexplored therapeutic scenario: the coadministration of tamoxifen and estradiol in women with breast cancer, particularly those at high risk of osteoporosis. Despite longstanding clinical reluctance to combine these agents due to potential metabolic interactions, our simulations, grounded in validated physicochemical parameters and dynamic modeling via GastroPlus®, provide new in silico evidence to re-examine this approach within a controlled, personalized medicine paradigm.
From a mechanistic perspective, tamoxifen and estradiol share hepatic metabolism pathways, most notably involving CYP3A4, which has historically raised concerns regarding competitive inhibition and altered drug exposure. Our results suggest, however, that such interactions may not be clinically relevant under standard dose ranges. Simulated pharmacokinetic parameters (AUC, Cmax, Tmax, and F) for tamoxifen remained stable across multiple estradiol dosing regimens and physiological profiles (premenopausal and postmenopausal women), with minimal deviation and well within expected ranges for inter-individual variability. This consistency suggests that coadministration may not compromise tamoxifen’s therapeutic exposure, a finding that contrasts with some earlier assumptions in clinical pharmacology.
Notably, the design of this study allowed the modeling of both single and repeated dosing, as well as population-level variability. The application of the PEAR model enabled precise simulation of age-specific physiological changes, while the population simulations (n = 10 and n = 20) incorporated variance in key parameters such as hepatic enzyme activity, body weight, and hormone levels. These layers of complexity are often absent in traditional pharmacokinetic studies, which typically rely on average values derived from small, homogenous cohorts. Our approach aligns with contemporary efforts to enhance predictive power and external validity in pharmacokinetic modeling by incorporating demographic and physiological diversity.
While no significant changes were observed in tamoxifen’s disposition, it is important to contextualize these results with caution. The absence of clinically relevant PK alterations in simulations does not automatically equate to safety or therapeutic compatibility in vivo. In silico methods, though powerful, are inherently constrained by the assumptions they embed, such as static enzyme activity, average organ function, and exclusion of co-pathologies or polypharmacy. Moreover, we recognize that while both tamoxifen and estradiol PBPK models were individually validated against clinical reference datasets, additional validation against multiple independent studies, including repeated-dose regimens, would further strengthen the robustness of our findings. Although this was beyond the primary scope of the present computational analysis, we have acknowledged this limitation and emphasize the importance of future work aimed at extending validation to diverse dosing scenarios and patient populations. In addition, the present models did not account for critical modulators of tamoxifen efficacy, such as CYP2D6 genetic polymorphisms or the production of active metabolites like endoxifen, which are known to influence therapeutic outcomes substantially.
In addition, estradiol’s regulatory effects on enzyme expression, particularly its potential to modulate the activity of metabolizing enzymes like SULTs or UGTs, were not included in the simulations. These biological dynamics, while complex, may prove essential for fully understanding long-term outcomes in combined hormone therapies. Therefore, although our PBPK models offer robust pharmacokinetic projections, they should be seen as complementary to, rather than replacements for, experimental validation.
From a broader clinical standpoint, these findings may prompt a re-evaluation of therapeutic strategies, particularly for patients in whom the benefits of hormone replacement (e.g., bone protection) must be weighed against oncological risks. As breast cancer survivorship increases, the management of comorbidities such as osteoporosis becomes more pressing. Being able to confidently simulate and predict drug behavior in such complex contexts is central to modern precision medicine.
Future research should aim to expand the scope of PBPK modeling in this domain by integrating real-world patient data, pharmacogenomic variables, and comorbidity indices. Experimental studies—both in vitro and clinical—are essential to validate the pharmacokinetic neutrality observed here and to examine pharmacodynamic endpoints. Further development of multi-scale models incorporating gene expression dynamics, hormonal cycles, and inter-organ interactions would also significantly enrich model realism and utility. This study provides a computationally grounded, mechanistically detailed starting point for future investigations into tamoxifen–estradiol coadministration. While further clinical validation is necessary, the findings open new avenues for safely optimizing complex oncologic and endocrine therapies through in silico precision tools.
Phenotype-resolved validation (e.g., CYP2D6 activity score stratification; metabolite kinetics) and broader covariates (e.g., hepatic impairment, enzyme regulation/induction, and polypharmacy) are outside the scope of the present study and are priorities for future work. Our finding of PK-DDI neutrality on parent tamoxifen should not be extrapolated to efficacy, since CYP2D6 polymorphisms and endoxifen formation, explicitly not modeled here, are known modulators of clinical response; these require phenotype-/metabolite-resolved analyses beyond the scope of this PK-focused study.
5. Conclusions
The results obtained showed that, after the doses of estradiol tested (0.5 mg, 1 mg and 2 mg) and 30 mg tamoxifen, there were no remarkable clinical alterations in the pharmacokinetic profiles of tamoxifen even under repeated dosing regimens and with longer population simulation (n = 10 and n = 20). The differences between the observed F, AUC and Cmax were minimal and within the ambit of inter-individual physiological variation, and were not outside the safety and efficacy limits. These results indicate that there is encouraging drug-to-drug pharmacokinetic compatibility.
It should be mentioned that this was the initial study to look at a potential pharmacokinetic interaction between tamoxifen and estradiol, so it was not possible to compare the results with published results directly. In silico simulations provide a good starting point for re-visiting therapeutic approaches once avoided due to fears of drug interactions. Although the PBPK models shown here are good, the results recorded here must be verified through controlled clinical trials in order to offer greater fidelity to real conditions and to enhance the potential of computer model-directed personalized medicine. However, it should be noted that this study adopts a completely computational approach and that it does not consider other routes of metabolization, i.e., those sulfotransferase-dependent, nor those of the estradiol influencing the expression of enzymes such as CYP2D6, nor whose activity is directly accountable for the production of active metabolites of tamoxifen (i.e., endoxifen). In addition, the study also involved healthy women and did not consider some clinical scenarios such as liver failure, polymedication or genetic variability in metabolism (CYP polymorphisms).
It is further established that extrapolation of in silico findings to real clinical application requires additional confirmation, namely through controlled clinical studies or in vitro studies that confirm assumptions of PBPK modeling. The inclusion of other clinical and physiological covariates, such as body mass index, nutritional status, comorbidities and use of concomitant therapy, will be essential to enhance the utility of such models in more realistic and individualized settings.
The present in silico work is PK-focused and does not incorporate estradiol-mediated enzyme regulation, CYP2D6 genetic variability, or active metabolite (endoxifen) kinetics. Conclusions are limited to parent-tamoxifen pharmacokinetics under average enzyme activity; phenotype-resolved and metabolite-focused validation remains a priori-ty for future work.
Author Contributions
Conceptualization, N.V.; methodology, B.G.; formal analysis, B.G. and N.V.; investigation, B.G.; writing—original draft preparation, B.G.; writing—review and editing, N.V.; supervision, N.V.; project administration, N.V.; funding acquisition, N.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financed by Fundo Europeu de Desenvolvimento Regional (FEDER) funds through the COMPETE 2020 Operational Programme for Competitiveness and Internationalisation (POCI), Portugal 2020, and by Portuguese funds through Fundação para a Ciência e a Tecnologia (FCT) in the framework of the projects IF/00092/2014/CP1255/CT0004, 2024.18026.PEX, PRR-09/C06-834I07/2024.P11721 and CHAIR in Onco-Innovation from the Faculty of Medicine, University of Porto (FMUP).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Acknowledgments
B.G. acknowledges CHAIR in Onco-Innovation/FMUP for funding her project.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AUC | Area under curve |
| BC | Breast cancer |
| BBB | Blood-brain barrier |
| BMD | Bone mineral density |
| Cmax | Maximum plasma concentration |
| Cmax liver | Maximum plasma concentration measured in liver |
| CYP | Cytochrome P450 |
| DCIS | Ductal cancer in situ |
| DDI | Drug-drug interaction |
| Diff. Coeff | Diffusion coefficient |
| ER | Estrogen receptor |
| Fa | Fraction absorbed of a drug |
| FDp | Fraction of drug concentration in the portal vein |
| fup | Fraction not bound in plasma |
| HER2 | Human epidermal growth factor receptor 2 |
| HR | Hormone receptor |
| Km | Michelis-Menten constant |
| PBPK | Physiological based pharmacokinetic |
| Peff | Human jejunum effective permeability |
| pKa | Ionization constant |
| Rbp | Blood-plasma concentration ratio |
| SERM | Selective estrogen receptor modulator |
| TAM | Tamoxifen |
| Tmax | Time to peak drug concentration |
| Vmax | Maximal rate of metabolism |
References
- Bento, M.J.; Leite, P.; Rita, S.; Revisão, C.; Gonçalves, A.F.; Sousa, A.; Rodrigues, C.; Teixeira, C.; Escorcio, C.; Teixeira, C.; et al. Título Registo Oncológico Nacional de Todos Os Tumores Na População Residente Em Portugal, Em 2020 Coordenador Do Registo Oncológico Nacional. Available online: https://ron.min-saude.pt/media/2223/ron-2020.pdf (accessed on 21 April 2025).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Galappaththi, S.P.L.; Smith, K.R.; Alsatari, E.S.; Hunter, R.; Dyess, D.L.; Turbat-Herrera, E.A.; Dasgupta, S. The Genomic and Biologic Landscapes of Breast Cancer and Racial Differences. Int. J. Mol. Sci. 2024, 25, 13165. [Google Scholar] [CrossRef] [PubMed]
- Obeagu, E.I.; Obeagu, G.U. Breast Cancer: A Review of Risk Factors and Diagnosis. Medicine 2024, 103, E36905. [Google Scholar] [CrossRef] [PubMed]
- Darida, M.; Rubovszky, G.; Kiss, Z.; Székely, B.; Madaras, B.; Horváth, Z.; Kocsis, J.; Sipőcz, I.; Várnai, M.; Balogh, É.; et al. Improvement in Breast Cancer Survival across Molecular Subtypes in Hungary between 2011 and 2020: A Nationwide, Retrospective Study. Front. Oncol. 2025, 15, 1465511. [Google Scholar] [CrossRef]
- Anampa, J.; Makower, D.; Sparano, J.A. Progress in Adjuvant Chemotherapy for Breast Cancer: An Overview. BMC Med. 2015, 13, 195. [Google Scholar] [CrossRef]
- Bhatia, N.; Thareja, S. Aromatase Inhibitors for the Treatment of Breast Cancer: An Overview (2019–2023). Bioorg Chem. 2024, 151, 107607. [Google Scholar] [CrossRef]
- Janni, W.; Untch, M.; Harbeck, N.; Gligorov, J.; Jacot, W.; Chia, S.; Boileau, J.F.; Gupta, S.; Mishra, N.; Akdere, M.; et al. Systematic Literature Review and Trial-Level Meta-Analysis of Aromatase Inhibitors vs Tamoxifen in Patients with HR+/HER2− Early Breast Cancer. Breast Off. J. Eur. Soc. Mastology 2025, 81, 104429. [Google Scholar] [CrossRef]
- Kanji, C.R.; Nyabadza, G.; Nhachi, C.; Masimirembwa, C. Pharmacokinetics of Tamoxifen and Its Major Metabolites and the Effect of the African Ancestry Specific CYP2D6*17 Variant on the Formation of the Active Metabolite, Endoxifen. J. Pers. Med. 2023, 13, 272. [Google Scholar] [CrossRef]
- Mezger, J. Effects of Tamoxifen on Bone Mineral Density in Postmenopausal Women with Breast Cancer: Comment. Aktuel Endokrinol. Stoffwechs. 1992, 13, 189. [Google Scholar]
- Burstein, H.J.; Lacchetti, C.; Anderson, H.; Buchholz, T.A.; Davidson, N.E.; Gelmon, K.E.; Giordano, S.H.; Hudis, C.A.; Solky, A.J.; Stearns, V.; et al. Adjuvant Endocrine Therapy for Women with Hormone Receptor-Positive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update on Ovarian Suppression. J. Clin. Oncol. 2016, 34, 1689–1701. [Google Scholar] [CrossRef]
- Fessele, K.L. Bone Health Considerations in Breast Cancer. Semin. Oncol. Nurs. 2022, 38, 151273. [Google Scholar] [CrossRef]
- Bichoo, R.A.; Mishra, A.; Lal, P.; Gyan, C.; Agarwal, G.; Agarwal, A.; Mishra, S.K. Quality of Life (QoL) in Postmenopausal Breast Cancer Patients Receiving Adjuvant Hormonal Therapy. Indian. J. Surg. 2021, 83, 461–467. [Google Scholar] [CrossRef]
- Kwon, M.; Kim, B.H.; Min, S.Y.; Chae, S. Effects of Anticancer Therapy on Osteoporosis in Breast Cancer Patients: A Nationwide Study Using Data from the National Health Insurance Service-National Health Information Database. J. Clin. Med. 2025, 14, 732. [Google Scholar] [CrossRef]
- Nisha, Y.; Dubashi, B.; Bobby, Z.; Sahoo, J.P.; Kayal, S. Effect of Cytotoxic Chemotherapy on Bone Health among Breast Cancer Patients. Does It Require Intervention? Support. Care Cancer 2021, 29, 6957–6972. [Google Scholar] [CrossRef]
- Hariri, L.; Rehman, A. Estradiol; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Hormone Replacement Therapy (HRT): Osteoporosis. Available online: https://theros.org.uk/information-and-support/osteoporosis/treatment/hormone-replacement-therapy/ (accessed on 21 April 2025).
- Hamoda, H.; Panay, N.; Pedder, H.; Arya, R.; Savvas, M.; Medical Advisory Council of the British Menopause Society. The British Menopause Society & Women’s Health Concern 2020 Recommendations on Hormone Replacement Therapy in Menopausal Women. Post Reprod. Health 2020, 26, 181–209. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.; Medical Advisory Council of the British Menopause Society. Prevention and Treatment of Osteoporosis in Women. Post Reprod. Health 2022, 29, 11. [Google Scholar] [CrossRef] [PubMed]
- Bagger, Y.Z.; Tankó, L.B.; Alexandersen, P.; Hansen, H.B.; Møllgaard, A.; Ravn, P.; Qvist, P.; Kanis, J.A.; Christiansen, C. Two to Three Years of Hormone Replacement Treatment in Healthy Women Have Long-Term Preventive Effects on Bone Mass and Osteoporotic Fractures: The PERF Study. Bone 2004, 34, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Rasha, F.; Sharma, M.; Pruitt, K. Mechanisms of Endocrine Therapy Resistance in Breast Cancer. Mol. Cell Endocrinol. 2021, 532, 111322. [Google Scholar] [CrossRef]
- Desta, Z.; Ward, B.A.; Soukhova, N.V.; Flockhart, D.A. Comprehensive Evaluation of Tamoxifen Sequential Biotransformation by the Human Cytochrome P450 System in Vitro: Prominent Roles for CYP3A and CYP2D6. J. Pharmacol. Exp. Ther. 2004, 310, 1062–1075. [Google Scholar] [CrossRef]
- Kim, D.; Oh, J.; Lee, H.S.; Jeon, S.; Park, W.C.; Yoon, C.I. Association between Tamoxifen and Incidence of Osteoporosis in Korean Patients with Ductal Carcinoma in Situ. Front. Oncol. 2023, 13, 1236188. [Google Scholar] [CrossRef]
- Qasim, Y.; Rafif, A.; Faisal, R.; Yaseen, A.; Abdulrazzaq Luqman Al-Hakeem, M.; Almajidi, Y.Q.; Faisal, R.R.; Al-Hakeem, M.A.L. GastroPlus® as Simulating/Conducting Software for Modeling Physiologically-Based Pharmacokinetics & Physiologically Based Biopharmaceutics. Maaen J. Med. Sci. 2025, 4, 10. [Google Scholar] [CrossRef]
- Chougule, M.; Kollipara, S.; Mondal, S.; Ahmed, T. A Critical Review on Approaches to Generate and Validate Virtual Population for Physiologically Based Pharmacokinetic Models: Methodologies, Case Studies and Way Forward. Eur. J. Clin. Pharmacol. 2024, 80, 1903–1922. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, F.; Guo, N.; Gu, Z.; Lin, H.; Xiang, X.; Shi, Y.; Han, B. Application of Physiologically Based Pharmacokinetics Modeling in the Research of Small-Molecule Targeted Anti-Cancer Drugs. Cancer Chemother. Pharmacol. 2023, 92, 253–270. [Google Scholar] [CrossRef] [PubMed]
- TAMOXIFENO FAMOZ_RCM. 2016. Available online: https://www.tecnimede.com/sites/default/files/portfolio/dosages/TAMOXIFENO%20FAMOZ_RCM.pdf (accessed on 21 April 2025).
- Stefanick, M.L. Estrogens and Progestins: Background and History, Trends in Use, and Guidelines and Regimens Approved by the US Food and Drug Administration. Am. J. Med. 2005, 118, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vera, L.; Yin, X.; Almoslem, M.; Romahn, K.; Cicali, B.; Lukacova, V.; Cristofoletti, R.; Schmidt, S. Comprehensive Physiologically Based Pharmacokinetic Model to Assess Drug-Drug Interactions of Phenytoin. Pharmaceutics 2023, 15, 2486. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, L.; Li, M.; Li, N.; Qi, F.; Wang, N. Predicting the Effects of Different Triazole Antifungal Agents on the Pharmacokinetics of Tamoxifen. AAPS PharmSciTech 2019, 20, 24. [Google Scholar] [CrossRef]
- Estradiol: Uses, Interactions, Mechanism of Action|DrugBank Online. Available online: https://go.drugbank.com/drugs/DB00783 (accessed on 7 July 2025).
- Yang, Y.; Manda, P.; Pavurala, N.; Khan, M.A.; Krishnaiah, Y.S.R. Development and Validation of in Vitro–in Vivo Correlation (IVIVC) for Estradiol Transdermal Drug Delivery Systems. J. Control. Release 2015, 210, 58–66. [Google Scholar] [CrossRef]
- Badawi, A.F.; Cavalieri, E.L.; Rogan, E.G. Role of Human Cytochrome P450 1A1, 1A2, 1B1, and 3A4 in the 2-, 4-, and 16alpha-Hydroxylation of 17beta-Estradiol. Metabolism 2001, 50, 1001–1003. [Google Scholar] [CrossRef]
- Tiryannik, I.; Heikkinen, A.T.; Gardner, I.; Onasanwo, A.; Jamei, M.; Polasek, T.M.; Rostami-Hodjegan, A. Static Versus Dynamic Model Predictions of Competitive Inhibitory Metabolic Drug–Drug Interactions via Cytochromes P450: One Step Forward and Two Steps Backwards. Clin. Pharmacokinet. 2025, 64, 155–170. [Google Scholar] [CrossRef]
- Step 3: Clinical Research | FDA. Available online: https://www.fda.gov/patients/drug-development-process/step-3-clinical-research#Clinical_Research_Phase_Studies (accessed on 25 June 2025).
- Marques, L.; Vale, N. Improving Individualized Salbutamol Treatment: A Population Pharmacokinetic Model for Oral Salbutamol in Virtual Patients. Pharmaceutics 2025, 17, 39. [Google Scholar] [CrossRef]
- Sier, J.H.; Thumser, A.E.; Plant, N.J. Linking Physiologically-Based Pharmacokinetic and Genome-Scale Metabolic Networks to Understand Estradiol Biology. BMC Syst. Biol. 2017, 11, 141. [Google Scholar] [CrossRef]
- Ning, L.; Gong, X.; Li, P.; Chen, X.; Wang, H.; Xu, J. Measurement and Correlation of the Solubility of Estradiol and Estradiol-Urea Co-Crystal in Fourteen Pure Solvents at Temperatures from 273.15 K to 318.15 K. J. Mol. Liq. 2020, 304, 112599. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).