Abstract
Air pollution, particularly from vehicular emissions, has emerged as a critical environmental health concern, contributing to a global estimated 7 million premature deaths annually. Diesel exhaust, a major component of urban air pollution, contains fine particulate matter and gases that evade respiratory filtration, penetrating deep into the lungs and triggering oxidative stress, inflammation, and immune dysregulation. Epidemiological and in vitro studies have linked diesel exhaust exposure to respiratory diseases such as asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, and lung cancer, with immunological mechanisms playing a central role. Diesel exhaust particles induce oxidative stress, impair macrophage phagocytosis, and skew T-cell polarization toward pro-inflammatory Th2 and Th17 responses, exacerbating chronic inflammation and tissue damage. Despite these insights, significant gaps remain in understanding the precise immunomodulatory pathways and long-term systemic effects of diesel exhaust exposure. While animal models and in vitro studies provide valuable data, they often fail to capture the complexity of human exposure and immune responses. Further research is needed to elucidate the mechanisms underlying diesel exhaust-induced immune dysregulation, particularly in vulnerable populations with pre-existing respiratory conditions. This review focuses on summarizing the current knowledge and identifying gaps that are essential for developing targeted interventions and policies to mitigate the adverse health impacts of diesel exhaust and improve respiratory health outcomes globally.
1. Introduction
Air pollution has recently become a significant environmental factor adversely impacting human health. According to the World Health Organization (WHO), in 2023, an estimated 7 million premature deaths occur worldwide annually due to air pollution. WHO reports indicate that over 99% of the urban population resides in areas where air pollution surpasses the recommended WHO limits [1]. Urban areas, particularly in low- and middle-income countries, are disproportionately affected due to rapid industrialization, urbanization, and increased reliance on fossil fuels for transportation and energy. Major sources of urban air pollution include vehicular emissions, industrial activities, construction, and the burning of biomass and coal for cooking and heating. In many cities, the concentration of pollutants such as particulate matter PM2.5 and NO2 far exceeds the WHO-recommended limits. For example, the WHO’s annual mean limit for PM2.5 is 5 µg/m3, but many cities, especially in South Asia, the Middle East, and Africa, regularly experience levels exceeding 50 µg/m3 [2]. Over the past few decades, the transportation sector has emerged as a major contributor to environmental pollution, particularly in both developed and developing countries, with consequential effects on the climate [3]. Apart from vehicular emissions playing a crucial role in exposing populations to these pollutants, emissions from motor vehicles significantly contribute to the overall burden of greenhouse gases [4]. Transport, specifically the road sector, accounts for approximately 16% of the 24% of global CO2 emissions produced by the transportation sector [5]. The escalating number of on-road vehicles has further positioned vehicular pollution as a major contributor to the growing issue of air pollution.
Together, these studies present diesel exhaust as a prominent anthropogenic pollutant on a global scale [6]. Diesel exhaust is a complex mixture of solid and liquid particles suspended in gaseous matter [7]. It consists of two components: soot and gases. The gaseous component in diesel exhaust consists mainly of various gases like CO, NOx, SOx, etc., and the diesel soot mainly consists of diesel exhaust particles (DEPs) and traces of heavy metals. DEPs include particles with sizes ranging from <0.1 μm to 10 μm in diameter. Ultra-fine particles (<0.1 μm) consist of carbon core and are associated with mutagens and polycyclic aromatic hydrocarbons (PAHs) [8]. Organic compounds that are found adsorbed to these particle surfaces are individually known to have harmful effects on human health, and many of them have been defined as mutagenic and carcinogenic [9]. A positive correlation between particulate pollution and adverse health effects, including increased respiratory disorders, has been seen in epidemiological studies [10]. Based on its toxicity, DEPs have been characterized as a major carcinogen by the WHO [11]. A recent pre-clinical investigation of human exposure to diesel exhaust, depicted increased nasal IL-6 concentration and fractional exhaled nitric oxide [12].
DEPs, a significant constituent of diesel exhaust, have been implicated in various adverse effects, including reduced lung function and lung inflammation [13]. In 2012, the International Agency for Research on Cancer designated diesel exhaust as a Group 1 human carcinogen based on a comprehensive case–control study [14]. This extensive investigation quantifies the risk of lung cancer associated with occupational exposure to diesel exhaust. The study revealed a proportional increase in lung cancer risk with higher respirable elemental carbon intensity. The risk was notably elevated for workers in the lowest quartile of exposure [14].
In summary, this review emphasizes the adverse effects of DEPs on lung function, inflammation, and the heightened risk of lung cancer, drawing attention to the necessity of targeted interventions. The paper also addresses the novel link between diesel exhaust, a major particulate matter source, and its potential association with the severity and mortality of pulmonary disease and its immune-modulatory effects. While providing valuable insights, the review underscores the need for further mechanistic studies to unravel the intricate pathways connecting particulate matter, diesel exhaust, and the complex dynamics of respiratory health and viral pandemics.
2. Pathogenic Mechanism Associated with Diesel Exhaust Particulates
Diesel exhaust particulate matter is a complex mixture of particles spanning nanometers to micrometers. Coarse particles with a diameter of 10 μm have the ability to reach the bronchiolar region of the lungs, where some are cleared by lung fluid, while others have been actively engulfed by macrophages, thereby restricting their entry to the lung epithelial barrier [15]. PM2.5 and PM1, known as fine and ultrafine particles, can evade phagocytosis and cross the lung epithelial membrane, reaching various body organs and leading to kidney failure, brain stroke, heart failure, etc. Research, including studies on susceptible populations, has demonstrated acute health effects of PM10, particularly at high concentrations, while limited impacts are observed in normal healthy individuals [16].
Diesel exhaust exposure initiates a cascade of molecular and cellular events that ultimately lead to the manifestation of pulmonary diseases. The process begins with the inhalation of DEPs, which deposit in the airways and alveoli. Ultrafine particles in diesel exhaust, characterized by their small size, could penetrate the lung alveolar region, compromising macrophage phagocytosis (Figure 1). These particles, composed of adsorbed PAHs, induce the generation of reactive oxygen species (ROS) in epithelial cells, macrophages, and dendritic cells [17]. The resulting oxidative stress causes lipid peroxidation and DNA damage, leading to cellular injury and the release of alarmins such as IL-25, IL-33, and thymic stromal lymphopoietin (TSLP) from damaged epithelial cells [18]. These alarmins eventually activate the innate immune cells, including macrophages and dendritic cells, which phagocytose DEPs and release pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α) and chemokines (e.g., CCL2, CXCL8) [19]. However, studies have indicated that the adverse effects of ultrafine particles may be linked, in part, to their inhibitory effects on phagocytosis, resulting in the failure of macrophage clearance [20].
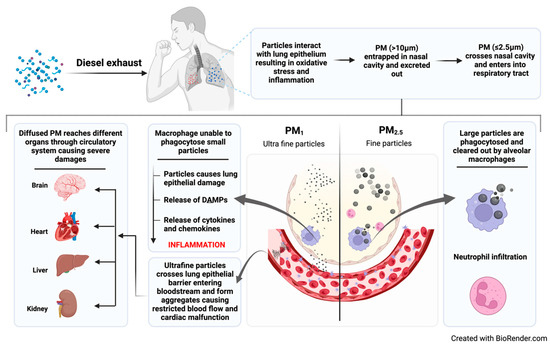
Figure 1.
Mechanistic action of particulate matter in alveolar sac.
The activation of innate immune cells triggers downstream signaling pathways, including NF-κB and MAPK (p38, JNK, ERK), which amplify the production of pro-inflammatory mediators [21]. Concurrently, cytokines such as IL-6 and IL-23 activate the STAT3 pathway, promoting the differentiation of T-helper 17 (Th17) cells, while IL-4 activates STAT6, driving Th2 polarization [22]. This skewed T-cell response is further exacerbated by the suppression of regulatory T-cells (Tregs), which normally maintain immune tolerance. The dominance of Th2 and Th17 responses leads to the recruitment of eosinophils and neutrophils, respectively, causing chronic airway inflammation in the airways [23]. Th2 cytokines (IL-4, IL-5, IL-13) promote eosinophil infiltration, IgE production, and mucus hypersecretion, while Th17 cytokines (IL-17) drive neutrophilic inflammation and tissue damage. Over time, the persistent inflammation and oxidative stress result in structural changes in the lungs, including airway remodeling, fibrosis, and alveolar destruction caused due to continuous particulate matter exposure [24]. This in turn triggers a complex interplay of oxidative stress, innate and adaptive immune activation, and chronic inflammation, culminating in the development and exacerbation of pulmonary diseases. This mechanistic understanding underscores the importance of mitigating diesel exhaust exposure to protect respiratory health.
Additionally, the dose–response relationship between diesel exhaust exposure and immune responses is influenced by both the concentration of DEPs and the duration of exposure, with these factors determining the magnitude and nature of the immune response. At low concentrations, short-term exposure typically triggers mild, transient inflammation characterized by the activation of epithelial cells and macrophages, accompanied by limited cytokine release (e.g., IL-6, TNF-α) [25]. In such cases, the immune response primarily involves innate immunity, with minimal T-cell polarization, and the immune system is often able to resolve the inflammation without long-term consequences [26]. However, even low-concentration exposure can exacerbate symptoms in individuals with pre-existing conditions like asthma, as their airways are already primed for hyperresponsiveness [27]. In contrast, high concentrations of diesel exhaust induce robust oxidative stress, leading to significant epithelial damage and the activation of dendritic cells and macrophages [17]. High-concentration exposure also drives strong Th2 and Th17 polarization, with suppressed regulatory T-cell (Treg) activity, leading to prominent eosinophilic and neutrophilic inflammation. This type of exposure increases the risk of acute exacerbations in asthma and COPD and may even initiate chronic inflammation in otherwise healthy individuals.
3. Inflammation and Diesel Exhaust
- Inflammatory pathways:
Diesel exhaust exposure is well-established as a potent inducer of inflammation, particularly in the respiratory system [28,29]. The inhalation of DEPs and gases initiates a cascade of events leading to the activation of various inflammatory pathways [30,31,32]. This activation of inflammatory pathways is caused by poor clearance of DEPs by macrophages, leading to accumulation in the respiratory system [33]. Eventually, a brief exposure to high levels of diesel exhaust leads to a clear systemic and pulmonary inflammatory response [34]. Key components of this diesel exhaust-induced inflammatory response include the release of inflammatory mediators, chemokines, and cytokines, creating a pro-inflammatory microenvironment in the lungs.
Chemokines and cytokines, pivotal in directing immune cell migration to sites of inflammation, play a crucial role in the immune response triggered by diesel exhaust [30,35]. Diesel exhaust exposure induces the release of pro-inflammatory cytokines such as interleukin-6 (IL-6), interleukin-8 (IL-8), and tumor necrosis factor-alpha (TNF-α), contributing to the establishment of a pro-inflammatory microenvironment within the respiratory system [31,32]. Simultaneously, diesel exhaust stimulates the production of chemokines, promoting the infiltration of immune cells into the respiratory tract [33,36]. Furthermore, diesel exhaust-induced activation of resident immune cells, including macrophages and epithelial cells, through mediators like ChemR23 and CCR2, underscores the complexity of diesel exhaust on inflammatory response modification [28,37,38].
- Oxidative Stress:
One of the intricate ways by which diesel exhaust exerts its inflammatory effects is through the generation of oxidative stress. The particles present in diesel exhaust comprise redox-active components that disturb the delicate balance between the production of ROS and the body’s antioxidant defense mechanisms [39]. This disruption in equilibrium results in oxidative stress, a phenomenon known to cause cellular damage and intensify the inflammatory response. The oxidative stress induced by diesel exhaust has notable repercussions on the functionality of mitochondria within lung cells, leading to impairment and increased apoptosis [40,41]. Mitochondrial dysfunction, in turn, exacerbates the generation of ROS, establishing a self-perpetuating cycle of oxidative stress and inflammation [42]. A recent study established a therapeutic role of resveratrol (potent antioxidant) in mice exposed to diesel exhaust [43]. This study showed treatment with resveratrol mitigates the progression of diesel exhaust-induced pulmonary fibrosis by regulating the SIRT1/FoxO3 signaling axis [43]. Oxidative stress induced by diesel exhaust exposure can lead to a concerning physiological consequence known as hypoxia, where oxygen availability in tissues becomes compromised. In the context of hypoxia, oxidative stress exacerbates inflammation and constriction of airways, compromising the exchange of oxygen and carbon dioxide in the lungs. Current studies have defined confounding evidence of the potential role of hypoxia on genetic alterations. The hypoxia-induced genetic variation plays a significant role in predisposing individuals to other health-related issues like hypertension, like people residing at high and low altitudes [44,45]. This genetic alteration in response to environmental stress may significantly increase pre-disposition to particulate matter exposure. Diesel exhaust contains a plethora of ROS and fine particulate matter that, upon inhalation, generate an oxidative burden within the respiratory system. Additionally, DEPs have the capacity to directly interact with cellular DNA, initiating genetic mutations and causing DNA damage [46]. This damage prompts cellular responses that not only contribute to inflammation but also carry potential long-term implications for respiratory health.
In summary, the various mechanisms through which diesel exhaust triggers inflammation involve the induction of oxidative stress, mitochondrial dysfunction, and direct interactions with cellular DNA, culminating in genetic mutations and damage. These intricate processes collectively underscore the multifaceted impact of diesel exhaust on modifying inflammatory responses within the respiratory system. The identified pathways highlight the need for ongoing research to delve deeper into the underlying mechanisms, providing a more comprehensive understanding of the health risks associated with environmental pollutant exposure. Such knowledge will be instrumental in shaping strategies to mitigate the adverse effects on respiratory health and overall well-being.
4. Immunomodulatory Impacts of Diesel Exhaust
Exposure to diesel exhaust profoundly affects various components of the immune system, influencing both innate and adaptive responses in various respiratory diseases (Figure 2). These immunomodulatory effects extend to the function of immune cells, reshaping their behavior in ways that can impact respiratory health. Noteworthy effects include impaired phagocytosis, dysregulated cytokine production, and compromised antigen presentation. As mentioned before, DEPs can impair the phagocytic capacity of macrophages, hindering their ability to engulf and eliminate pathogens efficiently [30]. Additionally, diesel exhaust-induced alterations in macrophage behavior extend to dysregulated cytokine production, with studies pointing towards upregulation of pro-inflammatory cytokines, such as IL-6 and TNF-α [33]. Moreover, exposure to diesel exhaust has been associated with compromised antigen presentation by macrophages, a critical process in initiating adaptive immune responses [47]. These modifications foster an environment conducive to sustained inflammation and heightened susceptibility to infections.
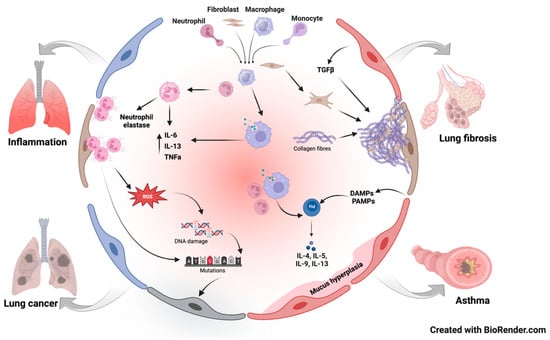
Figure 2.
Pathways involved in diesel exhaust-induced respiratory diseases.
Similarly, diesel exhaust profoundly affects neutrophils, critical components of the innate immune system. DEPs disrupt neutrophil chemotaxis, impairing their migration to infection sites and compromising pathogen clearance. Recent studies demonstrated exposure to diesel leads to an increase in neutrophil infiltration, releasing a serine protease called neutrophil elastase [48]. This neutrophil elastase causes irreversible cellular damage to the underlying basement membrane and lung architecture. The study also highlights a reduced level of Elafin, a known inhibitor of neutrophil elastase, in response to diesel exhaust. Additionally, diesel exhaust exposure heightens the production of ROS by neutrophils, potentially inducing oxidative stress and inflammation. Diesel exhaust inhibits neutrophil apoptosis and phagocytosis by macrophages, prolonging their survival and contributing to persistent inflammation within the respiratory system.
Furthermore, DEPs also act as adjuvants, amplifying immune responses to environmental allergens, and studies show that when DEPs are co-exposed with allergens, they promote Th2-biased responses in the lungs [49]. This occurs partly because of maturation of dendritic cells enhances antigen presentation to naïve T cells and triggers secretion of Th2-polarizing cytokines [50], while organic compounds in DE, like polycyclic aromatic hydrocarbons (PAHs), further amplify this effect by activating the aryl hydrocarbon receptor (AhR), a transcription factor that modulates T-cell fate, intensifying the allergic immune response. In addition to these Th2 effects, DE exposure can drive Th17 differentiation, characterized by IL-17 production, through inflammatory signals like IL-6 and TGF-β, which rise in response to DE-induced epithelial damage and macrophage activation, making Th17 cells, known for their pro-inflammatory nature, key contributors to tissue damage, especially in chronic exposure scenarios [51].
The shift toward Th17 polarization can perpetuate chronic inflammation and tissue damage, as IL-17 recruit’s neutrophils that release proteases, harming lung tissue and promoting fibrosis, a hallmark of these diseases [52]. This is worsened by the potential loss of immune tolerance, as reduced Treg function allows autoreactive T cells to escape regulation, possibly contributing to autoimmunity in conditions like systemic lupus erythematosus or rheumatoid arthritis with lung involvement [53,54], where environmental triggers like DE could exacerbate the diseases phenotype. The exact outcome of DE on T-cell polarization depends on factors like exposure duration, concentration, and individual variables such as genetics or pre-existing conditions, with acute exposure likely boosting Th2 responses and chronic exposure potentially shifting toward Th17 or mixed profiles, affecting urban populations near heavy traffic most severely, as research data tie DE to higher asthma rates and possibly autoimmune disease flares [55,56]. In conclusion, diesel exhaust alters T-cell polarization by promoting Th2 and Th17 responses while potentially suppressing Tregs, primarily through oxidative stress, adjuvant effects, and cytokine shifts, leading to worsened allergic inflammation and airway dysfunction in asthma. Diesel exhaust particles (DEPs) have been found to skew T-cell polarization toward a Th2 and Th17 phenotype; this is mediated by the ability of DEPs to induce oxidative stress and activate signaling pathways such as NF-κB and MAPK in an immune cell population, which in turn produce cytokines like IL-4, IL-5, IL-13, and IL-17 [57]. Major cytokines or chemokines involved in diesel exhaust-induced inflammation are listed in Table 1

Table 1.
Major cytokines, chemokines, and signaling pathways involved in diesel exhaust-induced inflammation.
Diesel exhaust-induced inflammation and modulation of immune cell activity pose a significant risk to the delicate balance of immune tolerance, leading to autoimmune diseases. The immunomodulatory effects triggered by diesel exhaust exposure may play a pivotal role in disrupting immune tolerance, potentially leading to the initiation or worsening of autoimmune conditions [55]. In the context of autoimmune interstitial lung diseases (AILDs), where the immune system erroneously targets lung tissue, resulting in persistent inflammation and scarring, diesel exhaust emerges as a potential contributor. Numerous studies suggest that diesel exhaust may foster the progression of AILDs by further disrupting immune homeostasis and sustaining prolonged inflammation within the respiratory system [33]. Understanding the nuanced relationship between diesel exhaust and immune dysregulation is essential for developing targeted approaches to mitigate the adverse effects on respiratory health.
In summary, the impact of diesel exhaust on immune cell function, particularly in macrophages and neutrophils, extends to far-reaching consequences for respiratory health. The alterations in immune cell function contribute to an increased susceptibility to respiratory infections and the exacerbation of pre-existing conditions. An in-depth exploration is warranted to unveil potential long-term consequences associated with chronic exposure, as well as to devise preventive strategies and therapeutic interventions. Recognizing these immunomodulatory effects is pivotal for advancing our comprehension of the intricate dynamics between diesel exhaust exposure and respiratory health, ultimately guiding efforts to safeguard individuals from the potential long-term implications of chronic exposure.
5. Intricate Interplay Between Diesel Exhaust and Respiratory Diseases
Diesel exhaust exposure has distinct and often more pronounced effects on individuals with pre-existing respiratory conditions such as asthma or chronic obstructive pulmonary disease (COPD) compared to healthy individuals. These differences arise due to the pre-existing inflammatory state, compromised lung function, and altered immune responses in these populations. For example, in individuals with asthma, DEPs exacerbate Th2-driven inflammation, which is already a hallmark of the disease [76]. DEPs amplify the production of Th2 cytokines such as IL-4, IL-5, and IL-13, leading to increased eosinophil infiltration, IgE production, and airway hyperresponsiveness [77]. In contrast, healthy individuals exposed to diesel exhaust may experience a milder Th2 response, as their immune systems are not primed for such reactivity. Additionally, DEPs can induce Th17 responses, which are particularly relevant in severe asthma phenotypes characterized by neutrophilic inflammation. This Th17 skewing is more pronounced in asthmatic individuals, potentially leading to corticosteroid resistance and worsening disease severity [78].
Comparative studies also highlight differences in systemic immune responses between healthy individuals and those with pre-existing conditions. For instance, diesel exhaust exposure in asthmatic or COPD patients can lead to systemic inflammation, as evidenced by elevated levels of C-reactive protein (CRP) and other inflammatory markers in the blood [79]. This systemic inflammation can contribute to comorbidities such as cardiovascular disease, which are already more prevalent in these populations. In contrast, healthy individuals may exhibit a more localized immune response confined to the lungs. Overall, the impact of diesel exhaust on immunity is significantly magnified in individuals with pre-existing respiratory conditions, underscoring the need for targeted interventions and stricter air quality regulations to protect these vulnerable populations. Various cytokines exert differential effects on the pathogenesis of pulmonary fibrosis with different molecular mechanisms, as listed in Table 2. The section below discusses in detail the pathological insights of diesel exhaust in various respiratory diseases.
Table 2.
Summarizing diesel exhaust contributes to different pulmonary diseases at the molecular level.
- Asthma:
Asthma is characterized by chronic airway inflammation, marked by mucus production, and results in airway hyperresponsiveness [83]. Over the last 50 years, there has been a notable surge in asthma prevalence in developed countries, with potential attributions to improved diagnostic capabilities and increased environmental pollution [84]. The existing literature extensively explores the correlation between air pollution and asthma, shedding light on its implications for pre-existing asthma conditions [27,85,86,87,88]. Studies involving patients demonstrate an accelerated manifestation of symptoms and increased incidence associated with elevated levels of particulate matter or DEPs [85]. The age of individuals during exposure emerges as a crucial factor influencing asthma incidence, with studies revealing a link between pollutant exposure during early life and subsequent asthma development [89]. In China, the prevalence of asthma among urban children has quadrupled in the past decade, emphasizing the global impact of environmental factors on childhood asthma. Moreover, early investigations on DEPs and childhood asthma reveal an inverse relationship between lung function and traffic intensity, as well as smoke concentration [90,91]. Studies with children having a family history of asthma further highlight the association of even low concentrations of particulate matter PM2.5 during early stages with asthma onset [92].
Mouse studies provide insights into the exacerbation of airway hyperresponsiveness and the development of pathogenic Th2/Th17 cells in response to DEP exposure to allergens [93]. Diesel exhaust exerts a substantial impact on asthma from an immunological perspective, as demonstrated in various studies, particularly in murine models [93,94]. This immunological response involves the release of pivotal cytokines, including IL-33, IL-25, TSLP, and IL-17a. These cytokines collectively contribute to the immunopathogenesis of asthma, fostering inflammatory reactions and enhancing airway hypersensitivity. IL-33, IL-25, and TSLP are implicated in activating Th2 cells, central players in allergic responses, while IL-17a is involved in recruiting inflammatory cells and initiating pro-inflammatory pathways [83]. The intricate interplay of these immunological responses induced by DEP exposure intensifies asthma symptoms and advances the progression of the condition. Cumulatively, existing studies underscore the significant impact of diesel exhaust on asthma prevalence, calling for comprehensive investigations into the multifaceted relationship for devising targeted interventions and therapeutic strategies, necessitating ongoing research.
- Chronic Obstructive Pulmonary Disease
Numerous review articles have comprehensively explored the connection between occupation and chronic obstructive pulmonary disease (COPD), assessing exposure risks in-depth [95,96,97,98,99,100]. Recent research underscores the association between COPD development and exposure to diverse airborne pollutants, establishing a positive correlation with air pollution. Urban pollution has been identified as a contributor, exacerbating responses in individuals already afflicted with COPD [101]. In individuals with COPD, diesel exhaust exposure further exacerbates the chronic inflammation and oxidative stress that underlie the disease. COPD is characterized by a predominance of Th1 and Th17 responses, which contribute to tissue destruction and emphysema [102]. DEPs enhance these responses by promoting the release of pro-inflammatory cytokines such as IL-1β, IL-6, and IL-17 while also suppressing anti-inflammatory regulatory T-cells (Tregs) [82]. This imbalance exacerbates lung damage and accelerates disease progression. In contrast, healthy individuals may experience a transient inflammatory response to diesel exhaust, but their immune systems are better equipped to resolve this inflammation without long-term consequences. Furthermore, the impaired mucociliary clearance and reduced antioxidant capacity in COPD patients make them more susceptible to the harmful effects of DEPs, leading to increased exacerbation rates and hospitalizations [103].
Inhalation of diesel exhaust components triggers inflammation in the respiratory tract, activating immune cells such as macrophages and neutrophils [104]. This immune cell activation leads to the release of pro-inflammatory cytokines and other mediators, fostering persistent inflammation in the airways, a hallmark of COPD [105]. Additionally, diesel exhaust induces oxidative stress due to the presence of ROS and oxidants, which can damage cellular components and exacerbate the inflammatory response. The activation of immune cells and the induction of epigenetic changes further contribute to the chronic inflammatory state associated with COPD [105]. DEPs have been shown to induce specific histone modifications that contribute to the dysregulation of genes involved in inflammation and oxidative stress, key processes in COPD. For example, exposure to DEPs has been linked to decreased histone acetylation (e.g., H3K9ac and H4K16ac) at the promoters of anti-inflammatory genes, leading to their transcriptional repression [106]. This suppression can exacerbate the inflammatory response in the airways, a hallmark of COPD [107]. Additionally, diesel exhaust has been associated with increased histone methylation (e.g., H3K4me3 and H3K27me3) at pro-inflammatory gene loci, further promoting the expression of inflammatory mediators and contributing to chronic lung damage [107]. The impact of diesel exhaust-induced histone modifications extends beyond immediate gene regulation, potentially leading to long-term epigenetic memory and sustained changes in gene expression patterns. These modifications can alter the recruitment of transcription factors and other chromatin-associated proteins, perpetuating a pro-inflammatory and oxidative state in the lungs. Moreover, diesel exhaust exposure can impair lung defense mechanisms, compromising ciliary function and mucociliary clearance, thereby increasing susceptibility to respiratory infections that can exacerbate COPD symptoms [107].
The recent literature provides statistical insights into the specific impact of diesel exhaust on COPD risk in the human population [108]. A case–control study involving 388 COPD patients unveils higher odds ratios, computed through logistic regression, associating COPD with diesel exhaust and other exposures [109]. Additionally, studies reveal a positive correlation between particulate matter (both coarse and fine particles) levels and increased COPD manifestation [110]. However, further research is essential to fully ascertain the culpability of diesel exhaust in the onset of COPD.
Further, in vitro studies focusing on diesel exhaust (DE) exposure reveal comparable activation of oxidative stress-related gene HMOX1, as well as markers of integrated stress response, CHOP, and GADD34, in cells sourced from COPD donors and controls [69]. Extensive investigations into the role of macrophages in COPD highlight their pivotal involvement in orchestrating pulmonary inflammation [111]. The adverse effects of DEPs on alveolar macrophage activation have been thoroughly examined [112,113]. Furthermore, studies utilizing motor vehicular exhaust demonstrate the release of various cytokines in airway cells capable of inducing COPD [114]. This multifaceted immunological impact underscores the importance of mitigating diesel exhaust exposure as a crucial public health strategy in the prevention and management of COPD.
- Pulmonary fibrosis
Pulmonary fibrosis is a progressive and irreversible lung disease characterized by a significant reduction in lung capacity, primarily attributed to the abnormal deposition of collagen and the activation of key cellular players such as fibroblasts, myofibroblasts, and macrophages [115]. These activated cells contribute to the excessive production of extracellular matrix components, further impairing lung function [116]. Extensive research has explored the correlation between particulate matter and lung fibrosis, revealing noteworthy findings. Particulate matter with a diameter of 10 μm or less (PM10) has been implicated in inducing an upregulation of platelet-derived growth factor (PDGF) [117]. Concurrently, PM10 activates NF-kB, a transcription factor known for inhibiting apoptosis and promoting cellular proliferation [118]. In murine models, exposure to PM10 leads to the differentiation of myofibroblasts and an enhanced production of collagen within the lungs. Moreover, PM10 exposure is associated with an increase in the expression of matrix metalloproteinases (MMPs) and E-cadherin, contributing to the fibrotic process [119].
Exposure to diesel exhaust is linked to adverse health effects, with a focus on the immunological aspects concerning pulmonary fibrosis. The PAHs and metals in diesel exhaust trigger an inflammatory response and oxidative stress in the lungs driven by the activation of immune cells like macrophages and neutrophils, potentially causing cellular damage and inflammation [120]. Prolonged exposure may activate fibroblasts, key players in tissue repair, leading to the excessive production of collagen and other matrix components associated with fibrosis. Diesel exhaust exposure has also been implicated in promoting epithelial–mesenchymal transition (EMT) in lung cells, a process contributing to tissue remodeling [121]. While these immunological responses provide insights into the potential mechanisms linking diesel exhaust to pulmonary fibrosis, the complex nature of this relationship warrants further research to comprehensively understand the underlying pathways and individual variations in susceptibility.
In experimental models of lung fibrosis using carbon nanoparticles, an augmentation in neutrophils and macrophages has been observed without a concurrent increase in eosinophils [122]. This discrepancy results in the exacerbation of pulmonary fibrosis characteristics, highlighting the complex and nuanced effects of particulate matter on the immune response and fibrotic pathways. DEPs, along with their fuel-borne catalyst by-product, cerium oxide, have been identified as potent activators of alveolar macrophages and lymphocytes. This activation triggers the release of proinflammatory cytokines, such as interleukin-12 (IL-12) by macrophages and interferon-gamma (IFN-γ) by lymphocytes [123]. Morphometric analysis further confirms the co-localization of collagen in lung tissues in response to the instillation of DEPs with cerium oxide, underscoring their role in promoting fibrotic changes [124]. The intricate interplay between particulate matter, immune activation, and collagen deposition underscores the complex pathophysiology of pulmonary fibrosis, shedding light on potential targets for therapeutic interventions aimed at mitigating the progression of this debilitating lung disease.
- Lung Cancer
Exposure to diesel exhaust has been associated with various adverse health effects, and emerging evidence links it to the development and progression of lung cancer [107]. The International Agency for Research on Cancer (IARC) categorized diesel exhaust as a Group 1 carcinogen in 2012. Numerous studies have investigated the link between exposure to diesel exhaust and the risk of developing lung cancer, often utilizing elemental carbon as an indicator [125]. Reviews, cohort studies, and epidemiological meta-analyses consistently support the notion that exposure to diesel exhaust is associated with an increased risk of lung cancer [126,127,128]. Specifically, individuals occupationally exposed to diesel exhaust exhibit a significantly higher risk compared to their unexposed counterparts. Cohort studies within the trucking industry reveal an elevated cancer risk with prolonged working years due to consistent exposure to diesel and other vehicle emissions [129,130]. The literature reports an 18% rise in lung cancer risk among drivers exposed to diesel exhaust [131].
Diesel exhaust may interfere with immune surveillance mechanisms, impairing the recognition and clearance of cancer cells by cytotoxic T cells and natural killer cells [132]. Current research has provided significant evidence of the T-cell population playing a crucial role in managing different tumors [133]. Prolonged exposure to diesel exhaust is associated with immunosuppressive effects, weakening the body’s defense mechanisms against cancer cells and promoting their evasion of immune surveillance. Furthermore, research highlights a multifactorial connection between diesel exhaust and cancer, with inflammation and toxicity playing crucial roles in tumor initiation [125]. Diesel exhaust-induced inflammation activates immune cells such as macrophages and neutrophils, releasing pro-inflammatory cytokines and ROS that contribute to DNA damage and the transformation of normal cells into cancerous cells [134,135]. The PAHs, nitro-PAHs, and other mutagenic compounds in DEPs, upon inhalation, deposit in the lungs and penetrate epithelial cells [127,136,137]. These compounds are metabolized by cytochrome P450 enzymes into reactive intermediates, which generate ROS. The resulting oxidative stress causes direct DNA damage, including double-strand breaks, base modifications, and the formation of DNA adducts [137]. This damage often leads to mutations in critical genes such as TP53 (a tumor suppressor gene) and KRAS (an oncogene), both of which are frequently implicated in lung cancer [138]. Rat model studies indicate that exposure to diesel exhaust can induce lung tumor formation through mechanisms involving oxidative stress and the creation of DNA adducts [139,140]. In vitro studies on human bronchiolar epithelial cells (HBECs) exposed to diesel exhaust particulates (DEP) reveal DEP-induced regulation of CYP1A1, IL-1β, PGE2, and PGF2α, potentially contributing to acute inflammation and carcinogenesis [141]. In addition to direct DNA damage, DEPs activate key pro-inflammatory signaling pathways, such as NF-κB and MAPK (p38, JNK, ERK) [142]. These pathways are triggered by the interaction of DEPs with pattern recognition receptors (e.g., TLR4) on epithelial cells and macrophages, leading to the release of pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α) and chemokines (e.g., CXCL8) [143]. The chronic inflammatory microenvironment created by these mediators promotes cell proliferation, inhibits apoptosis, and supports angiogenesis, all of which are hallmarks of cancer. Furthermore, DEPs can induce epigenetic modifications, such as DNA methylation and histone acetylation, which silence tumor suppressor genes or activate oncogenes, contributing to malignant transformation [106,107]. Diesel exhaust also prompts significant neutrophil infiltration and elevates Leukotriene B4 (LTB4) levels in the lungs, with LTB4-dependent inflammation implicated in promoting lung tumor growth [135]. These findings collectively underscore the intricate and comprehensive mechanisms through which diesel exhaust may contribute to the development of lung cancer.
Overall, diesel exhaust exposure is linked to epigenetic changes that influence the expression of genes involved in immune regulation and tumor suppression. This may contribute to the dysregulation of immune responses, increasing susceptibility to lung cancer. Diesel exhaust exposure shapes the tumor microenvironment by promoting the recruitment of immunosuppressive cells, such as myeloid-derived suppressor cells and regulatory T cells, fostering conditions conducive to tumor growth. Individual genetic factors may also play a role in determining susceptibility to the effects of diesel exhaust exposure, influencing an individual’s risk of developing lung cancer. Understanding these immunological insights is crucial for developing preventive strategies and therapeutic interventions to mitigate the impact of diesel exhaust exposure on lung cancer development, highlighting the importance of reducing environmental exposure to diesel exhaust and other air pollutants to protect public health.
6. Conclusions
Emissions from combustion engines, particularly those used in transportation, represent a major contributor to environmental pollution, significantly affecting densely populated areas. Fine particles, measuring less than 4 μm, evade standard filtration mechanisms, reaching deep into the lungs and either settling in the alveoli or crossing into surrounding tissues. This process initiates a cascade of inflammatory responses, disrupts immune balance, and compromises the body’s ability to maintain tolerance, potentially leading to autoimmune disorders and heightened vulnerability to infections. The involvement of key immune cells, such as neutrophils and macrophages, underscores the complex interplay between these particles and the immune system, which is central to the development of chronic respiratory conditions, including asthma, COPD, and malignancies.
The impact of diesel exhaust on respiratory diseases extends to the realm of immunology, as the intricate interplay between the pollutants and the immunological alterations in different respiratory diseases (Figure 3). Chronic inflammation not only contributes to the manifestation and exacerbation of respiratory diseases but also disrupts the delicate balance of immune regulation. Prolonged exposure to diesel exhaust has been shown to compromise the immune system’s ability to mount an effective defense against respiratory infections, rendering individuals more susceptible to pathogens. Furthermore, the oxidative stress induced by diesel pollutants may adversely affect immune cell function, impairing the body’s capacity to combat diseases. Understanding the immunological implications of diesel exhaust exposure is essential for devising effective strategies to protect respiratory health and enhance the immune response in populations at risk.

Figure 3.
Immuno-pathological changes in lung associated diesel exhaust associated pulmonary diseases.
Efforts to understand the link between these emissions and respiratory ailments have largely depended on population-based and laboratory studies. While these approaches yield important data, they often fall short of capturing the full scope of systemic immune effects or the specific toxicity of such emissions. Animal models, with their complex biological networks, provide a more holistic view, revealing how exposure influences disease susceptibility and progression. However, much remains unknown about the precise mechanisms driving immune dysregulation and disease onset. Advancing research in this area is vital to uncover these pathways and develop effective strategies to counteract the detrimental health impacts of prolonged exposure to these pollutants.
Author Contributions
N.S.: conceptualization, writing—original draft, reviewing, and editing. S.S.: reviewing and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Neira, M. Air Pollution: The Invisible Health Threat. 2023. Available online: https://www.who.int/news-room/feature-stories/detail/air-pollution--the-invisible-health-threat (accessed on 8 February 2025).
- Guttikunda, S.; Ka, N. Evolution of India’s PM2.5 pollution between 1998 and 2020 using global reanalysis fields coupled with satellite observations and fuel consumption patterns. Environ. Sci. Atmos. 2022, 2, 1502–1515. [Google Scholar] [CrossRef]
- Nunes, L.J.R. The Rising Threat of Atmospheric CO2: A Review on the Causes, Impacts, and Mitigation Strategies. Environments 2023, 10, 66. [Google Scholar] [CrossRef]
- Abdullah, Z.; Keeley, A.R.; Coulibaly, T.Y.; Managi, S. The impact of fuel cell vehicles deployment on road transport greenhouse gas emissions through 2050: Evidence from 15 G20 countries. J. Environ. Manag. 2024, 370, 122660. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Lu, Y.; Zhou, Y.; He, G.; Song, S.; Yang, S.; Wang, R.; Wang, S.; Han, G.; Yi, X.; et al. Carbon flow through continental-scale ground logistics transportation. iScience 2023, 26, 105792. [Google Scholar] [CrossRef]
- Falcon-Rodriguez, C.I.; Osornio-Vargas, A.R.; Sada-Ovalle, I.; Segura-Medina, P. Aeroparticles, Composition, and Lung Diseases. Front. Immunol. 2016, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Ristovski, Z.D.; Miljevic, B.; Surawski, N.C.; Morawska, L.; Fong, K.M.; Goh, F.; Yang, I.A. Respiratory health effects of diesel particulate matter. Respirology 2012, 17, 201–212. [Google Scholar] [CrossRef]
- Burtscher, H. Measurement and characteristics of combustion aerosols with special consideration of photoelectric charging and charging by flame ions. J. Aerosol Sci. 1992, 23, 549–595. [Google Scholar] [CrossRef]
- Adonis, M.; Gil, L. Polycyclic aromatic hydrocarbon levels and mutagenicity of inhalable particulate matter in Santiago, Chile. Inhal. Toxicol. 2000, 12, 1173–1183. [Google Scholar] [CrossRef]
- Pope, C.A., 3rd. Particulate air pollution and lung function. Am. J. Respir. Crit. Care Med. 2014, 190, 485–486. [Google Scholar] [CrossRef]
- National Toxicology Program. 15th Report on Carcinogens. Rep. Carcinog. 2021, 15, roc15. [Google Scholar] [CrossRef]
- Orach, J.; Rider, C.F.; Yan Yuen, A.C.; Schwartz, C.; Mookherjee, N.; Carlsten, C. Controlled Diesel Exhaust Exposure Induces a Concentration-dependent Increase in Airway Inflammation: A Clinical Trial. Ann. Am. Thorac. Soc. 2023, 20, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Barone-Adesi, F.; Dent, J.E.; Dajnak, D.; Beevers, S.; Anderson, H.R.; Kelly, F.J.; Cook, D.G.; Whincup, P.H. Long-Term Exposure to Primary Traffic Pollutants and Lung Function in Children: Cross-Sectional Study and Meta-Analysis. PLoS ONE 2015, 10, e0142565. [Google Scholar] [CrossRef]
- Silverman, D.T.; Samanic, C.M.; Lubin, J.H.; Blair, A.E.; Stewart, P.A.; Vermeulen, R.; Coble, J.B.; Rothman, N.; Schleiff, P.L.; Travis, W.D.; et al. The Diesel Exhaust in Miners study: A nested case-control study of lung cancer and diesel exhaust. J. Natl. Cancer Inst. 2012, 104, 855–868. [Google Scholar] [CrossRef]
- Losacco, C.; Perillo, A. Particulate matter air pollution and respiratory impact on humans and animals. Environ. Sci. Pollut. Res. Int. 2018, 25, 33901–33910. [Google Scholar] [CrossRef] [PubMed]
- Kyung, S.Y.; Jeong, S.H. Particulate-Matter Related Respiratory Diseases. Tuberc. Respir. Dis. 2020, 83, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Y.; Jin, H.; Lu, Q. Effect of polycyclic aromatic hydrocarbons on immunity. J. Transl. Autoimmun. 2022, 5, 100177. [Google Scholar] [CrossRef]
- Lu, H.F.; Zhou, Y.C.; Yang, L.T.; Zhou, Q.; Wang, X.J.; Qiu, S.Q.; Cheng, B.H.; Zeng, X.H. Involvement and repair of epithelial barrier dysfunction in allergic diseases. Front. Immunol. 2024, 15, 1348272. [Google Scholar] [CrossRef]
- De Grove, K.C.; Provoost, S.; Brusselle, G.G.; Joos, G.F.; Maes, T. Insights in particulate matter-induced allergic airway inflammation: Focus on the epithelium. Clin. Exp. Allergy 2018, 48, 773–786. [Google Scholar] [CrossRef]
- Renwick, L.C.; Donaldson, K.; Clouter, A. Impairment of alveolar macrophage phagocytosis by ultrafine particles. Toxicol. Appl. Pharmacol. 2001, 172, 119–127. [Google Scholar] [CrossRef]
- Wang, J.; Huang, J.; Wang, L.; Chen, C.; Yang, D.; Jin, M.; Bai, C.; Song, Y. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-kappaB signaling pathway. J. Thorac. Dis. 2017, 9, 4398–4412. [Google Scholar] [CrossRef]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.H.; Whitehead, G.S.; Nakano, H.; Free, M.E.; Kolls, J.K.; Cook, D.N. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 2009, 180, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.Y.; Kim, G.D. Particulate Matter-Induced Emerging Health Effects Associated with Oxidative Stress and Inflammation. Antioxidants 2024, 13, 1256. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Jin, Y.; Carlsten, C. Inflammatory health effects of indoor and outdoor particulate matter. J. Allergy Clin. Immunol. 2018, 141, 833–844. [Google Scholar] [CrossRef]
- Glencross, D.A.; Ho, T.R.; Camina, N.; Hawrylowicz, C.M.; Pfeffer, P.E. Air pollution and its effects on the immune system. Free Radic. Biol. Med. 2020, 151, 56–68. [Google Scholar] [CrossRef]
- Guarnieri, M.; Balmes, J.R. Outdoor air pollution and asthma. Lancet 2014, 383, 1581–1592. [Google Scholar] [CrossRef]
- Grytting, V.S.; Chand, P.; Låg, M.; Øvrevik, J.; Refsnes, M. The pro-inflammatory effects of combined exposure to diesel exhaust particles and mineral particles in human bronchial epithelial cells. Part. Fibre Toxicol. 2022, 19, 14. [Google Scholar] [CrossRef]
- Wang, H.; Duan, H.; Meng, T.; Yang, M.; Cui, L.; Bin, P.; Dai, Y.; Niu, Y.; Shen, M.; Zhang, L.; et al. Local and Systemic Inflammation May Mediate Diesel Engine Exhaust–Induced Lung Function Impairment in a Chinese Occupational Cohort. Toxicol. Sci. 2018, 162, 372–382. [Google Scholar] [CrossRef]
- Bao, Y.; Zhu, X. Role of Chemokines and Inflammatory Cells in Respiratory Allergy. J. Asthma Allergy 2022, 15, 1805–1822. [Google Scholar] [CrossRef]
- Ren, H.; Lu, J.; Ning, J.; Su, X.; Tong, Y.; Chen, J.; Ding, Y. Exposure to fine particulate matter induces self-recovery and susceptibility of oxidative stress and inflammation in rat lungs. Environ. Sci. Pollut. Res. 2020, 27, 40262–40276. [Google Scholar] [CrossRef]
- Ma, Q.Y.; Huang, D.Y.; Zhang, H.J.; Wang, S.; Chen, X.F. Exposure to particulate matter 2.5 (PM2.5) induced macrophage-dependent inflammation, characterized by increased Th1/Th17 cytokine secretion and cytotoxicity. Int. Immunopharmacol. 2017, 50, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Arora, N. Diesel exhaust exposure in mice induces pulmonary fibrosis by TGF-beta/Smad3 signaling pathway. Sci. Total Environ. 2022, 807, 150623. [Google Scholar] [CrossRef]
- Salvi, S.; Blomberg, A.; Rudell, B.; Kelly, F.; Sandström, T.; Holgate, S.T.; Frew, A. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am. J. Respir. Crit. Care Med. 1999, 159, 702–709. [Google Scholar] [CrossRef]
- Oliveira, A.C.; Richards, E.M.; Raizada, M.K. Pulmonary hypertension: Pathophysiology beyond the lung. Pharmacol. Res. 2020, 151, 104518. [Google Scholar] [CrossRef]
- Fahy, O.; Hammad, H.; Sénéchal, S.; Pestel, J.; Tonnel, A.B.; Wallaert, B.; Tsicopoulos, A. Synergistic effect of diesel organic extracts and allergen Der p 1 on the release of chemokines by peripheral blood mononuclear cells from allergic subjects: Involvement of the map kinase pathway. Am. J. Respir. Cell Mol. Biol. 2000, 23, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Provoost, S.; De Grove, K.C.; Fraser, G.L.; Lannoy, V.J.; Tournoy, K.G.; Brusselle, G.G.; Maes, T.; Joos, G.F. Pro- and Anti-Inflammatory Role of ChemR23 Signaling in Pollutant-Induced Inflammatory Lung Responses. J. Immunol. 2016, 196, 1882–1890. [Google Scholar] [CrossRef]
- Provoost, S.; Maes, T.; Joos, G.F.; Tournoy, K.G. Monocyte-derived dendritic cell recruitment and allergic T(H)2 responses after exposure to diesel particles are CCR2 dependent. J. Allergy Clin. Immunol. 2012, 129, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.H.; Gibb, M.; Kramer, A.T.; Sayes, C.M. Peripheral (lung-to-brain) exposure to diesel particulate matter induces oxidative stress and increased markers for systemic inflammation. Environ. Res. 2023, 231 Pt 3, 116267. [Google Scholar] [CrossRef]
- Piao, M.J.; Ahn, M.J.; Kang, K.A.; Ryu, Y.S.; Hyun, Y.J.; Shilnikova, K.; Zhen, A.X.; Jeong, J.W.; Choi, Y.H.; Kang, H.K.; et al. Particulate matter 2.5 damages skin cells by inducing oxidative stress, subcellular organelle dysfunction, and apoptosis. Arch. Toxicol. 2018, 92, 2077–2091. [Google Scholar] [CrossRef]
- Xin, Y.; Wan, B.; Yang, Y.; Cui, X.J.; Xie, Y.C.; Guo, L.H. Perfluoroalkyl acid exposure induces protective mitochondrial and endoplasmic reticulum autophagy in lung cells. Arch. Toxicol. 2018, 92, 3131–3147. [Google Scholar] [CrossRef]
- Guo, Z.; Hong, Z.; Dong, W.; Deng, C.; Zhao, R.; Xu, J.; Zhuang, G.; Zhang, R. PM2.5-Induced Oxidative Stress and Mitochondrial Damage in the Nasal Mucosa of Rats. Int. J. Environ. Res. Public Health 2017, 14, 134. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Nagar, E.; Gautam, A.; Kapoor, H.; Arora, N. Resveratrol mitigates miR-212-3p mediated progression of diesel exhaust-induced pulmonary fibrosis by regulating SIRT1/FoxO3. Sci. Total Environ. 2023, 902, 166063. [Google Scholar] [CrossRef]
- Kumar, R.; Kohli, S.; Ali, Z.; Duhan, K.; Ram, R.; Gupta, M.; Tyagi, S.; Mohammad, G.; Pasha, M.A.Q. CYBA (p22phox) variants associate with blood pressure and oxidative stress markers in hypertension: A replication study in populations of diverse altitudes. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2015, 38, 498–506. [Google Scholar] [CrossRef]
- Kohli, S.; Kumar, R.; Gupta, M.; Tyagi, S.; Pasha, M.A.Q. Impact of interactions between risk alleles on clinical endpoints in hypertension. Heart Asia 2016, 8, 83. [Google Scholar] [CrossRef]
- Ritz, C.; Ruminski, W.; Hougaard, K.S.; Wallin, H.; Vogel, U.; Yauk, C.L. Germline mutation rates in mice following in utero exposure to diesel exhaust particles by maternal inhalation. Mutat. Res. 2011, 712, 55–58. [Google Scholar] [CrossRef]
- Ural, B.B.; Caron, D.P.; Dogra, P.; Wells, S.B.; Szabo, P.A.; Granot, T.; Senda, T.; Poon, M.M.L.; Lam, N.; Thapa, P.; et al. Inhaled particulate accumulation with age impairs immune function and architecture in human lung lymph nodes. Nat. Med. 2022, 28, 2622–2632. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Nagar, E.; Arora, N. Diesel exhaust exposure impairs recovery of lung epithelial and cellular damage in murine model. Mol. Immunol. 2023, 158, 1–9. [Google Scholar] [CrossRef]
- Fahy, O.; Senechal, S.; Pene, J.; Scherpereel, A.; Lassalle, P.; Tonnel, A.B.; Yssel, H.; Wallaert, B.; Tsicopoulos, A. Diesel exposure favors Th2 cell recruitment by mononuclear cells and alveolar macrophages from allergic patients by differentially regulating macrophage-derived chemokine and IFN-gamma-induced protein-10 production. J. Immunol. 2002, 168, 5912–5919. [Google Scholar] [CrossRef] [PubMed]
- Liotta, F.; Frosali, F.; Querci, V.; Mantei, A.; Fili, L.; Maggi, L.; Mazzinghi, B.; Angeli, R.; Ronconi, E.; Santarlasci, V.; et al. Human immature myeloid dendritic cells trigger a TH2-polarizing program via Jagged-1/Notch interaction. J. Allergy Clin. Immunol. 2008, 121, 1000–1005.e8. [Google Scholar] [CrossRef]
- van Voorhis, M.; Knopp, S.; Julliard, W.; Fechner, J.H.; Zhang, X.; Schauer, J.J.; Mezrich, J.D. Exposure to atmospheric particulate matter enhances Th17 polarization through the aryl hydrocarbon receptor. PLoS ONE 2013, 8, e82545. [Google Scholar] [CrossRef]
- Sun, L.; Wang, L.; Moore, B.B.; Zhang, S.; Xiao, P.; Decker, A.M.; Wang, H.L. IL-17: Balancing Protective Immunity and Pathogenesis. J. Immunol. Res. 2023, 2023, 3360310. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.Y.; Li, H.C.; Jiang, X.J.; Jiang, Z.Q.; Zhang, Y.; Zhang, H.Y.; Zhang, S.X. Exploring the therapeutic potential of regulatory T cell in rheumatoid arthritis: Insights into subsets, markers, and signaling pathways. Biomed. Pharmacother. 2024, 174, 116440. [Google Scholar] [CrossRef]
- Horwitz, D.A. Regulatory T cells in systemic lupus erythematosus: Past, present and future. Arthritis Res. Ther. 2008, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, C.A.; Owens, L.A.; Gallo, M.E.; Hoffmann, E.J.; Afrazi, A.; Han, M.; Fechner, J.H.; Schauer, J.J.; Bradfield, C.A.; Mezrich, J.D. Differential effects of diesel exhaust particles on T cell differentiation and autoimmune disease. Part. Fibre Toxicol. 2018, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Pandya, R.J.; Solomon, G.; Kinner, A.; Balmes, J.R. Diesel exhaust and asthma: Hypotheses and molecular mechanisms of action. Environ. Health Perspect. 2002, 110 (Suppl. 1), 103–112. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Hyun, Y.M.; Kim, S.H.; Ko, M.K.; Park, C.O.; Hyun, J.W. Particulate matter induces inflammatory cytokine production via activation of NFkappaB by TLR5-NOX4-ROS signaling in human skin keratinocyte and mouse skin. Redox Biol. 2019, 21, 101080. [Google Scholar] [CrossRef]
- Brandt, E.B.; Biagini Myers, J.M.; Acciani, T.H.; Ryan, P.H.; Sivaprasad, U.; Ruff, B.; LeMasters, G.K.; Bernstein, D.I.; Lockey, J.E.; LeCras, T.D.; et al. Exposure to allergen and diesel exhaust particles potentiates secondary allergen-specific memory responses, promoting asthma susceptibility. J. Allergy Clin. Immunol. 2015, 136, 295–303 e7. [Google Scholar] [CrossRef]
- Diaz-Sanchez, D.; Dotson, A.R.; Takenaka, H.; Saxon, A. Diesel exhaust particles induce local IgE production in vivo and alter the pattern of IgE messenger RNA isoforms. J. Clin. Investig. 1994, 94, 1417–1425. [Google Scholar] [CrossRef]
- Pourazar, J.; Frew, A.J.; Blomberg, A.; Helleday, R.; Kelly, F.J.; Wilson, S.; Sandstrom, T. Diesel exhaust exposure enhances the expression of IL-13 in the bronchial epithelium of healthy subjects. Respir. Med. 2004, 98, 821–825. [Google Scholar] [CrossRef]
- Zhao, C.N.; Xu, Z.; Wu, G.C.; Mao, Y.M.; Liu, L.N.; Qian, W.; Dan, Y.L.; Tao, S.S.; Zhang, Q.; Sam, N.B.; et al. Emerging role of air pollution in autoimmune diseases. Autoimmun. Rev. 2019, 18, 607–614. [Google Scholar] [CrossRef]
- Bengalli, R.; Longhin, E.; Marchetti, S.; Proverbio, M.C.; Battaglia, C.; Camatini, M. The role of IL-6 released from pulmonary epithelial cells in diesel UFP-induced endothelial activation. Environ. Pollut. 2017, 231 Pt 2, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Joos, G.; Boon, L.; Tournoy, K.; Provoost, S.; Maes, T. Role of tumor necrosis factor-alpha and its receptors in diesel exhaust particle-induced pulmonary inflammation. Sci. Rep. 2017, 7, 11508. [Google Scholar] [CrossRef]
- Singh, N.; Nagar, E.; Roy, D.; Arora, N. NLRP3/GSDMD mediated pyroptosis induces lung inflammation susceptibility in diesel exhaust exposed mouse strains. Gene 2024, 918, 148459. [Google Scholar] [CrossRef]
- Nadeau, K.; McDonald-Hyman, C.; Noth, E.M.; Pratt, B.; Hammond, S.K.; Balmes, J.; Tager, I. Ambient air pollution impairs regulatory T-cell function in asthma. J. Allergy Clin. Immunol. 2010, 126, 845–852 e10. [Google Scholar] [CrossRef] [PubMed]
- Fahy, O.; Tsicopoulos, A.; Hammad, H.; Pestel, J.; Tonnel, A.B.; Wallaert, B. Effects of diesel organic extracts on chemokine production by peripheral blood mononuclear cells. J. Allergy Clin. Immunol. 1999, 103, 1115–1124. [Google Scholar] [CrossRef]
- Bach, N.S.; Lag, M.; Ovrevik, J. Toll like receptor-3 priming alters diesel exhaust particle-induced cytokine responses in human bronchial epithelial cells. Toxicol. Lett. 2014, 228, 42–47. [Google Scholar] [CrossRef]
- Yang, L.; Cohn, L.; Zhang, D.H.; Homer, R.; Ray, A.; Ray, P. Essential role of nuclear factor kappaB in the induction of eosinophilia in allergic airway inflammation. J. Exp. Med. 1998, 188, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Zarcone, M.C.; van Schadewijk, A.; Duistermaat, E.; Hiemstra, P.S.; Kooter, I.M. Diesel exhaust alters the response of cultured primary bronchial epithelial cells from patients with chronic obstructive pulmonary disease (COPD) to non-typeable Haemophilus influenzae. Respir. Res. 2017, 18, 27. [Google Scholar] [CrossRef]
- Hashimoto, S.; Gon, Y.; Takeshita, I.; Matsumoto, K.; Jibiki, I.; Takizawa, H.; Kudoh, S.; Horie, T. Diesel exhaust particles activate p38 MAP kinase to produce interleukin 8 and RANTES by human bronchial epithelial cells and N-acetylcysteine attenuates p38 MAP kinase activation. Am. J. Respir. Crit. Care Med. 2000, 161, 280–285. [Google Scholar] [CrossRef]
- Vitucci, E.C.M.; Simmons, A.E.; Martin, E.M.; McCullough, S.D. Epithelial MAPK signaling directs endothelial NRF2 signaling and IL-8 secretion in a tri-culture model of the alveolar-microvascular interface following diesel exhaust particulate (DEP) exposure. Part. Fibre Toxicol. 2024, 21, 15. [Google Scholar] [CrossRef]
- Zimmermann, N.; Mishra, A.; King, N.E.; Fulkerson, P.C.; Doepker, M.P.; Nikolaidis, N.M.; Kindinger, L.E.; Moulton, E.A.; Aronow, B.J.; Rothenberg, M.E. Transcript signatures in experimental asthma: Identification of STAT6-dependent and -independent pathways. J. Immunol. 2004, 172, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Tal, T.L.; Graves, L.M.; Gilmour, I.; Linak, W.; Reed, W.; Bromberg, P.A.; Samet, J.M. Diesel exhaust particulate-induced activation of Stat3 requires activities of EGFR and Src in airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L422–L429. [Google Scholar] [CrossRef]
- Cao, D.; Bromberg, P.A.; Samet, J.M. Diesel particle-induced transcriptional expression of p21 involves activation of EGFR, Src, and Stat3. Am. J. Respir. Cell Mol. Biol. 2010, 42, 88–95. [Google Scholar] [CrossRef]
- Xu, C.; Chen, S.; Deng, Y.; Song, J.; Li, J.; Chen, X.; Chang, P.; Yao, L.; Tang, H. Distinct roles of PI3Kdelta and PI3Kgamma in a toluene diisocyanate-induced murine asthma model. Toxicology 2021, 454, 152747. [Google Scholar] [CrossRef] [PubMed]
- Bassol, S.; Ulloa-Aguirre, A.; Perez, A.E.; Mendez, J.P.; Perez-Palacios, G. 5-alpha steroid reductase and 17-beta steroid oxidoreductase activity in human fibroblasts. Comparison between cells of normal individuals and patients with different types of male pseudohermaphroditism. Rev. Investig. Clin. 1984, 36, 231–235. [Google Scholar]
- Brandt, E.B.; Kovacic, M.B.; Lee, G.B.; Gibson, A.M.; Acciani, T.H.; Le Cras, T.D.; Ryan, P.H.; Budelsky, A.L.; Khurana Hershey, G.K. Diesel exhaust particle induction of IL-17A contributes to severe asthma. J. Allergy Clin. Immunol. 2013, 132, 1194–1204.e2. [Google Scholar] [CrossRef] [PubMed]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; Dupont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef]
- Aksu, F.; Capan, N.; Aksu, K.; Ofluoglu, R.; Canbakan, S.; Yavuz, B.; Akin, K.O. C-reactive protein levels are raised in stable Chronic obstructive pulmonary disease patients independent of smoking behavior and biomass exposure. J. Thorac. Dis. 2013, 5, 414–421. [Google Scholar] [CrossRef]
- Li, N.; Xia, T.; Nel, A.E. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic. Biol. Med. 2008, 44, 1689–1699. [Google Scholar] [CrossRef]
- Weng, C.M.; Lee, M.J.; He, J.R.; Chao, M.W.; Wang, C.H.; Kuo, H.P. Diesel exhaust particles up-regulate interleukin-17A expression via ROS/NF-kappaB in airway epithelium. Biochem. Pharmacol. 2018, 151, 1–8. [Google Scholar] [CrossRef]
- Gu, X.Y.; Chu, X.; Zeng, X.L.; Bao, H.R.; Liu, X.J. Effects of PM2.5 exposure on the Notch signaling pathway and immune imbalance in chronic obstructive pulmonary disease. Environ. Pollut. 2017, 226, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Lambrecht, B.N. The basic immunology of asthma. Cell 2021, 184, 2521–2522. [Google Scholar] [CrossRef]
- Reddel, H.K.; Bacharier, L.B.; Bateman, E.D.; Brightling, C.E.; Brusselle, G.G.; Buhl, R.; Cruz, A.A.; Duijts, L.; Drazen, J.M.; FitzGerald, J.M.; et al. Global Initiative for Asthma Strategy 2021: Executive Summary and Rationale for Key Changes. Am. J. Respir. Crit. Care Med. 2022, 205, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Chanel, O.; Perez, L.; Kunzli, N.; Medina, S.; Aphekom, Group. The hidden economic burden of air pollution-related morbidity: Evidence from the Aphekom project. Eur. J. Health Econ. 2016, 17, 1101–1115. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.Y.; Ding, H.; Jiang, L.N.; Chen, S.W.; Zheng, J.P.; Qiu, M.; Zhou, Y.X.; Chen, Q.; Guan, W.J. Association between Air Pollutants and Asthma Emergency Room Visits and Hospital Admissions in Time Series Studies: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0138146. [Google Scholar] [CrossRef]
- Jacquemin, B.; Siroux, V.; Sanchez, M.; Carsin, A.E.; Schikowski, T.; Adam, M.; Bellisario, V.; Buschka, A.; Bono, R.; Brunekreef, B.; et al. Ambient air pollution and adult asthma incidence in six European cohorts (ESCAPE). Environ. Health Perspect. 2015, 123, 613–621. [Google Scholar] [CrossRef]
- Gehring, U.; Beelen, R.; Eeftens, M.; Hoek, G.; de Hoogh, K.; de Jongste, J.C.; Keuken, M.; Koppelman, G.H.; Meliefste, K.; Oldenwening, M.; et al. Particulate matter composition and respiratory health: The PIAMA Birth Cohort study. Epidemiology 2015, 26, 300–309. [Google Scholar] [CrossRef]
- To, T.; Zhu, J.; Stieb, D.; Gray, N.; Fong, I.; Pinault, L.; Jerrett, M.; Robichaud, A.; Menard, R.; van Donkelaar, A.; et al. Early life exposure to air pollution and incidence of childhood asthma, allergic rhinitis and eczema. Eur. Respir. J. 2020, 55, 1900913. [Google Scholar] [CrossRef]
- van Vliet, P.; Knape, M.; de Hartog, J.; Janssen, N.; Harssema, H.; Brunekreef, B. Motor vehicle exhaust and chronic respiratory symptoms in children living near freeways. Environ. Res. 1997, 74, 122–132. [Google Scholar] [CrossRef]
- Wjst, M.; Reitmeir, P.; Dold, S.; Wulff, A.; Nicolai, T.; von Loeffelholz-Colberg, E.F.; von Mutius, E. Road traffic and adverse effects on respiratory health in children. BMJ 1993, 307, 596–600. [Google Scholar] [CrossRef]
- Carlsten, C.; Dybuncio, A.; Becker, A.; Chan-Yeung, M.; Brauer, M. Traffic-related air pollution and incident asthma in a high-risk birth cohort. Occup. Environ. Med. 2011, 68, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Bolcas, P.E.; Brandt, E.B.; Zhang, Z.; Biagini Myers, J.M.; Ruff, B.P.; Khurana Hershey, G.K. Vitamin D supplementation attenuates asthma development following traffic-related particulate matter exposure. J. Allergy Clin. Immunol. 2019, 143, 386–394.e383. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.M.; Wang, C.H.; Lee, M.J.; He, J.R.; Huang, H.Y.; Chao, M.W.; Chung, K.F.; Kuo, H.P. Aryl hydrocarbon receptor activation by diesel exhaust particles mediates epithelium-derived cytokines expression in severe allergic asthma. Allergy 2018, 73, 2192–2204. [Google Scholar] [CrossRef]
- Chapman, K.R.; Mannino, D.M.; Soriano, J.B.; Vermeire, P.A.; Buist, A.S.; Thun, M.J.; Connell, C.; Jemal, A.; Lee, T.A.; Miravitlles, M.; et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur. Respir. J. 2006, 27, 188–207. [Google Scholar] [CrossRef]
- Driscoll, T.; Nelson, D.I.; Steenland, K.; Leigh, J.; Concha-Barrientos, M.; Fingerhut, M.; Pruss-Ustun, A. The global burden of non-malignant respiratory disease due to occupational airborne exposures. Am. J. Ind. Med. 2005, 48, 432–445. [Google Scholar] [CrossRef]
- Balmes, J.R. Occupational contribution to the burden of chronic obstructive pulmonary disease. J. Occup. Environ. Med. 2005, 47, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Viegi, G.; Di Pede, C. Chronic obstructive lung diseases and occupational exposure. Curr. Opin. Allergy Clin. Immunol. 2002, 2, 115–121. [Google Scholar] [CrossRef]
- Burge, P.S. Occupation and chronic obstructive pulmonary disease (COPD). Eur. Respir. J. 1994, 7, 1032–1034. [Google Scholar] [CrossRef]
- Becklake, M.R. Occupational exposures: Evidence for a causal association with chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 1989, 140 Pt 2, S85–S91. [Google Scholar] [CrossRef]
- Blanc, P.D. Occupation and COPD: A brief review. J. Asthma 2012, 49, 2–4. [Google Scholar] [CrossRef]
- Barnes, P.J.; Burney, P.G.J.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F.M. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Prim. 2015, 1, 15076. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.A., 3rd; Ezzati, M.; Dockery, D.W. Fine-particulate air pollution and life expectancy in the United States. N. Engl. J. Med. 2009, 360, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Sydbom, A.; Blomberg, A.; Parnia, S.; Stenfors, N.; Sandstrom, T.; Dahlen, S.E. Health effects of diesel exhaust emissions. Eur. Respir. J. 2001, 17, 733–746. [Google Scholar] [CrossRef]
- Chung, K.F.; Adcock, I.M. Multifaceted mechanisms in COPD: Inflammation, immunity, and tissue repair and destruction. Eur. Respir. J. 2008, 31, 1334–1356. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Afthab, M.; Hambo, S.; Kim, H.; Alhamad, A.; Harb, H. Particulate matter-induced epigenetic modifications and lung complications. Eur. Respir. Rev. 2024, 33, 240129. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, P.; Park, D.; Lee, Y.C. Recent Insights into Particulate Matter (PM(2.5))-Mediated Toxicity in Humans: An Overview. Int. J. Environ. Res. Public Health 2022, 19, 7511. [Google Scholar] [CrossRef]
- Weinmann, S.; Vollmer, W.M.; Breen, V.; Heumann, M.; Hnizdo, E.; Villnave, J.; Doney, B.; Graziani, M.; McBurnie, M.A.; Buist, A.S. COPD and occupational exposures: A case-control study. J. Occup. Environ. Med. 2008, 50, 561–569. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Q.; Krewski, D.; Shi, Y.; Burnett, R.T.; McGrail, K. Influence of relatively low level of particulate ar pollution on hospitalization for COPD in elderly people. Inhal. Toxicol. 2004, 16, 21–25. [Google Scholar] [CrossRef]
- Barnes, P.J. Chronic obstructive pulmonary disease. N. Engl. J. Med. 2000, 343, 269–280. [Google Scholar] [CrossRef]
- Hiura, T.S.; Li, N.; Kaplan, R.; Horwitz, M.; Seagrave, J.C.; Nel, A.E. The role of a mitochondrial pathway in the induction of apoptosis by chemicals extracted from diesel exhaust particles. J. Immunol. 2000, 165, 2703–2711. [Google Scholar] [CrossRef] [PubMed]
- Hiura, T.S.; Kaszubowski, M.P.; Li, N.; Nel, A.E. Chemicals in diesel exhaust particles generate reactive oxygen radicals and induce apoptosis in macrophages. J. Immunol. 1999, 163, 5582–5591. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Liao, B.; Pu, J.; Li, C.; Zheng, M.; Huang, L.; Zhou, Y.; Zhao, D.; Li, B.; Ran, P. Exposure to Ambient Particulate Matter Induced COPD in a Rat Model and a Description of the Underlying Mechanism. Sci. Rep. 2017, 7, 45666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef]
- Bonner, J.C.; Rice, A.B.; Lindroos, P.M.; O’Brien, P.O.; Dreher, K.L.; Rosas, I.; Alfaro-Moreno, E.; Osornio-Vargas, A.R. Induction of the lung myofibroblast PDGF receptor system by urban ambient particles from Mexico City. Am. J. Respir. Cell Mol. Biol. 1998, 19, 672–680. [Google Scholar] [CrossRef]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef]
- Morales-Barcenas, R.; Chirino, Y.I.; Sanchez-Perez, Y.; Osornio-Vargas, A.R.; Melendez-Zajgla, J.; Rosas, I.; Garcia-Cuellar, C.M. Particulate matter (PM(1)(0)) induces metalloprotease activity and invasion in airway epithelial cells. Toxicol. Lett. 2015, 237, 167–173. [Google Scholar] [CrossRef]
- Faber, S.C.; McNabb, N.A.; Ariel, P.; Aungst, E.R.; McCullough, S.D. Exposure Effects Beyond the Epithelial Barrier: Transepithelial Induction of Oxidative Stress by Diesel Exhaust Particulates in Lung Fibroblasts in an Organotypic Human Airway Model. Toxicol. Sci. 2020, 177, 140–155. [Google Scholar] [CrossRef]
- Lee, H.; Hwang-Bo, H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Park, C.; Hong, S.H.; Kim, G.Y.; Song, K.S.; Hyun, J.W.; et al. Diesel particulate matter2.5 promotes epithelial-mesenchymal transition of human retinal pigment epithelial cells via generation of reactive oxygen species. Environ. Pollut. 2020, 262, 114301. [Google Scholar] [CrossRef]
- Saputra, D.; Yoon, J.H.; Park, H.; Heo, Y.; Yang, H.; Lee, E.J.; Lee, S.; Song, C.W.; Lee, K. Inhalation of carbon black nanoparticles aggravates pulmonary inflammation in mice. Toxicol. Res. 2014, 30, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Y.; Young, S.H.; Mercer, R.R.; Barger, M.; Schwegler-Berry, D.; Ma, J.K.; Castranova, V. Interactive effects of cerium oxide and diesel exhaust nanoparticles on inducing pulmonary fibrosis. Toxicol. Appl. Pharmacol. 2014, 278, 135–147. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Ilar, A.; Plato, N.; Lewne, M.; Pershagen, G.; Gustavsson, P. Occupational exposure to diesel motor exhaust and risk of lung cancer by histological subtype: A population-based case-control study in Swedish men. Eur. J. Epidemiol. 2017, 32, 711–719. [Google Scholar] [CrossRef]
- Finkelman, F.D.; Yang, M.; Orekhova, T.; Clyne, E.; Bernstein, J.; Whitekus, M.; Diaz-Sanchez, D.; Morris, S.C. Diesel exhaust particles suppress in vivo IFN-gamma production by inhibiting cytokine effects on NK and NKT cells. J. Immunol. 2004, 172, 3808–3813. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K.; Loridas, S. Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Environ. Res. Public Health 2013, 10, 3886–3907. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Husgafvel-Pursiainen, K. Air pollution and cancer: Biomarker studies in human populations. Carcinogenesis 2005, 26, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Garshick, E.; Laden, F.; Hart, J.E.; Rosner, B.; Davis, M.E.; Eisen, E.A.; Smith, T.J. Lung cancer and vehicle exhaust in trucking industry workers. Environ. Health Perspect. 2008, 116, 1327–1332. [Google Scholar] [CrossRef]
- Garshick, E.; Laden, F.; Hart, J.E.; Davis, M.E.; Eisen, E.A.; Smith, T.J. Lung cancer and elemental carbon exposure in trucking industry workers. Environ. Health Perspect. 2012, 120, 1301–1306. [Google Scholar] [CrossRef]
- Tsoi, C.T.; Tse, L.A. Professional drivers and lung cancer: A systematic review and meta-analysis. Occup. Environ. Med. 2012, 69, 831–836. [Google Scholar] [CrossRef]
- Pierdominici, M.; Maselli, A.; Cecchetti, S.; Tinari, A.; Mastrofrancesco, A.; Alfe, M.; Gargiulo, V.; Beatrice, C.; Di Blasio, G.; Carpinelli, G.; et al. Diesel exhaust particle exposure in vitro impacts T lymphocyte phenotype and function. Part. Fibre Toxicol. 2014, 11, 74. [Google Scholar] [CrossRef]
- Sharma, S.; Singh, N.; Turk, A.A.; Wan, I.; Guttikonda, A.; Dong, J.L.; Zhang, X.; Opyrchal, M. Molecular insights into clinical trials for immune checkpoint inhibitors in colorectal cancer: Unravelling challenges and future directions. World J. Gastroenterol. 2024, 30, 1815–1835. [Google Scholar] [CrossRef]
- Smyth, T.; Jaspers, I. Diesel exhaust particles induce polarization state-dependent functional and transcriptional changes in human monocyte-derived macrophages. Am. J. Physiol. Lung Cell Mol. Physiol. 2024, 326, L83–L97. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, T.; Xiong, Y.; Lv, J.; Cui, X.; He, R. Diesel exhaust particle promotes tumor lung metastasis via the induction of BLT1-mediated neutrophilic lung inflammation. Cytokine 2018, 111, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Dybdahl, M.; Risom, L.; Bornholdt, J.; Autrup, H.; Loft, S.; Wallin, H. Inflammatory and genotoxic effects of diesel particles in vitro and in vivo. Mutat. Res. 2004, 562, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Steiner, S.; Bisig, C.; Petri-Fink, A.; Rothen-Rutishauser, B. Diesel exhaust: Current knowledge of adverse effects and underlying cellular mechanisms. Arch. Toxicol. 2016, 90, 1541–1553. [Google Scholar] [CrossRef]
- Azad, N.; Rojanasakul, Y.; Vallyathan, V. Inflammation and lung cancer: Roles of reactive oxygen/nitrogen species. J. Toxicol. Environ. Health B Crit. Rev. 2008, 11, 1–15. [Google Scholar] [CrossRef]
- Iwai, K.; Adachi, S.; Takahashi, M.; Moller, L.; Udagawa, T.; Mizuno, S.; Sugawara, I. Early oxidative DNA damages and late development of lung cancer in diesel exhaust-exposed rats. Environ. Res. 2000, 84, 255–264. [Google Scholar] [CrossRef]
- Nagy, E.; Zeisig, M.; Kawamura, K.; Hisamatsu, Y.; Sugeta, A.; Adachi, S.; Moller, L. DNA adduct and tumor formations in rats after intratracheal administration of the urban air pollutant 3-nitrobenzanthrone. Carcinogenesis 2005, 26, 1821–1828. [Google Scholar] [CrossRef]
- Rynning, I.; Neca, J.; Vrbova, K.; Libalova, H.; Rossner, P., Jr.; Holme, J.A.; Gutzkow, K.B.; Afanou, A.K.J.; Arnoldussen, Y.J.; Hruba, E.; et al. In Vitro Transformation of Human Bronchial Epithelial Cells by Diesel Exhaust Particles: Gene Expression Profiling and Early Toxic Responses. Toxicol. Sci. 2018, 166, 51–64. [Google Scholar] [CrossRef]
- Ma, C.; Wang, J.; Luo, J. Activation of nuclear factor kappa B by diesel exhaust particles in mouse epidermal cells through phosphatidylinositol 3-kinase/Akt signaling pathway. Biochem. Pharmacol. 2004, 67, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Saeed, A.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).