
Targeted Treatment Approaches for Gastrointestinal Metastases of Malignant Melanoma: Clinical Insights and Overcoming Drug Resistance



Abstract
1. Introduction
2. Clinical Insights and Management Strategies of Malignant Melanoma in GIT
- Enhancing methods to classify patient risk.
- Improving predictions of disease prognosis.
- Refining treatment strategies.
- Better managing treatment-related side effects.
- Developing more effective surveillance methods for patients diagnosed with advanced melanoma [26].
3. Mechanisms of Drug Therapy Resistance of Malignant Melanoma in GIT
3.1. Immunotherapy for Melanoma—Current State
3.2. Melanoma Cell Plasticity: A Key Feature of Therapy Resistance
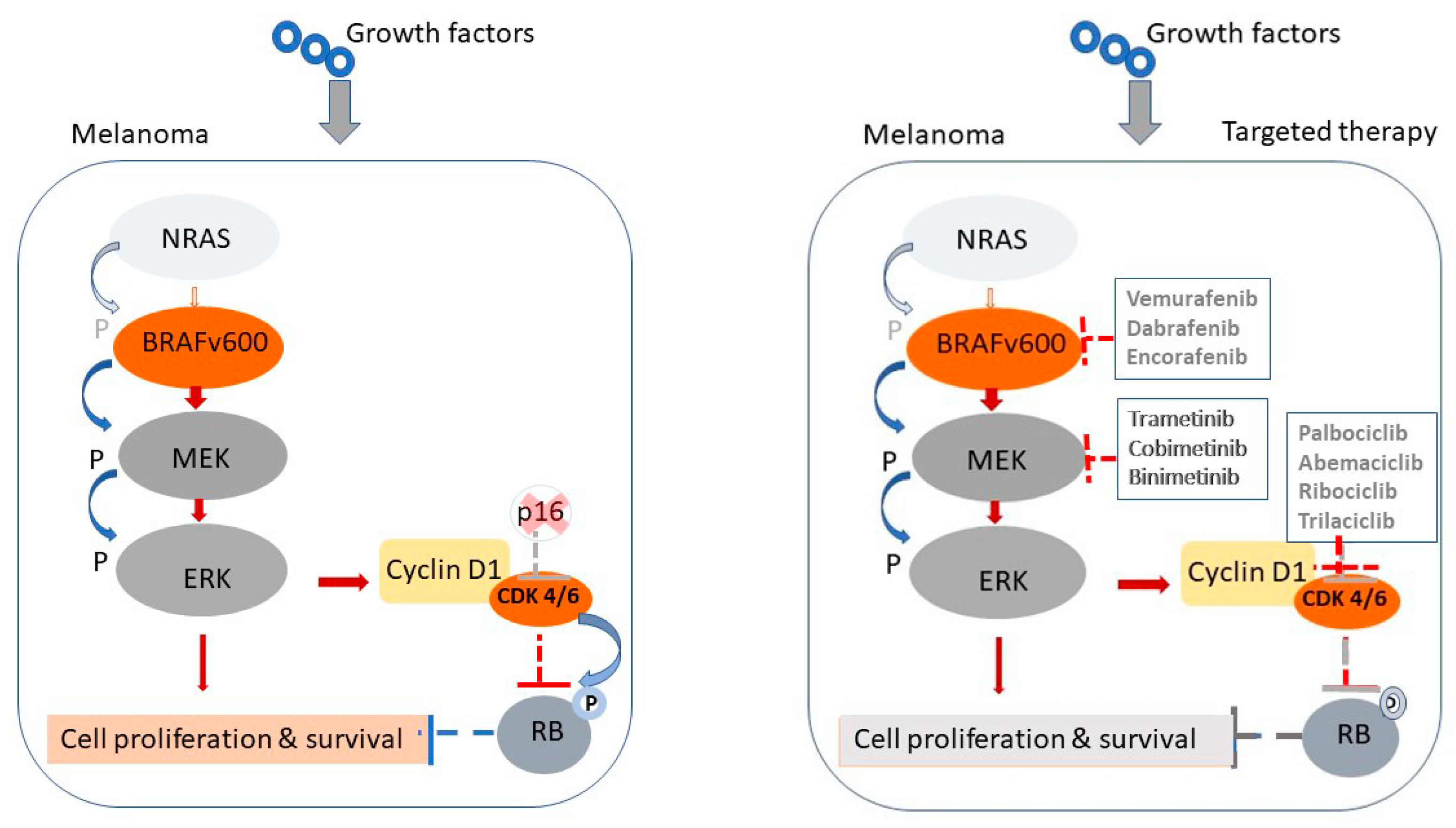
3.3. Mechanisms of Resistance to Targeted Therapy
3.4. Autophagy, ER Stress, and miRNA-Mediated Resistance Mechanisms
3.5. Mechanical Phenotypic Plasticity in Melanoma Therapy Resistance
3.6. Immunological Mechanisms of Primary Resistance to ICI Treatment
- JAK1/STAT Pathway Mutation: A loss of function mutation in the Janus kinase 1 (JAK1) or Signal transducer and activator of transcription (STAT) pathway impairs downstream signal transduction and interferon-gamma (IFN-γ) effects on melanoma cells.
- Beta 2 Microglobulin (B2M) Mutation: Mutations in beta 2 microglobulin (B2M) lead to defective B2M and major histocompatibility complex I (MHC I) molecules, thus preventing the priming of cytotoxic CD8+ T cells.
- Inhibitory Signal Overexpression: Overexpression of PD-L1, LAG-3, and T-cell immunoglobulin and mucin domain 3 (TIM-3) promotes inhibitory signals in effector T cells, resulting in their anergy [51].
3.7. Secondary (Acquired) Resistance
3.8. Treatment Sequencing
4. Strategies for Overcoming Immunotherapy Resistance
- The composition and metabolites of the gut microbiome significantly regulate malignant transformation, metastasis, and anti-tumor immunity [64].
- In a study involving 124 patients with metastatic acral or mucosal melanoma treated with anti-PD-1 monotherapy, researchers identified T cell-inflamed gene expression profiles, PD-L1 expression, tumor mutational burden (TMB), immune cell infiltration, and RAS and WNT signaling as potential biomarkers of resistance [65].
- CP-GEP can identify high-risk stage I/II melanoma patients, with five-year relapse-free survival rates of 77.8% for CP-GEP high-risk patients compared to 93% for low-risk patients. This model can potentially replace sentinel lymph node biopsy (SLNB) [66].
- Targeting androgen receptors (ARs) may reduce drug resistance in melanoma. Inhibition of AR expression or activity can blunt gene expression changes, suppress proliferation, promote CD8+ T cell infiltration, and enhance cancer cell killing [67].
- Strategies to overcome resistance include understanding and modulating the interactions between cancer cells and the immune system, improving patient selection through biomarkers, and combining immunotherapies with other treatments to enhance efficacy [69].
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BRAF | B-Rapidly Accelerated Fibrosarcoma protein |
| BRAFi | BRAF inhibitors |
| CRC | Colorectal cancer |
| CT | Computed tomography |
| CTLA-4 | cytotoxic T-lymphocyte antigen 4 |
| ECM | Extracellular matrix |
| GI | Gastrointestinal |
| GIT | Gastrointestinal tract |
| GTPase | Guanosine Triphosphatase |
| ICIs | Immune checkpoint inhibitors |
| JAK 1 | Janus kinase 1 |
| LAG-3 | Lymphocyte Activation Gene-3 |
| MAPK | Mitogen-activated protein kinase |
| MEK | Mitogen-Activated Protein Kinase kinase |
| MEKi | MEK inhibitors |
| MITF | Microphthalmia-associated Transcription Factor |
| MM | Malignant melanoma |
| NCSC | Neural crest stem-like cell |
| NF1 | Neurofibromin 1 |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| PD-1 | Programmed Cell Death Protein 1 |
| PET | Positron Emission Tomography |
| PI3K-AKT | Phosphoinositide 3-Kinase/Protein Kinase B |
| TT | Targeted therapies |
| TILs | Tumor-infiltrating lymphocytes |
| TME | Tumor microenvironment |
| STAT | Signal transducer and activator of transcription |
| RTK | Receptor tyrosine kinase |
| WHO | World Health Organization |
References
- Long, G.V.; Swetter, S.M.; Menzies, A.M.; Gershenwald, J.E.; Scolyer, R.A. Cutaneous melanoma. Lancet 2023, 402, 485. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, K.; Robbins, K.J.; Perone, J.; Hinojosa, F.L.; Chen, D.; Jones, S.; Kaufman, C.K.; Weiser, R.; Fields, R.C.; Tyler, D.S. Malignant melanoma: Evolving practice management in an era of increasingly effective systemic therapies. Curr. Probl. Surg. 2022, 59, 101030. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.E.; Bastian, B.C.; Cree, I.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch. Pathol. Lab. Med. 2020, 144, 500. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.K.; Lubner, M.G.; Menias, C.O.; Mellnick, V.M.; Kennedy, T.A.; Bhalla, S.; Pickhardt, P.J. Clinical and imaging features of noncutaneous melanoma. AJR Am. J. Roentgenol. 2017, 208, 942–959. [Google Scholar] [CrossRef]
- Duarte, A.F.; Sousa-Pinto, B.; Azevedo, L.F.; Barros, A.M.; Puig, S.; Malvehy, J.; Haneke, E.; Correia, O. Clinical ABCDE rule for early melanoma detection. European journal of dermatology. Eur. J. Dermatol. 2021, 31, 771–778. [Google Scholar] [CrossRef]
- Grob, J.J.; Bonerandi, J.J. The ‘ugly duckling’ sign: Identification of the common characteristics of nevi in an individual as a basis for melanoma screening. Arch. Dermatol. 1998, 134, 103. [Google Scholar] [CrossRef]
- MacKie, R.M. Clinical recognition of early invasive malignant melanoma. BMJ 1990, 301, 1005. [Google Scholar] [CrossRef]
- Dobre, E.-G.; Surcel, M.; Constantin, C.; Ilie, M.A.; Caruntu, A.; Caruntu, C.; Neagu, M. Skin Cancer Pathobiology at a Glance: A Focus on Imaging Techniques and Their Potential for Improved Diagnosis and Surveillance in Clinical Cohorts. Int. J. Mol. Sci. 2023, 24, 1079. [Google Scholar] [CrossRef]
- Krishnan, T.; Menzies, A.M.; Roberts-Thomson, R. Recent advancements in melanoma management. Intern Med J. 2021, 51, 327–333. [Google Scholar] [CrossRef]
- Falk Delgado, A.; Zommorodi, S.; Falk Delgado, A. Sentinel Lymph Node Biopsy and Complete Lymph Node Dissection for Melanoma. Curr. Oncol. Rep. 2019, 21, 54. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Switzer, B.; Puzanov, I.; Skitzki, J.J.; Hamad, L.; Ernstoff, M.S. Managing Metastatic Melanoma in 2022: A Clinical Review. JCO Oncol. Pract. 2022, 18, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Serrao, E.M.; Costa, A.M.; Ferreira, S.; McMorran, V.; Cargill, E.; Hough, C.; Shaw, A.S.; O’carrigan, B.; Parkinson, C.A.; Corrie, P.G.; et al. The different faces of metastatic melanoma in the gastrointestinal tract. Insights Imaging 2022, 13, 161. [Google Scholar] [CrossRef] [PubMed]
- Kohoutova, D.; Worku, D.; Aziz, H.; Teare, J.; Weir, J.; Larkin, J. Malignant Melanoma of the Gastrointestinal Tract: Symptoms, Diagnosis, and Current Treatment Options. Cells 2021, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef]
- Adair, C.; Ro, J.Y.; Sahin, A.A.; El-Naggar, A.K.; Ordónēz, N.G.; Ayala, A.G. Malignant Melanoma Metastatic to Gastrointestinal Tract: A Clinicopathologic Study. Int. J. Surg. Pathol. 1994, 2, 3–9. [Google Scholar] [CrossRef]
- Gilg, M.M.; Gröchenig, H.-P.; Schlemmer, A.; Eherer, A.; Högenauer, C.; Langner, C. Secondary tumors of the GI tract: Origin, histology, and endoscopic findings. Gastrointest. Endosc. 2018, 88, 151–158. [Google Scholar] [CrossRef]
- Bozhkov, V.; Chernopolsky, P. Malignant melanoma–metastases in GIT: Report of 4 cases and literature review. J. IMAB 2024, 30, 5533–5537. [Google Scholar] [CrossRef]
- Filippi, L.; Bianconi, F.; Schillaci, O.; Spanu, A.; Palumbo, B. The Role and Potential of 18F-FDG PET/CT in Malignant Melanoma: Prognostication, Monitoring Response to Targeted and Immunotherapy, and Radiomics. Diagnostics 2022, 12, 929. [Google Scholar] [CrossRef]
- Lo Mastro, A.; Grassi, R.; Reginelli, A.; Russo, A.; Urraro, F.; Belfiore, M.P.; Sandomenico, F.; Iovino, M.; Picascia, O.; Montella, M.; et al. Gastrointestinal metastatic melanoma: Imaging findings and review of literature. J Med. Imaging Intervent. Radiol. 2024, 11, 2. [Google Scholar] [CrossRef]
- Holmberg, C.J.; Alwan, G.; Ny, L.; Olofsson Bagge, R.; Katsarelias, D. Surgery for gastrointestinal metastases of malignant melanoma—A retrospective exploratory study. World J. Surg. Oncol. 2019, 17, 123. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Menon, R.; Iyer, R. Ileo-ileal intussusception secondary to malignant metastatic melanoma: A case report. Int. Surg. J. 2024, 11, 854–856. [Google Scholar] [CrossRef]
- Tatlidil, R.; Mandelkern, M. FDG-PET in the detection of gastrointestinal metastases in melanoma. Melanoma Res. 2001, 11, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Othman, A.E.; Eigentler, T.K.; Bier, G.; Pfannenberg, C.; Bösmüller, H.; Thiel, C.; Garbe, C.; Nikolaou, K.; Klumpp, B. Imaging of gastrointestinal melanoma metastases: Correlation with surgery and histopathology of resected specimen. Eur. Radiol. 2017, 27, 2538–2545. [Google Scholar] [CrossRef]
- Batus, M.; Waheed, S.; Ruby, C.; Petersen, L.; Bines, S.D.; Kaufman, H.L. Optimal management of metastatic melanoma: Current strategies and future directions. Am. J. Clin. Dermatol. 2013, 14, 179–194. [Google Scholar] [CrossRef]
- Bozzini, C.; Busti, F.; Marchi, G.; Vianello, A.; Cerchione, C.; Martinelli, G.; Girelli, D. Anemia in patients receiving anticancer treatments: Focus on novel therapeutic approaches. Front. Oncol. 2024, 14, 1380358. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Switzer, B.; Piperno-Neumann, S.; Lyon, J.; Buchbinder, E.; Puzanov, I. Evolving Management of Stage IV Melanoma. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e397478. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Hodi, F.S.; Fisher, D.E. From genes to drugs: Targeted strategies for melanoma. Nat. Rev. Cancer 2012, 12, 349–361, Erratum in Nat. Rev. Cancer 2020, 20, 757. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Diazzi, S.; Tartare-Deckert, S.; Deckert, M. The mechanical phenotypic plasticity of melanoma cell: An emerging driver of therapy cross-resistance. Oncogenesis 2023, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of melanoma: Facts and hopes. Clin. Cancer Res. 2019, 25, 5191–5201. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef]
- Zuo, Q.; Liu, J.; Huang, L.; Qin, Y.; Hawley, T.; Seo, C.; Merlino, G.; Yu, Y. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 2018, 37, 3275–3289. [Google Scholar] [CrossRef]
- Cesi, G.; Philippidou, D.; Kozar, I.; Kim, Y.J.; Bernardin, F.; Van Niel, G.; Wienecke-Baldacchino, A.; Felten, P.; Letellier, E.; Dengler, S.; et al. A new ALK isoform transported by extracellular vesicles confers drug resistance to melanoma cells. Mol. Cancer 2018, 17, 145. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, X.; Cai, Z.; Zhou, J.; Cao, R.; Zhao, Y.; Chen, Z.; Wang, D.; Ruan, W.; Zhao, Q.; et al. A novel class of microRNA-recognition elements that function only within open reading frames. Nat. Struct. Mol. Biol. 2018, 25, 1019–1027. [Google Scholar] [CrossRef]
- Fattore, L.; Ruggiero, C.F.; Pisanu, M.E.; Liguoro, D.; Cerri, A.; Costantini, S.; Capone, F.; Acunzo, M.; Romano, G.; Nigita, G.; et al. Reprogramming miRNAs global expression orchestrates development of drug resistance in BRAF mutated melanoma. Cell Death Differ. 2019, 26, 1267–1282. [Google Scholar] [CrossRef] [PubMed]
- Zingg, D.; Sommer, L. Rare, yet relevant tumor cells—A new twist to melanoma cell plasticity. Pigment Cell Melanoma Res. 2018, 31, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., II; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef]
- Smith, M.P.; Rowling, E.J.; Miskolczi, Z.; Ferguson, J.; Spoerri, L.; Haass, N.K.; Sloss, O.; McEntegart, S.; Arozarena, I.; von Kriegsheim, A.; et al. Targeting endothelin receptor signalling overcomes heterogeneity driven therapy failure. EMBO Mol. Med. 2017, 9, 1011–1029. [Google Scholar] [CrossRef]
- Perego, M.; Maurer, M.; Wang, J.X.; Shaffer, S.; Müller, A.C.; Parapatics, K.; Li, L.; Hristova, D.; Shin, S.; Keeney, F.; et al. A slow-cycling subpopulation of melanoma cells with highly invasive properties. Oncogene 2018, 37, 302–312. [Google Scholar] [CrossRef]
- Carreira, S.; Goodall, J.; Denat, L.; Rodriguez, M.; Nuciforo, P.; Hoek, K.S.; Testori, A.; Larue, L.; Goding, C.R. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006, 20, 3426–3439. [Google Scholar] [CrossRef]
- Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Eckert, R.L. Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells. Oncotarget 2017, 8, 110257–110272. [Google Scholar] [CrossRef]
- Kim, I.; Heilmann, S.; Kansler, E.; Zhang, Y.; Zimmer, M.; Ratnakumar, K.; Bowman, R.L.; Simon-Vermot, T.; Fennell, M.; Garippa, R.; et al. Microenvironment-derived factors driving metastatic plasticity in melanoma. Nat. Commun. 2017, 8, 14343. [Google Scholar] [CrossRef]
- Vukadin, S.; Khaznadar, F.; Kizivat, T.; Vcev, A.; Smolic, M. Molecular Mechanisms of Resistance to Immune Checkpoint Inhibitors in Melanoma Treatment: An Update. Biomedicines 2021, 9, 835. [Google Scholar] [CrossRef] [PubMed]
- Machiraju, D.; Schäfer, S.; Hassel, J.C. Potential Reasons for Unresponsiveness to Anti-PD1 Immunotherapy in Young Patients with Advanced Melanoma. Life 2021, 11, 1318. [Google Scholar] [CrossRef] [PubMed]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef]
- Thornton, J.; Chhabra, G.; Singh, C.K.; Guzmán-Pérez, G.; Shirley, C.A.; Ahmad, N. Mechanisms of Immunotherapy Resistance in Cutaneous Melanoma: Recognizing a Shapeshifter. Front. Oncol. 2022, 12, 880876. [Google Scholar] [CrossRef]
- Velikova, T.; Krastev, B.; Lozenov, S.; Gencheva, R.; Peshevska-Sekulovska, M.; Nikolaev, G.; Peruhova, M. Antibiotic-Related Changes in Microbiome: The Hidden Villain behind Colorectal Carcinoma Immunotherapy Failure. Int. J. Mol. Sci. 2021, 22, 1754. [Google Scholar] [CrossRef]
- Savoia, P.; Zavattaro, E.; Cremona, O. Clinical Implications of Acquired BRAF Inhibitors Resistance in Melanoma. Int. J. Mol. Sci. 2020, 21, 9730. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Haist, M.; Stege, H.; Kuske, M.; Bauer, J.; Klumpp, A.; Grabbe, S.; Bros, M. Combination of immune-checkpoint inhibitors and targeted therapies for melanoma therapy: The more, the better? Cancer Metastasis Rev. 2023, 42, 481–505. [Google Scholar] [CrossRef]
- Haist, M.; Stege, H.; Ebner, R.; Fleischer, M.I.; Loquai, C.; Grabbe, S. The Role of Treatment Sequencing with Immune-Checkpoint Inhibitors and BRAF/MEK Inhibitors for Response and Survival of Patients with BRAFV600-Mutant Metastatic Melanoma-A Retrospective, Real-World Cohort Study. Cancers 2022, 14, 2082. [Google Scholar] [CrossRef]
- Boutros, A.; Croce, E.; Ferrari, M.; Gili, R.; Massaro, G.; Marconcini, R.; Arecco, L.; Tanda, E.T.; Spagnolo, F. The treatment of advanced melanoma: Current approaches and new challenges. Crit. Rev. Oncol. Hematol. 2024, 196, 104276. [Google Scholar] [CrossRef]
- Finn, L.; Markovic, S.N.; Joseph, R.W. Therapy for metastatic melanoma: The past, present, and future. BMC Med. 2012, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Semin. Oncol. 2015, 42, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Wherry, E.J. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell 2016, 165, 272–275. [Google Scholar] [CrossRef]
- Lozenov, S.; Krastev, B.; Nikolaev, G.; Peshevska-Sekulovska, M.; Peruhova, M.; Velikova, T. Gut Microbiome Composition and Its Metabolites Are a Key Regulating Factor for Malignant Transformation, Metastasis and Anti-tumor Immunity. Int. J. Mol. Sci. 2023, 24, 5978. [Google Scholar] [CrossRef]
- Shui, I.M.; Liu, X.Q.; Zhao, Q.; Kim, S.T.; Sun, Y.; Yearley, J.H.; Choudhury, T.; Webber, A.L.; Krepler, C.; Cristescu, R.; et al. Baseline and post-treatment biomarkers of resistance to anti-PD-1 therapy in acral and mucosal melanoma: An observational study. J. Immunother. Cancer 2022, 10, e004879. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Amaral, T.; Sinnberg, T.; Chatziioannou, E.; Niessner, H.; Leiter, U.; Keim, U.; Forschner, A.; Dwarkasing, J.; Tjien-Fooh, F.; Wever, R.; et al. Identification of stage I/II melanoma patients at high risk for recurrence using a model combining clinicopathologic factors with gene expression profiling (CP-GEP). Eur. J. Cancer 2023, 182, 155–162. [Google Scholar] [CrossRef]
- Samarkina, A.; Youssef, M.K.; Ostano, P.; Ghosh, S.; Ma, M.; Tassone, B.; Proust, T.; Chiorino, G.; Levesque, M.P.; Goruppi, S.; et al. Androgen receptor is a determinant of melanoma targeted drug resistance. Nat. Commun. 2023, 14, 6498. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Almawash, S. Revolutionary Cancer Therapy for Personalization and Improved Efficacy: Strategies to Overcome Resistance to Immune Checkpoint Inhibitor Therapy. Cancers 2025, 17, 880. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maio, M.; Lewis, K.; Demidov, L.; Mandalà, M.; Bondarenko, I.; Ascierto, P.A. Adjuvant vemurafenib in resected, BRAFV600 mutation-positive melanoma (BRIM8): A randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol. 2018, 19, 510–520, Erratum in Lancet Oncol. 2018, 19, e184. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Kudchadkar, R.R.; De Conti, R.C.; Thebeau, M.S.; Czupryn, M.P.; Tetteh, L.; Eysmans, C.; Richards, A.; Schell, M.J.; Fisher, K.J.; et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin. Cancer Res. 2015, 21, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221, Erratum in Nature 2018, 555, 402. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Details | Ref. |
|---|---|---|
| EMT | Melanoma cells tend to switch their molecular and cellular phenotypes, like epithelial-to-mesenchymal transition (EMT), to evade drug treatment. This switching involves expressing the master transcription factor microphthalmia-associated transcription factor (MITF) and the receptor tyrosine kinase AXL. | [31] |
| MITF-Linked Phenotype Switching | Melanoma tumors are highly heterogeneous, with subpopulations of cells expressing high or low levels of MITF, which typically respond well to targeted therapies. However, during drug treatment, tumors can shift to a MITF^high or AXL^high slow-proliferating state, enhancing their resistance to treatment. | [31] |
| MITF/AXL Ratio and Phenotype Switching | A low MITF/AXL ratio is associated with BRAFi-resistance in melanoma. This involves a switch from a differentiated, proliferative, MITF^high/AXL^low state to a de-differentiated, invasive, MITF^low/AXL^high phenotype. | [43,44,45,46] |
| Mechanism | Details | Ref. |
|---|---|---|
| Differentiation/De-differentiation | During invasion and metastasis formation, melanoma cells adopt a de-differentiated phenotype characterized by low pigmentation and reduced proliferation. Upon reaching secondary sites and forming metastatic growths, they revert to a differentiated, highly pigmented, proliferative phenotype. | [47] |
| Metabolic Rewiring | Metabolic switches in response to drug resistance involve MITF and Jumonji AT-Rich Interactive Domain 1B (JARID1B), a histone demethylase that mediates chromatin remodeling. This allows slow-proliferating cells to remain largely undamaged by treatment. | [48,49] |
| YAP-Mediated Phenotype Switch | In cancer, reduced Hippo signaling leads to the accumulation of Yes-associated protein (YAP) and Transcriptional co-activator with PDZ-binding motif (TAZ) (YAP/TAZ) complexes in the nucleus, enhancing cell proliferation and survival via increased Extracellular signal-regulated Kinases 1 and 2 (ERK1/2) activity. | [50] |
| Melanoma Characteristics | Tumor Immunogenicity | Antigen Presentation and T Cell Priming | T Cell Trafficking and TME Infiltration by TILs | T cell Cytotoxic Effect on TM Cell |
|---|---|---|---|---|
| Mechanisms of primary resistance | Low Tumor Mutational Burden (TMB), antigen loss | Impaired Dendritic cell maturation (TME secretion of IL-6 and IL-10, lipid accumulation, IL-35) | Vascular Endothelial Growth Factor (VEGF) and Angiopoietin-2 (ANG2) overexpression | Tumor-Infiltrating Lymphocyte (TIL) inhibition by Tregs |
| Strategies to overcome primary resistance | CTLA-4 inhibition; radiotherapy + ICI | Anti-IL-35 | Anti-VEGF + ICI, anti-VEGF + anti-ANG2, C-X-C Motif Chemokine Receptor 3 (CXCR3) upregulation | Treg suppression: sunitinib, cytotoxic T-cell therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velikova, T.; Konaktchieva, M.; Peruhova, M. Targeted Treatment Approaches for Gastrointestinal Metastases of Malignant Melanoma: Clinical Insights and Overcoming Drug Resistance. Targets 2025, 3, 21. https://doi.org/10.3390/targets3020021
Velikova T, Konaktchieva M, Peruhova M. Targeted Treatment Approaches for Gastrointestinal Metastases of Malignant Melanoma: Clinical Insights and Overcoming Drug Resistance. Targets. 2025; 3(2):21. https://doi.org/10.3390/targets3020021
Chicago/Turabian StyleVelikova, Tsvetelina, Marina Konaktchieva, and Milena Peruhova. 2025. "Targeted Treatment Approaches for Gastrointestinal Metastases of Malignant Melanoma: Clinical Insights and Overcoming Drug Resistance" Targets 3, no. 2: 21. https://doi.org/10.3390/targets3020021
APA StyleVelikova, T., Konaktchieva, M., & Peruhova, M. (2025). Targeted Treatment Approaches for Gastrointestinal Metastases of Malignant Melanoma: Clinical Insights and Overcoming Drug Resistance. Targets, 3(2), 21. https://doi.org/10.3390/targets3020021