Abstract
The methods utilized in the drug discovery pipeline routinely combine machine learning and deep learning algorithms to enhance the outputs. The generation of a drug target, through virtual screening and computational analysis of databases used for target discovery, has increased the reliability of the machine learning and deep learning incorporated techniques. Recent technological advances in human immunology have provided improved tools that allow a better understanding of the biological and molecular mechanisms leading to the protective human immune response to pathogens, inspiring new strategies for vaccine design. Immunoinformatics approaches are more beneficial, and thus there is a demand for modern technologies such as reverse vaccinology, structural vaccinology, and system approaches in developing potential vaccine candidates. System theory, defined as a set of machine learning, control theory, and optimization-based methods applied to networked systems, provides a unifying framework for modeling and analyzing biological complexity. In this review, we explore the application of such computational methods at every stage of the therapeutic pipeline, including lead discovery, optimization, and dosing, as well as vaccine target prediction and immunogen design. Here, we summarize the system theoretic methods which provide insights into developed approaches and their applications in rational drug discovery and vaccine formulations. The approaches ranged in the review yield accurate predictions and insights. This review is intended to serve as a resource for researchers seeking to understand, adopt, or build upon system theoretic techniques in drug and vaccine development, offering both conceptual foundations and practical directions.
1. Introduction
Drug discovery is a multidimensional process that requires assessing parameters such as the safety and efficacy of natural and synthetic compounds during candidate selection [1]. Drug discovery is a lengthy and complex process typically divided into four main stages: (i) target identification and validation, (ii) compound screening and lead optimization, (iii) preclinical evaluation, and (iv) clinical trials [2,3,4]. Drug discovery is a multivariable optimization problem that can be performed on supercomputers using a reliable scoring function, which quantifies the binding affinity or inhibitory potential of a drug-like compound [5]. Drug discovery comprises multiple stages, including target identification, lead discovery, lead optimization, ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiling, and clinical trials [6]. Chronic diseases require long-term drug treatment [7,8].
Drug discovery screening is performed through Target Drug Discovery (TDD), Phenotypic Drug Discovery (PDD), Molecular Dynamics (MD) and disease-related molecular networks. Disease-related molecular networks use a network bio-informatics approach to repurpose drugs. Protein networks can be built based on the knowledge of pathways, protein–protein interactions, and graph theory [9]. TDD screening is fully reliant on the drug target validity inside the human body. The targets then undergo a pool of optimization techniques to discover measures for effectiveness, and toxicity. The target candidate with the highest favorable properties becomes the suitable candidate for further clinical trials [10]. However, TDD is not an efficient approach for complex diseases, where the target candidate is difficult to categorize [11].
In comparison to TDD, PDD begins with the examination and analysis of drug candidates to determine the presence, absence or quantity of one or more components in the drug target. Later steps include target identification and validation, followed by optimizing and selecting the target candidate [10]. Some of the gaps that researchers [11] discussed were the non-uniformity in PDD applications in the bio-pharma industry. The MD technique is the interaction of genes involved in the symptoms of a disease [12]. Genomics data, gene expression data, or data directly collected from the scientific literature are some of the methods used to identify disease-related genes. MD simulations are employed to identify potential drug-binding sites on target proteins, estimate binding free energies between targets and drug molecules, and elucidate drug action mechanisms [13,14].
Drug design approaches relying solely on simple protein interactions are insufficient to meet the current clinical safety requirements [15]. Consequently, integrating diverse data types and sources, known as data fusion techniques, which combine structural, genetic, and pharmacological information from the molecular to the organismal level, will be essential for discovering safer and more effective drugs [16]. Drug repurposing (also called repositioning or re-tasking) is a technique for identifying novel usage for approved drugs that are generally not considered for the medical market. The latest era of AI and network medicine proposes applications of information science for specifying diseases, medicines, and recognizing targets with the least amount of error [17].
Vaccination is one of the most powerful and cost-effective tools in modern medicine. Vaccine development is a multidisciplinary process involving a molecular understanding of host–pathogen interactions, antigen selection to elicit an immune response, formulation design, and preclinical and clinical testing to ensure optimal therapeutic efficacy and safety in humans [18]. A resurgence in vaccine research is underway, driven by increasing recognition from healthcare authorities of their public health impact and cost-effectiveness [19]. Vaccination remains the most effective strategy for reducing infectious disease-related morbidity and mortality [20,21]. Vaccines derived from live-attenuated pathogens typically induce strong, long-lasting immunity, though feasibility may be limited by manufacturing or safety concerns [22], as attenuated viruses can eventually revert to a virulent phenotype [23,24]. The DNA vaccine, on the other hand, appears to be a safer alternative because it does not contain any pathogen, but its use is limited because it is effective only for those pathogens that have proteins as immunogens [25]. Ideally, vaccination elicits an immune response equivalent to or better than that of natural infection, providing long-term protection that not only prevents disease but also limits pathogen transmission, contributing to herd immunity [26].
The application of systems biology to vaccinology is beginning to reveal mechanisms by which vaccines elicit immune responses and offers strategies for predicting vaccine immunogenicity [27]. Structural vaccinology has been employed to enhance the biochemical properties of vaccine candidates and may increase the immunogenicity of protein antigens identified through reverse vaccinology [28]. This approach leverages protein structural information to design immunogens and holds potential for developing vaccines against traditionally challenging targets [29].
In this review paper, we are discussing the various system theoretic methods that are involved in drug discovery and vaccine formulation. The review is divided into two parts. In the first part, the system theoretic ways for the drug discovery pipeline are mentioned. These include the methods for target discovery, virtual screening, and drug optimization. In the second part, a review is performed for the system theoretic ways for the formulation of vaccines. These include the methods involved in the reverse, structural, and system vaccinology. Table 1 shows the marketed drugs discovered using system theoretical methods. The preprint of this paper is located at [30].

Table 1.
Marketed drugs developed using system theoretic methods.
2. System Theoretic Ways of Target Discovery
One of the most critical steps in drug development is target identification and validation. A target can refer to various biological entities, including proteins, genes, or RNA. An ideal target should be efficacious, safe, and druggable [41]. Identifying appropriate targets and validating them through mechanism-of-action studies increases the likelihood of success in drug discovery and helps anticipate the potential side effects related to target modulation [41,42]. During the target identification phase, various techniques are employed to detect and isolate targets, characterize their functions, and determine their relevance to disease processes [43,44]. In the target validation phase, to select targets that are most likely to be useful in the development of new drugs, scientists must analyze and compare each drug target to others on the basis of their relationships with a specific disease and their abilities to regulate and influence biological compounds and chemical compounds in the body [43,45,46]. Figure 1 illustrates the different system theoretic ways involved in identifying the targets. In this section, we will discuss the various system theoretic approaches involved in the first step of drug discovery pipeline, i.e., target identification.
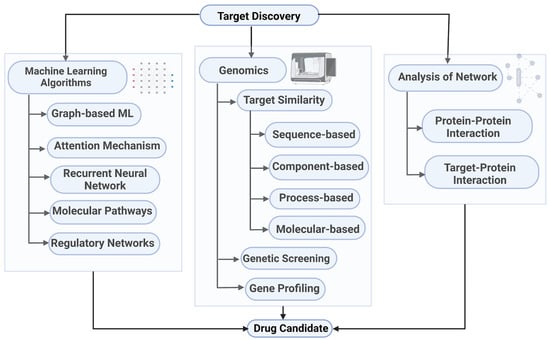
Figure 1.
System theoretic ways of target discovery [47,48,49,50]. Created with BioRender.com.
Graph-based machine learning methods utilize the network’s topological structure to classify node properties, predict edge existence, and identify communities [51]. A graph can be represented as a quadruple , where V is a finite set of nodes, E is a set of edges (which may be directed or undirected, and weighted or unweighted), denotes the set of node features, and denotes the set of edge features [52]. Given a set of n labeled graphs , the goal of graph classification can be to learn a function , where is the space of graphs and is the set of possible graph labels [53]. In node classification, given a graph with known labels , the objective can be to learn a function to predict labels for unlabeled nodes in the graph [54,55]. Link prediction refers to the task of inferring missing or future edges in a graph, which may involve predicting the existence or weight of edges between node pairs [56]. These methods assume that nodes with similar functional or structural roles are more likely to be connected [57]. Gene profiling combines gene expression data, such as mRNA, with chemical analysis to identify targets [58]. This approach assumes that deleting genes encoding a target protein produces the same inhibitory effect as active compounds. Targets are identified by comparing expression profiles, which indicate protein synthesis or expression, with those obtained after compound exposure [59]. Several approaches also compare biological data on regulatory networks, molecular pathways, and cellular phenotypes with corresponding profiles of bioactive molecules and targets to identify common patterns in biological responses and drug activity [60].
Genetic screening is a well-established strategy for identifying drug targets [61,62]. A computational method to predict drug targets estimates the likelihood that two drugs share a common target based on the similarity of their clinical side effects, assuming that similar side effects reflect a shared mechanism of action [63]. Computational approaches for identifying the protein targets of small-molecule drugs are known as compound–protein interaction (CPI) methods [64]. Tsubaki et al. [65] proposed an end-to-end representation learning approach that combines graph neural networks for compounds and convolutional neural networks for proteins. The method integrates the representations of both entities and employs an attention mechanism to enable visualization and interpretability, even with real-valued representations. MDeePred [66] uses a deep neural network to predict binding affinities between small-molecule compounds and target proteins, using amino acid sequences as input. The output is a quantitative prediction of the binding affinity between the input compound and the protein [66].
Zeng et al. [49] developed a network-based deep learning framework, deepDTnet, for identifying the molecular targets of known drugs. This method integrates 15 types of chemical, genomic, phenotypic, and cellular networks to learn low-dimensional, informative vector representations for drugs and targets, capturing biologically and pharmacologically relevant features. Kumari et al. [67] proposed a sequence-based method to distinguish human drug target proteins from non-drug targets using features such as amino acid composition, property group composition, and dipeptide composition. To address class imbalance, SMOTE (Synthetic Minority Over-sampling Technique) was applied to equalize the ratio between drug targets and non-drug targets [68]. Feature selection was performed using the Rotation Forest algorithm and the ReliefF technique [69,70]. Recurrent neural networks with Long Short-Term Memory (LSTM) units have been used to generate large sets of novel molecules with similar physicochemical properties. These models use transfer learning to optimize active molecules toward specific biological targets by iteratively updating a language model and evaluating candidates with a target prediction model (TPM) [71]. DEMETER is an analytical framework designed to separate the on-target and off-target effects of RNAs using a nonlinear regression model based on conditional inference trees and incorporating gene expression, copy number variation, and somatic mutations [72].
3. System Theoretic Ways of Drug Discovery
In drug discovery, the initial and most critical step is identifying appropriate targets, such as genes or proteins, that are implicated in disease pathophysiology, followed by the search for drugs or drug-like molecules capable of modulating these targets [73]. Biomolecular simulations using multiscale models enable the examination of the structural and thermodynamic properties of target proteins across different levels, facilitating the identification of binding sites and the elucidation of drug action mechanisms [74]. This section focuses on various system theoretic approaches applied in the second stage of the drug discovery pipeline, namely, virtual screening.
Drug discovery has traditionally relied on libraries of small molecules to identify therapeutic agents, but new modalities such as genetically encoded cyclic peptide libraries (including phage display [75], mRNA display [76], and split-intein circular ligation of peptides and proteins [77]) are gaining prominence as molecular scaffolds in drug discovery, especially for inhibiting protein–protein interactions [78]. Advances in computational power and techniques in computational chemistry have accelerated the adoption of Computer-Aided Drug Design (CADD), which now constitutes a major component of the drug discovery pipeline [58]. CADD is applied in hit identification, lead discovery, and lead optimization, and is broadly categorized into structure-based and ligand-based approaches [79]. Structure-based CADD utilizes the three-dimensional structure of the target protein to predict binding potential, while ligand-based CADD uses data from known active and inactive compounds to estimate the activity of new candidates [58] (see Figure 2). Wei et al. [80] propose a model named BioKG-CMI, which uses the sequence information of circRNAs and miRNAs to generate spatial proximity, sequence representations from a 12-layer BERT-based Transformer model [81], and a biological knowledge graph, and fuses these features using an AdaBoost classifier to predict potential circRNA–miRNA interactions (CMIs). Liang et al. [82] present MNDCDA, which combines multisource data with graph convolutional network (GCNs) as neighborhood-aware embedding models to capture structural information about circRNAs and diseases, and with MLPs as deep feature projection networks to learn high-order feature interactions and nonlinear relationships for predicting circRNA–disease associations, and is validated using the CircR2Disease dataset [83], where 25 out of 30 predicted pairs have been experimentally validated in wet lab studies.
Virtual screening is used to search chemical libraries for potential drug candidates based on predicted binding sites on target proteins [84,85,86]. The goal is to screen a large set of ligands to identify a smaller subset for purchase and experimental validation [85]. Commonly used virtual screening tools include Gold [87], DOCK [88], Glide [89,90], FlexX [91], FRED [92], and LigandFit [93].

Figure 2.
Methods of virtual screening [94,95,96]: When the three-dimensional structure of the target is unavailable, ligand-based approaches are employed. These strategies use structure–activity data from known active compounds to identify candidates for experimental testing [97]. Ligand-based methods include similarity and substructure searches, quantitative structure–activity relationships (QSAR), pharmacophore modeling, and three-dimensional shape matching [98]. Among these, similarity searching and pharmacophore mapping are widely used [95,99,100].
Figure 2.
Methods of virtual screening [94,95,96]: When the three-dimensional structure of the target is unavailable, ligand-based approaches are employed. These strategies use structure–activity data from known active compounds to identify candidates for experimental testing [97]. Ligand-based methods include similarity and substructure searches, quantitative structure–activity relationships (QSAR), pharmacophore modeling, and three-dimensional shape matching [98]. Among these, similarity searching and pharmacophore mapping are widely used [95,99,100].

Similarity-searching algorithms are used to identify and annotate DNA or protein targets. Based on genomic data, proteins can be inferred from their DNA sequences through similarity search techniques [101]. A pharmacophore represents the ensemble of electronic features required for optimal interaction with a specific target protein and for modulating its biological activity [95,102]. Structure-based approaches utilize the three-dimensional structure of a biological target to dock candidate molecules and rank them according to predicted binding affinity or complementarity to the binding site [103]. Access to the target’s 3D structure facilitates virtual screening through molecular docking and scoring methods [96]. Molecular docking involves two main steps: sampling and scoring. Sampling searches for the optimal ligand pose within the receptor binding site, while scoring evaluates this pose by estimating the binding affinity [96]. Combining multiple computational approaches is often advantageous for drug design [96].
Machine learning (ML) is increasingly used in early-stage drug discovery, driven by the growing availability of relevant experimental data [104]. These datasets include bioactivity measurements of chemically characterized molecules against non-molecular targets, as well as binding affinities against molecular targets [104]. This trend has been supported by the development of community resources such as ChEMBL [105], PubChem [106], and PDBbind [107], which curate and enable the reuse of such datasets for predictive modeling. Another contributing factor is the widespread availability of high-quality, well-documented implementations of various ML algorithms, including approaches such as deep learning [108] and conformal prediction [109].
Greedy network cluster forecasts disease–drug relationships in drug–disease networks and protein interactions in protein–protein networks [110]. In order to project the drug–target interactions for detecting and discovering new drugs, researchers utilized a representation method named large-scale information embedding (LINE). This method assisted them in identifying the behavioral details, i.e., the association of drug nodes with protein nodes inside the network. Later, the amalgamation of these details represented the drug–target interaction pairs. Finally, a random forest classification (RF) method was utilized for training and projection [111]. Costa et al. [112] developed a decision tree-based meta-classifier trained on datasets comprising morbid and druggable genes, network topological features, and tissue expression profiles.
The Naive Bayesian classifier estimates the probability of biological activity based on molecular descriptors by calculating the product of the probabilities of individual fragments present in active molecules; the same process is applied for predicting inactivity [113]. One example of this approach is the Prediction of Activity Spectra for Substances (PASS) program, which simultaneously predicts hundreds of biological activity types for any drug-like compound [114]. PASS uses a variant of the Naive Bayes algorithm to estimate drug activity from query structures based on a prior probability distribution [15]. The predictions are derived from structure–activity relationships in a training set of over 30,000 known biologically active compounds [114]. DigSee employs a Bayesian classifier to identify genes and diseases, extract biological events between them, and rank supporting evidence sentences [115]. Ryu et al. [116] showed that Bayesian inference enables more reliable predictions with quantitative uncertainty estimates. They developed a Bayesian GCN for molecular property prediction, demonstrating improved performance in classifying bioactivity and toxicity through predictive uncertainty. Liang et al. [117] proposed a Bayesian neural network (BNN) to identify drug-sensitive genes and predict molecular inhibitory activity using the ChEMBL dataset. Beker et al. [118] applied BNNs to assess drug-likeness, showing that the Bayesian error distribution across classifiers can achieve 93 percent accuracy in distinguishing drug-like from non-drug-like compounds. Rongting Yue and Abhishek Dutta [119] used a graph convolutional network on a drug–target interaction network, with edges of low sensitivity removed, to repurpose drugs for Zika virus and COVID-19 (Betamethasone phosphate and Bizelesin for Zika virus, and Chloroquine, Heparin Disaccharide, and Resveratrol for COVID-19). Abhishek Dutta [120] used a fully connected deep learning network to predict the mechanism of action of novel drugs given in vitro gene expression and cell viability data. Hsiao et al. [121] applied a matrix-based backpropagation technique for graph convolutional networks to a link prediction task on a reduced drug–drug interaction network of COVID-19, showing that the output sensitivity with respect to the input feature is lower for distant nodes than for those that are closer to each other.
Quantitative structure–activity relationship (QSAR) modeling is an effective approach for investigating and leveraging the relationship between chemical structure and biological activity in the development of novel drug candidates [122]. Ramsundar et al. [123] analyzed the potential of deep neural networks to replace random forest (RF) models without compromising predictive performance. DeepChem was utilized to test the performance of two alternative deep learning architectures: multitask networks and progressive networks. The simple multitask deep architecture remained as a robust deep architecture for QSAR datasets and showed performance boosts over random forest methods. Preuer et al. [124] investigated the ability of deep neural networks (DNNs) to handle multimodal inputs for predicting drug combination synergy effects, to evaluate generative models for molecules, and to identify indicative substructures in QSAR predictions. For predicting the effects of drug synergy, descriptors of both drugs and genetic features were described. A new evaluation metric was proposed to compare the generated molecules to real-world molecules based on a chemically and biologically grounded representation using DNN for drug discovery.
BeFree is a text-mining system that applies natural language processing (NLP) methods to identify drug–disease, gene–disease and target–drug associations. Automated text-mining approaches were aimed at finding relationships between biomedical entities, with a special focus on genes and their associated diseases [125]. Olivecrona et al. [126] used RNNs to expand the chemical space by fine-tuning a sequence-based generative model to design compounds with improved solubility, pharmacokinetic properties, and bioactivity. The method fine-tunes an RNN pre-trained on the ChEMBL database using a user-defined scoring function to guide the generation of desirable compounds. Popova et al. [127] introduced ReLeaSE (Reinforcement Learning for Structural Evolution), a strategy for de novo molecular design. ReLeaSE consists of two deep neural networks: a generative model that produces chemically valid molecules and a predictive model that evaluates the output. The generative model functions as the agent, while the predictive model evaluates each generated molecule and provides feedback by assigning rewards or penalties.
Zhavoronkov et al. [128] developed Generative Tensorial Reinforcement Learning (GENTRL), a machine learning method for de novo drug design that prioritizes synthetic feasibility, effectiveness against a biological target, and novelty relative to existing molecules in the literature and patent space. Lin et al. [129] proposed DR-A (Dimensionality Reduction with Adversarial Variational Autoencoder), a data-driven method for dimensionality reduction based on a variant of generative adversarial networks. Wu and Wei [130] introduced the Element Specific Topological Descriptor (ESTD) for predicting small molecule toxicity. They enhanced ESTDs with physical descriptors derived from established physical models and combined them with advanced machine learning algorithms, including two deep neural networks and two ensemble methods: random forest (RF) and gradient boosting decision tree (GBDT). Yang et al. [131] developed a multitask framework called Macau for large-scale drug screening and the interpretation of drug–cell line interactions. Using Bayesian multitask multi-relation learning, they modeled interactions between drug targets and signaling pathway activation, with gene expression data as molecular inputs and nominal drug targets as drug inputs. This approach aimed to uncover the drug mechanisms of action through the analysis of target–pathway relationships.
Sanchez-Lengeling et al. [132] developed the Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC), an extension of the Objective-Reinforced Generative Adversarial Networks (ORGANs) framework. This method integrates a generative adversarial network (GAN), which produces chemically plausible and diverse molecular structures, with reinforcement learning (RL), which biases the generation toward compounds exhibiting desired chemical properties. Putin et al. [133] introduced a deep neural network architecture named the Reinforced Adversarial Neural Computer (RANC) for the de novo design of small-molecule organic compounds. RANC leverages a differentiable neural computer (DNC) to generate molecules consistent with the distribution of chemical descriptors and SMILES string lengths in the training dataset, using both GAN and RL components. Prykhodko et al. [134] proposed LatentGAN, a deep learning model that combines an autoencoder with a GAN for de novo molecular generation. LatentGAN is capable of producing both random drug-like compounds and target-biased molecules, and sampling from the trained model yields a significant proportion of novel structures.
Large language models (LLMs), including specialized architectures such as Geneformer [135] and ESM [136], as well as general-purpose models like GPT-4, have been increasingly adopted to support various stages of drug discovery, from disease mechanism analysis and target identification to molecular generation and clinical trial optimization [137]. For instance, SpatialPPIv2 [138] combines pretrained protein language models (e.g., ProtT5-XL-UniRef50 [139], ProtBert [139], and ESM-2 [140]) with graph attention networks to accurately predict protein–protein interactions using both sequence and structural features, outperforming prior methods like FoldDock [141] and Struct2Graph [142] across multiple benchmarks. Likewise, Peng et al. [143] enhanced effector protein prediction by integrating ESM-2 [140] embeddings with diverse biological features such as structural tokens, functional annotations, and omics-derived profiles using a contrastive learning framework. Contrastive Learning of Language Embedding and Biological Features (CLEF) achieves state-of-the-art performance in identifying type III, IV, and VI secreted effectors across multiple pathogens and facilitates the discovery of virulence factors and protein–protein interactions, representing a multimodal System Theoretic methodology bridging experimental and computational domains. Similarly, FusOn-pLM [144], a fusion oncoprotein-specific language model fine-tuned from ESM-2 [140] on a curated database of over 44,000 fusion sequences (FusOn-DB), employs cosine-scheduled masked language modeling to generate biologically meaningful embeddings that surpass baseline methods in predicting puncta formation, subcellular localization, intrinsic disorder, and drug resistance mutations, thereby supporting fusion-targeted therapeutic development within a System Theoretic framework.
3.1. Software Utilized in Drug Discovery
The most commonly used open-source docking software is developed to address the flexibilities of proteins, larger percentages of these programs do not factor in full receptor flexibilities [145]. DOCK software tools are used for many features like binding modes prediction protein complexes, search databases of ligands, search databases of bind protein–ligands, check protein–protein, and protein–DNA complexes [146,147]. AutoDock is a suite of free open-source software for the computational docking and virtual screening of small molecules to macro-molecular receptors [85]. It is a beneficial docking software, where the docking score of a known 3D structure with a target protein can be obtained in terms of affinity. This docking score gives insight regarding how a new molecule binds to the active site of the target [5,148,149,150]. AutoDock Vina is a highly optimized tool for molecular docking, offering fast and effective performance with well-validated default settings [151]. It is suitable for most systems, while the original AutoDock is preferred for cases requiring methodological customization [85]. The AutoDock suite, including its source code, is freely available and widely used in drug discovery and related research [85]. Table 2 provides the details on different computational software tools.
Table 2.
Public online drug–target interaction software (accessed on 16 June 2025).
3.2. Databases Utilized in Drug Discovery
Predictive computational software has contributed to the identification of molecular targets. In the future, the continued use of computational tools and databases for modeling molecular interactions and predicting key features and parameters will support the development of promising drug candidates [1].
Presently, there are a number of public online drug–target interaction databases, like DrugBank [158], STITCH [159], KEGG [160] and ChEMBL [161], that all gather vital information about drugs and their interacting targets. These databases support the development of new methods for studying drug–target interactions, and many computational models rely on known interactions within these resources to predict novel drug–target associations [162]. PubChem serves as a major repository of published biomedical data, and its mining can aid in identifying potential targets for various diseases [163]. Once a suitable target has been identified and validated, the subsequent step is to identify drugs or drug-like molecules that can interact with the target to produce the desired therapeutic effect [164].
The Library of Integrated Network-based Cellular Signatures (LINCS) L1000 includes L1000CDS2, a data-driven, open-access search engine that identifies drugs capable of reversing the expression of differentially expressed genes, making it a valuable resource for drug discovery [165,166]. The Protein Data Bank (PDB) is also widely used for analyzing protein–ligand interactions to identify potential inhibitors of target proteins [167,168]. Table 3 provides the details on different databases.
Table 3.
Public online drug–target interaction databases (accessed on 16 June 2025).
3.3. Case Studies of System Theoretic Ways for the Discovery of Small-Molecule Drugs
In [187], RI-962 was identified as a potent and selective receptor interacting protein kinase 1 (RIPK1) inhibitor using a conditional recurrent neural network architecture (cRNN) with Long Short-Term Memory (LSTM), pretrained on approximately 16 million molecules from the ZINC12 database [188] and fine-tuned on 1030 RIPK1-targeted compounds; from 79,323 generated molecules, a three-step virtual screening pipeline comprising Murcko scaffold and substructure exclusion based on known RIPK1 inhibitors, drug-likeness filtering, and pharmacophore-based screening yielded 23,925 candidates, from which eight synthetically accessible molecules were selected for chemical synthesis and bioactivity evaluation, with RI-962 demonstrating the highest inhibitory activity against RIPK1 with an IC50 of 35.0 nM.
In [189], Ribociclib, topiroxostat, amodiaquine, and gefitinib were identified as potential JAK2 inhibitors using the GraphConvMol model from the DeepChem library, which was trained on the JAK2 dataset from the DUD-E database to distinguish active compounds from decoys based on SMILES representations; following virtual screening, the top 20 predicted FDA-approved drugs were subjected to molecular docking using tofacitinib as a reference inhibitor, and their binding interactions with JAK2 active site residues were further validated through Discovery Studio and UCSF Chimera. Experimental JAK2 kinase assays demonstrated that all four candidate drugs exhibited significant enzymatic inhibition at 25 nM, comparable to the known IC50 of tofacitinib, suggesting that these previously unreported drugs may serve as novel JAK2 inhibitors.
4. System Theoretic Ways of Drug Optimization
The discovery of drug candidates typically begins with target identification and validation [190]. Confirmed hits exhibiting concentration-dependent activity are validated by analyzing their structures and clustering patterns. Top-ranked clusters then undergo hit-to-lead optimization to identify leads suitable as starting points for further development [191]. Once the hit compounds are selected, they enter an optimization phase aimed at generating a refined set of improved candidates, referred to as leads [58]. Drug-like hits derived from high throughput screening provide good starting points for optimization [192]. High-throughput screening used to identify lead candidates for drug development usually yields compounds with binding affinities to their intended targets [193]. In this section, we will discuss the various system theoretic approaches involved in the last step of the drug discovery pipeline, i.e., optimizing the leads derived from the previous virtual screening step. Figure 3 describes the workflow of the drug optimization process through ML predictive models.
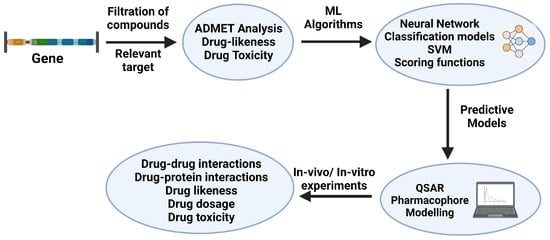
Figure 3.
Drug optimization workflow (adapted from [3,194,195,196]). The filtration of compounds of a target is performed through various ML algorithms and scoring functions for the drug likeness, toxicity, and ADMET (absorption, distribution, metabolism, excretion, and toxicity) predicting tools. Created with BioRender.com.
Olivecrona et al. defined an agent in the Markov decision process that can interact with an incomplete representation of the environment [126]. It can be optimized using an inverse QSAR, which aims to map a favorable region in terms of predicted activity to the corresponding molecular structures [197,198]. Multitask DNNs are employed in lead identification and optimization, as they can integrate diverse biological data through multiple output nodes [199]. Jiménez et al. [200] proposed KDEEP, a 3D convolutional neural network designed to predict binding affinities. This method uses a 3D representation of proteins and ligands based on van der Waals radii, with each atom assigned to one of eight pharmacophoric-like property channels, including hydrophobic, hydrogen bond donor or acceptor, aromatic, ionic (positive or negative), metallic, and excluded volume. Wei et al. [201] introduced TopologyNet, a multichannel topological neural network that combines element-specific persistent homology (ESPH) with deep convolutional networks to predict protein–ligand binding affinities and protein stability upon mutation. ESPH encodes 3D molecular structures into 1D topological invariants while preserving biological information in a multichannel image-like format. Feinberg et al. [202] developed PotentialNet, a graph neural network that separates the derivation of bonded atom features from inter-molecular information propagation. The model comprises three stages: covalent-only propagation, combined covalent and non-covalent propagation, and ligand-level graph processing. Stepniewska-Dziubinska et al. [203] introduced Pafnucy, a deep neural network used in structure-based ligand discovery. It functions as a scoring model for virtual screening and as an affinity predictor, and can also be applied during docking to optimize ligand poses.
The Partial Least Squares Regression (PLSR) modeling approach was utilized to handle large independent variables with a minimal sample size. A pathway-based filtering step was used to reduce the gene signature set without compromising model performance, resulting in high accuracy and strong drug specificity [204]. Trott et al. [151] introduced AutoDock Vina, a molecular docking and virtual screening tool that employs an advanced gradient-based local optimization algorithm. The gradient computation provides directional guidance from a single function evaluation, enhancing optimization efficiency. Ashtawy et al. [205] reviewed the protein-based drug design, where ligands are positioned within the receptor’s active site. In molecular docking, multiple binding poses are assessed using a scoring function (SF), which quantitatively estimates the binding free energy of each pose. The output is a ranked list of ligands based on predicted binding scores. Random forest, geometrical features, and the diverse core set of 2007 PDBbind were considered to assess the performance of ML SFs on targets. Ballester et al. [206] proposed a scoring function (RF-Score) that utilizes random forest to implicitly capture binding effects. The RF-Score is particularly useful as a re-scoring method and can support virtual screening and lead optimization.
Wang et al. [207] proposed a scoring function called Feature Functional Theory–Binding Predictor (FFT-BP), which is based on the principles of representability, feature–function relationships, and similarity. FFT-BP uses some physical features for ranking the nearest neighbors via microscopic features. Nguyen et al. [208] introduced the Algebraic Graph Learning Score (AGL-Score), which encodes high-dimensional physical and biological information into low-dimensional representations. The AGL-Score was validated for scoring, ranking, docking, and screening power using benchmark datasets including CASF-2007, CASF-2013, and CASF-2016. Cang et al. [209] examined the impact of featurization, the process of converting 3D biomolecular structures into features, on scoring function performance. They applied topological descriptors and machine learning models such as k-nearest neighbors, ensemble trees, and deep neural networks for protein–ligand binding prediction and small molecule screening. Nguyen and Wei [210] introduced Differential Geometry-based Geometric Learning (DG-GL), a method that encodes chemical, biological, and physical information into low-dimensional manifolds and applies differential geometry to generate latent representations. Boyles et al. [211] demonstrated that adding diverse, readily computable ligand-based features improves the ranking accuracy of machine learning scoring functions in predicting protein–ligand binding affinities.
5. System Theoretic Methods in Vaccine Formulation
A vaccine is a biological product that can be used to safely induce an immune response, conferring protection against infection and/or disease upon subsequent exposure to a pathogen [212]. Vaccines are being developed and approved for various pathogens, with ongoing studies aimed at improving efficacy through the testing of novel adjuvants and the rational identification of antigen formulations and pathogen components [213,214,215]. Improved outcomes have also been achieved by modifying delivery strategies [216]. The primary goal of vaccination is to confer long-term protection to individuals susceptible to disease [27]. Computational tools (see Table 4) are increasingly used to design vaccines for emerging diseases. The incorporation of mass spectrometry-derived eluted ligand data alongside traditional binding affinity data has significantly enhanced their predictive power, particularly in identifying naturally processed and presented peptides [217]. Like many machine learning models, NetMHCpan’s performance can be affected by biases present in its training data [218]. Importantly, NetMHCpan does not account for hydrophobicity, which is a key biochemical factor in peptide binding, resulting in cases where it predicts highly hydrophobic peptides as strong binders, even though biochemical evidence suggests otherwise [219]. Vaccine development requires a solid understanding of immunology and the integration of diverse disciplines, including cell biology, physical chemistry, and computational science [220]. Conventional approaches to vaccine development are time-consuming, often identify only highly expressed antigens that may not confer protection, and are limited when the pathogen cannot be cultured in vitro.
Reverse vaccinology addresses these challenges by leveraging the pathogen’s genome sequence to identify potential antigens [221]. Structural vaccinology enhances the biochemical properties of vaccine candidates and can improve the immunogenicity of protein antigens identified through reverse vaccinology. Structural insight is particularly valuable for designing vaccines against viral pathogens [28]. Systems biology aims to integrate and analyze data on the components of biological systems (e.g., genes, proteins, and cells) to model and predict system behavior. In vaccinology, systems biology is applied to understand the mechanisms by which vaccines induce immunity and to predict vaccine immunogenicity and efficacy [27].
5.1. Reverse Vaccinology
The reverse vaccinology approach begins with the genomic sequence of a pathogen and uses computational analysis to predict antigens that are most likely to serve as vaccine candidates [221] (see Figure 4). This section discusses system theoretic methods applied in vaccine formulation using reverse vaccinology techniques.
Table 4.
Software/tools used for vaccine formulation (accessed on 16 June 2025).
Table 4.
Software/tools used for vaccine formulation (accessed on 16 June 2025).
| Resources | Vaccinology | Description | URL | Authors |
|---|---|---|---|---|
| VaxiJen v3.0 | Reverse | Web-server which is an alignment independent prediction of protective antigens | https://www.ddg-pharmfac.net/vaxijen3/home/ | [222] |
| VacSol | Reverse | Software which automates vaccine candidate prediction process for the identification vaccine candidates against the proteome of bacterial pathogens | https://sourceforge.net/projects/vacsol/ | [223] |
| Vacceed | Reverse | Configurable and scalable framework designed to automate the process of high-throughput in silico vaccine candidate discovery for pathogens | https://github.com/sgoodswe/vacceed/releases | [224] |
| Protegen | System | Web-based central database and analysis system that curates, stores and analyzes protective antigens | https://www.violinet.org/protegen | [225] |
| CoronaVIR | System | Web-based resource developed to maintain predicted and existing information on coronavirus SARS-CoV-2 | https://webs.iiitd.edu.in/raghava/coronavir/ | [226] |
| AntigenDB | Structural | Database entry contains information regarding the sequence, structure, origin, etc. of an antigen available | https://webs.iiitd.edu.in/raghava/antigendb/ | [227] |
| DBCOVP | Structural | Manually-curated, web-based resource to provide extensive information on the complete repertoire of structural virulent glycoproteins from coronavirus genome | http://covp.immt.res.in/ | [18,228,229] |
| COVIEdb | Structural | Database provides details on potential B/T-cell epitopes for SARS-CoV, SARS-CoV-2, and MERS-CoV to provide potential targets for coronaviruses vaccine development | https://pgx.zju.edu.cn/coviedb/ | [229,230] |
5.1.1. Computational Tool-Based Frameworks for Reverse Vaccinology
Bowman et al. [231] developed a training dataset consisting of 136 bacterial protective antigens and 136 non-antigens. This dataset was used to train support vector machine (SVM) classifiers to distinguish protective antigens from non-antigens, evaluated using cross-validation. Ong et al. [232] introduced Vaxign-ML, a supervised ML classifier for predicting bacterial protective antigens (BPAs). To determine the most effective ML model, five algorithms were tested using the biological and physicochemical features extracted from curated training data. Performance was evaluated using nested five-fold cross-validation and leave-one-pathogen-out validation to ensure unbiased assessment and the ability to predict candidates for emerging pathogens. Heinson et al. [233] enhanced this approach by implementing nested cross-validation, removing bias in negative data selection, expanding the training dataset by approximately one-third, and incorporating updated protein annotation tools. Their SVM classifier demonstrated the ability to detect protection-related signals by comparing curated BPA data with randomly permuted datasets. Ong et al. [234] presented Vaxign2, a comprehensive web-based tool that integrates both filtering-based and ML-based prediction frameworks. Vaxign2 also supports post-prediction analyses, including epitope prediction, population coverage assessment, and functional annotation. Doytchinova and Flower [222] developed VaxiJen, an alignment-independent web server that classifies antigens using the auto cross-covariance (ACC) transformation of protein sequences into feature vectors representing principal amino acid properties. Basso et al. [235] applied ARACNe (Algorithm for the Reconstruction of Accurate Cellular Networks) to reverse engineer gene expression data. ARACNe identifies statistically significant gene–gene co-regulation through mutual information and removes indirect interactions using the data-processing inequality from information theory. Dalsass et al. [236] conducted a comparative study of six reverse vaccinology tools (NERVE, Vaxign, VaxiJen, Jenner-Predict, Bowman–Heinson, and VacSol) using a curated set of bacterial protective antigens from 11 pathogens.

Figure 4.
Reverse vaccinology [237,238,239,240]. The approach of reverse vaccinology is described as the screening and detection of suitable candidates of proteins essential for vaccination that are performed through various machine learning classification approaches. The prioritized candidates are then utilized in designing and formulating vaccines, which are later tested in the clinical trials phase. Created with BioRender.com.
Figure 4.
Reverse vaccinology [237,238,239,240]. The approach of reverse vaccinology is described as the screening and detection of suitable candidates of proteins essential for vaccination that are performed through various machine learning classification approaches. The prioritized candidates are then utilized in designing and formulating vaccines, which are later tested in the clinical trials phase. Created with BioRender.com.

5.1.2. Computational Tool-Based Frameworks for Reverse Vaccinology of Bacterial Vaccines
Vivona et al. [241] introduced NERVE (New Enhanced Reverse Vaccinology Environment), a fully automated system for identifying bacterial vaccine candidates from completely sequenced genomes. NERVE ranks candidates and stores all proteomic data in a structured database for downstream analysis. He et al. [240] enhanced NERVE by introducing Vaxign, a web-accessible tool that includes MHC class I and II binding predictions. Rizwan et al. [223] developed VacSol, a scalable and configurable tool that automates in silico vaccine candidate identification from bacterial proteomes. Its outputs are available in five formats based on the input proteome sequence. Jaiswal et al. [242] created Jenner-Predict, a tool for predicting protein vaccine candidates by targeting host–pathogen interactions and known functional protein domains. It prioritizes predicted candidates for downstream experimental validation. Masignani et al. [243] analyzed the genome of Neisseria meningitidis serogroup B (MenB) to identify surface-exposed antigens suitable for vaccine design. Maiden et al. [244] highlighted the transformative role of genome sequencing in meningococcal vaccine development, emphasizing the genetic and antigenic diversity revealed by sequence-based studies. Bianconi et al. [245] used reverse vaccinology and bioinformatic tools to analyze the genome of Pseudomonas aeruginosa, identifying 52 candidate antigens, of which 30 were successfully expressed. Nagpal et al. [246] employed immunoinformatics to screen antigenic proteins from 14 pathogenic bacteria. To prevent self-reactivity, predicted epitopes were mapped to the human proteome, yielding 21 virulence-associated, essential proteins across five species as optimal vaccine targets.
5.1.3. Case Studies: Reverse Vaccinology for Acinetobacter baumannii
Acinetobacter baumannii is a Gram-negative, multidrug-resistant opportunistic pathogen responsible for ventilator-associated pneumonia, bloodstream infections, and wound sepsis, particularly in immunocompromised and hospitalized individuals [247]. Moriel et al. [248] employed an integrated reverse vaccinology and proteomics strategy to identify subunit vaccine candidates against multidrug-resistant A. baumannii by combining in silico prediction tools (PSORTb, SignalP, PHYRE2, and Pfam) with the experimental profiling of outer membrane vesicles and secretomes via HPLC-MS/MS, yielding 42 conserved, surface-exposed, and structurally soluble antigens, primarily lipoproteins, adhesins, and toxins, suitable for inclusion in a multi-component subunit vaccine to combat hospital-acquired infections caused by this pathogen. Due to its remarkable ability to persist in harsh environmental conditions and rapidly acquire antibiotic resistance determinants, Acinetobacter baumannii was classified by the World Health Organization in 2017 as a “critical priority pathogen” requiring urgent development of new therapeutic interventions [249]. Ahmad and Azam [250] introduced a virulome-based reverse vaccinology framework to identify conserved, non-allergenic 9-mer epitopes for a peptide-based subunit vaccine targeting multidrug-resistant Acinetobacter baumannii, integrating subcellular localization tools (PSORTb, CELLO, and CELLO2GO), epitope prediction platforms (VaxiJen, ProPred I/II, MHCPred, and VirulentPred), allergenicity and conservation screening (SORTALLER and CLC Viewer), 3D modeling (I-TASSER, ModWeb, Phyre2, and Swiss-Model), and docking simulations with DRB1*0101 using GalaxyPepDock and UCSF Chimera, resulting in the identification of epitopes “FYLNDQPVS” (EpsA) and “LQNNTRRMK” (CsuB), which exhibited high MHC affinity, structural stability, and broad conservation across 34 strains. Shahid et al. [251] applied reverse vaccinology to design a chimeric subunit vaccine by performing a pan-genome analysis of 246 clinical isolates to identify the conserved core proteins, followed by subtractive proteomics and virulence filtering, using tools such as PSORTb (for subcellular localization), VaxiJen and ANTIGENpro (for antigenicity), and IEDB (for B- and T-cell epitope prediction) to select five antigenic targets, with final multi-epitope constructs modeled using PEP-FOLD, refined via molecular docking with ClusPro, and evaluated for binding energy using MM/GBSA and immunogenicity using the C-ImmSim simulator. Beiranvand et al. [252] adopted a genomics-driven reverse vaccinology approach to identify B-cell-targeted subunit vaccine candidates against various Acinetobacter baumannii serotypes by incorporating PSORTb (for protein localization), Vaxign and VaxiJen (for antigenicity screening), ccSOL omics and Protein-sol (for solubility prediction), IEDB and BepiPred-2.0 (for B-cell epitope prediction), and PRED-TMBB (for outer membrane topology), resulting in the identification of five highly conserved, surface-exposed proteins (Pfsr, LptE, OmpH, CarO, and FimF) as promising subunit vaccine targets, with LptE exhibiting the highest average antigenicity score (1.043) and epitope surface accessibility. Xu et al. [253] proposed a novel multi-target mRNA-based vaccine against Acinetobacter baumannii by integrating conserved antigens with CTL, HTL, and LBL epitopes into three candidate constructs using adjuvants (CTB, RS09, and -defensin 3), employing a system theoretic pipeline that utilized NetCTL, VaxiJen, AllerTOP, SignalP, PSIPRED, RNAfold, and GROMACS for epitope prediction, structure modeling, and stability validation, with vaccine–receptor interactions assessed via HADDOCK, binding energies calculated using MM-PBSA, and immune simulations performed with C-ImmSim confirming the robust activation of adaptive and memory responses.
5.2. Structural Vaccinology
Structural vaccinology aims to selectively present the conserved determinants of complex and variable antigens. Distinguishing antigenic structures that elicit protective versus disease-enhancing immune responses is critical to avoid vaccine-mediated disease exacerbation (see Figure 5). By engineering structurally stable antigens suitable for combination vaccines, immunization regimens can be simplified [254]. Docking-based virtual screening techniques can also identify novel compounds from collections of approved or clinical-stage drugs. Structural vaccinology incorporates methods such as X-ray crystallography, nuclear magnetic resonance (NMR), and electron microscopy (EM) to resolve antibody–antigen complexes [255]. This section focuses on system theoretic approaches within structural vaccinology for vaccine design.
5.2.1. Antigen Identification and Structural Methods
Khan et al. [256] described a combined immunoinformatics and molecular strategy for vaccine development. Publicly available bioinformatics tools were used to identify pathogen peptides and reduce the list of candidate epitopes, which then served as input for molecular experiments validating their functional roles. Yoder and Dormitzer [257] identified the VP5* antigen domain as an autonomously folding unit, noting that its solubility when expressed independently makes it a promising vaccine component. Optimizing vaccine antigens requires understanding both the structural determinants of immunogenicity and their biochemical stability.

Figure 5.
Structural vaccinology [220,258,259,260]. The structural vaccinology approach provides drug repositioning candidates and targets for further in vitro and in vivo studies and trials. Computational drug repurposing is an effective approach to identify novel drug–target interactions using the drugs already known to be safe, which provides the advantages of significantly reducing the time for drug development and a reduced failure rate. Created with BioRender.com.
Figure 5.
Structural vaccinology [220,258,259,260]. The structural vaccinology approach provides drug repositioning candidates and targets for further in vitro and in vivo studies and trials. Computational drug repurposing is an effective approach to identify novel drug–target interactions using the drugs already known to be safe, which provides the advantages of significantly reducing the time for drug development and a reduced failure rate. Created with BioRender.com.

5.2.2. Epitope Prediction and Mapping Approaches
Saha and Raghava [261] developed an artificial neural network model for predicting continuous B-cell epitopes from protein sequences. The model, trained on 700 experimentally validated epitopes and 700 random peptides, achieved 66% accuracy—comparable to its performance on an independent set of 187 epitopes excluded from training [262]. Andersen et al. [263] proposed DiscoTope, which combines hydrophilicity, amino acid composition, and relative solvent accessibility to predict discontinuous epitopes. DiscoTope was trained on 76 X-ray structures of antibody–protein complexes. EL-Manzalawy et al. [264] explored two machine learning approaches for predicting linear B-cell epitopes of variable length. The first used sequence kernels to score similarity between sequences, while the second mapped sequences to fixed-length feature vectors. They introduced a subsequence kernel-based method to improve the prediction of flexible-length epitopes. DeGoot et al. [265] presented a transallelic prediction model for peptide–MHC-II binding affinity, combining peptide sequences with MHC structural data.
The Pepitope server [266] is a web-based tool for predicting discontinuous epitopes based on affinity-selected peptide sequences. It assumes that selected peptides mimic the genuine epitope in physicochemical properties and spatial arrangement. If the antigen’s 3D structure is known, these peptides can be computationally mapped to the corresponding epitope. Mayrose et al. [267] introduced PepSurf, an algorithm for aligning affinity-selected peptides onto the surface graph of a known antigen structure. Each peptide is mapped to optimal paths in the surface graph, followed by clustering to infer the most likely epitope region.
5.2.3. Antigen–Antibody Interaction Analysis
Kringelum et al. [268] developed a framework to characterize antigen–antibody interfaces, focusing on the epitope region. This framework enabled the quantitative analysis of epitope shape, directionality, and amino acid composition relative to the rest of the antigen and antibody surfaces.
Jones and Thornton [269] proposed a scoring method that predicts protein–protein interaction sites by assigning probabilities to surface patches. Their method achieved successful predictions for 66% of the evaluated antigen structures.
Ofran et al. [270] developed PEASE, a web server that predicts antibody-specific epitopes based on antibody sequence. The predictions are presented at both the residue and patch levels, with user-tunable precision–recall trade-offs. The output is available in text, HTML, and graphical formats, and can be visualized on 3D antigen structures.
Sela-Culang et al. [271] used antibody sequence data to identify discontinuous epitopes. Their method leverages residue-pairing preferences and structural interface features. Validation was performed through experimental epitope mapping against previously uncharacterized antigens, confirming high predictive performance and reducing methodological bias through complementary approaches.
Simek et al. [272] screened HIV-1-infected individuals from diverse populations to identify elite neutralizers. Using linear regression models, they selected a reduced panel and identified 1% as elite neutralizers—an essential resource for isolating broadly neutralizing antibodies for HIV-1 vaccine design.
Duhovny et al. [273] developed a protein–protein docking algorithm inspired by object recognition and image segmentation in computer vision. It evaluates docking models based on local shape features and assigns scores to identify the most plausible antibody–antigen complex. The algorithm performs better in “bound” docking (where antibody and antigen originate from the same complex) than in “unbound” settings.
5.2.4. Case Study: RSV Subunit Vaccine via Structure-Guided Design
The development of a prefusion-stabilized subunit vaccine for Respiratory Syncytial Virus (RSV) exemplifies structure-guided antigen design informed by antigen–antibody interactions and computational modeling. McLellan et al. [274] resolved the crystal structure of the RSV F glycoprotein in complex with a neutralizing antibody, identifying antigenic site Ø specific to the prefusion conformation. They engineered the DS-Cav1 immunogen by introducing a disulfide bond (S155C–S290C) and cavity-filling mutations (S190F, V207L) to stabilize the prefusion state. Krarup et al. [275] extended this approach by introducing additional stabilizing mutations (E161P, S215P, and N67I), targeting flexible regions of the protein and improving thermal stability and antigenic integrity. Joyce et al. [276] further refined the design through iterative structure-based improvement, including fusion peptide deletion, genetic fusion of F1–F2 subunits, and disulfide bond insertions to develop DS2, a next-generation immunogen with enhanced thermal stability and immunogenicity in preclinical models. Moin et al. [277] applied an in silico vaccine design pipeline that included B-cell and T-cell epitope prediction using IEDB tools, antigenicity and allergenicity profiling via VaxiJen and AllerTOP, structural modeling with AlphaFold2, molecular docking using ClusPro and HADDOCK, Molecular Dynamics simulations with GROMACS, binding free energy estimation via MM-PBSA, and immune response simulation using the C-ImmSim server. Their study demonstrates how multi-step computational workflows can support rational RSV subunit vaccine design by predicting structure, binding, and immunogenic potential prior to experimental validation.
5.3. Systems Vaccinology
Systems vaccinology has recently emerged as an interdisciplinary field that integrates high-dimensional data, network modeling, and predictive analytics within the context of vaccinology [278]. It encompasses the perturbation of biological systems through vaccination, followed by system-wide monitoring, data integration, network inference, and the development of predictive rules describing immune responses to vaccination [279,280] (see Figure 6). In this section, we will discuss the various system theoretic approaches involved in vaccine formulation through systems vaccinology techniques.
Berry et al. [281] conducted transcriptomic analysis of blood samples from asymptomatic individuals infected with Mycobacterium tuberculosis, identifying a 393-gene signature enriched in interferon signaling that correlated with the radiological severity of disease. A k-nearest neighbors (KNN) algorithm was used to classify individuals with active tuberculosis. Vahey et al. [282] explored gene expression profiles predictive of individual responses to malaria vaccines. The differential expression of immunoproteasome pathway genes was identified prior to pathogen challenge in individuals who responded to vaccination.
5.3.1. High-Throughput Profiling and Predictive Modeling
Brown et al. [283] demonstrated the use of SVMs to classify genes into functional categories based on microarray expression data, offering predictions for unannotated yeast genes. Díaz-Uriarte and Alvarez de Andrés [284] proposed a random forest-based gene selection method that identifies small, non-redundant gene sets with strong predictive power. This approach is competitive with existing feature selection methods in microarray-based classification. The MicroArray Quality Control (MAQC) consortium [285] evaluated the reproducibility and robustness of microarray-based predictive models. In the MAQC-II project, participating teams built models without prior knowledge of biological endpoints and validated them using blinded test sets to simulate real-world clinical applications.
5.3.2. Network Inference and Systems Analysis
Amit et al. [286] introduced a systematic perturbation strategy to investigate transcriptional regulatory networks in mouse dendritic cells in response to pathogen stimulation. Their approach identified 125 regulatory genes and produced a network model comprising 24 core regulators and 76 fine-tuners, providing insight into how pathogen-sensing pathways achieve specificity. Systems biology approaches have also been applied to identify dynamic gene regulatory circuits that control inflammatory responses. For example, Litvak et al. [287] used the mathematical modeling of transcriptional regulation, followed by experimental validation, to reveal circuits that distinguish between transient and sustained receptor signaling.

Figure 6.
Systems vaccinology [288,289]. Machine learning approaches can be utilized within systems vaccinology. The input data are derived from the clinical data records, manual curation, and text-mining approaches on people, and are fed to systems algorithms for overall vaccination cohorts. The output predicts the status of immunogenicity (response of antibody and protection from pathogen). Created with BioRender.com.
Figure 6.
Systems vaccinology [288,289]. Machine learning approaches can be utilized within systems vaccinology. The input data are derived from the clinical data records, manual curation, and text-mining approaches on people, and are fed to systems algorithms for overall vaccination cohorts. The output predicts the status of immunogenicity (response of antibody and protection from pathogen). Created with BioRender.com.

Wang et al. [290] developed and validated MINDy (Modulator Inference by Network Dynamics), a method for identifying genes that modulate transcription factor (TF) activity post-translationally. These modulators may include kinases, transcriptional co-factors, or upstream signaling proteins that affect TF localization, stability, or DNA-binding activity without altering TF mRNA levels.
Lynn et al. [291] presented InnateDB, a database designed for systems-level analysis of the innate immune response. It facilitates the rapid interpretation of large-scale gene expression datasets, the identification of perturbed pathways, and the analysis of molecular interactions. Huttenhower et al. [292] introduced HEFalMp (Human Experimental/Functional Mapper), a Bayesian integration platform that enables the interactive exploration of functional networks in human cellular biology.
Abhishek Dutta [293] used an impulsive feedback control strategy and systems to model the wave dynamics of COVID-19 and demonstrates how to suppress the second wave peak, thereby reducing mortality. Abhishek Dutta [294] presented the systematic pathological model learning of COVID-19 dynamics, followed by derivative-free multi-objective optimization, yielding a remdesivir regimen with lower toxicity than and comparable efficacy to the standard dosing of 200 mg on day 1 followed by 100 mg/day from days 2 to 10. Rongting Yue and Abhishek Dutta [295] designed a Koopman-based Impulsive Model Predictive Controller for BCG vaccine dosing using a nonlinear bladder cancer model with four states—BCG concentration ( c.f.u/mL), activated immune cells (), infected tumor cells (), and uninfected tumor cells ()—showing that the uninfected tumor cell population decreases as desired, and the control objectives are achieved within 10% model uncertainty.
5.3.3. Epitope Selection Strategies
A critical step in epitope-based vaccine design is the optimal selection of T-cell epitopes. Toussaint et al. [296] proposed an integer linear programming approach for epitope selection that maximizes coverage of target antigens and population diversity. Their framework outperformed traditional heuristics and genetic algorithms. Fatima et al. [297] designed a subunit multi-epitope peptide vaccine (MEBPV) against Rift Valley Fever virus (RVFV). The selected epitopes, derived from both B and T cells, were validated through molecular docking and simulations to assess receptor binding stability and conformational dynamics.
5.3.4. A Case Study of Using Systems Vaccinology for Vaccine Formulation
In [298], a multi-epitope norovirus vaccine pipeline was developed integrating the Human Calicivirus Typing Tool (sequence collection), EMBOSS-transeq [299] (translation), ClustalX 2.1 [300] (alignment and consensus), Modeller 10.4 [301] (3D structure modeling), CE-BLAST [302] (B-cell epitope similarity), IEDB with NetMHCpan 4.1 EL [217] and IEDB 2.22 [303] (MHC-I/II epitope prediction), AlgPred v2.0 [304] and AllergenFP v1.1 [305] (allergenicity), VaxiJen v2.0 [222] (antigenicity), ToxinPred2 [306](toxicity), SOLpro [307] (solubility), ProtParam [308] (physicochemical properties), DeepTMHMM [309] (transmembrane topology), PSIPRED 4.0 [310] (secondary structure), GalaxyWEB [311] and Robetta [312] (structure refinement), ProSA-web [313] (structure validation), ClusPro 2.0 [314] and PDBsum [315] (molecular docking with TLR7 and HLA alleles), AlphaFold 3.0 [316] (epitope structure prediction), and C-ImmSim [317] (immune simulation), resulting in four vaccine constructs (Vac-VP1, Vac-VP2, Vac-VP1-VP2, and Vac-B), which demonstrated favorable in silico properties including predicted immunogenicity, safety, and structural integrity, and in vivo elicited robust cross-genotype IgG and IgA responses in mice, with Vac-B and Vac-VP1-VP2 showing comparable or enhanced responses relative to the GII.4C virus-like particle control.
6. Discussion and Perspective
This review highlights the theoretical foundations and recent advancements in machine learning (ML) techniques that are widely applied in drug discovery. These computational approaches have become increasingly popular due to their accessibility through open-source platforms [318]. As a result, a growing number of data-driven ML models have been developed and shown to be effective in identifying new starting points for the drug discovery process [104].
ML and deep learning (DL) approaches offer opportunities to enhance efficiency across the drug discovery and development pipeline, including target identification, validation, drug design, and optimization for disease diagnosis [319]. However, identifying target genes solely based on gene expression remains challenging, as many target genes remain stable following drug treatment [320]. To address this limitation, some studies integrate protein–protein interaction networks with gene perturbation data, under the assumption that interfered genes are in closer network proximity to the actual targets [321]. Network-based methods often incorporate protein–protein interactions alongside metabolic network data to enhance prediction accuracy [322].
Neural networks have also been applied in virtual drug screening to complement high-throughput screening. However, the approach can be time-consuming. To overcome this challenge, multitask neural networks have been employed to improve screening efficiency by simultaneously learning across multiple tasks [323]. Effective docking algorithms require both accurate and fast scoring functions. Therefore, the design of scoring functions and optimization or search algorithms are critical components in simulating protein–ligand interactions [324].
In the context of vaccine development, this review also examined the computational and systems-theoretic methods used in reverse, structural, and systems vaccinology. The findings underscore the importance of immune profiling and systems-level measurements in building predictive modeling tools that facilitate rational vaccine design [325]. Vaccines remain essential in reducing disease-related morbidity and mortality. They not only prevent the onset of infectious diseases but also reduce disease severity and associated toxicity [220].
Reverse vaccinology offers the advantage of bypassing the limitations associated with in vitro culturing and traditional antigen identification methods. It enables the rapid and cost-effective selection of promising vaccine candidates from large antigenic repertoires using genome-based analysis [326]. However, it cannot reliably predict conformational epitopes or account for post-translational modifications [327].
Structural vaccinology, particularly relevant for viral pathogens, allows the rational design of immunogens based on structural insights. The success of this approach depends on the ability to apply high-throughput methods to generate and evaluate diverse antigen structures [254]. The integration of tools from human immunology and structural biology offers a new, multidisciplinary pathway for antigen discovery [328]. However, it is limited by the need for high-quality structural data, which is often unavailable in early studies, and it relies on computationally intensive modeling [329].
Systems vaccinology represents a transformative approach that combines computational modeling with high-throughput immune profiling to predict vaccine responses. These methods provide insights into the molecular networks that govern immune responses and help guide the design of vaccines that induce robust and durable protection across diverse populations [330]. However, it requires complex data integration, is sensitive to dataset bias and variability, and lacks good experimental models that recapitulate the diversity of the human immune response [331].
Traditional vaccine development relies heavily on culturing pathogens, followed by attenuation or inactivation, and the empirical testing of immunogenicity [332]. While this approach has led to many successful vaccines (e.g., for polio and measles), it is time-consuming, labor-intensive, and often ineffective against highly variable pathogens [333]. In contrast, reverse vaccinology begins with the pathogen genome and applies computational tools to predict antigen candidates without the need for culturing, enabling rapid and high-throughput antigen identification. Structural vaccinology leverages the high-resolution 3D structural data of antigens and antigen–antibody complexes to guide immunogen design, enabling faster development, improved safety, and enhanced efficacy, particularly against highly variable pathogens. Systems vaccinology integrates omics profiling (e.g., transcriptomics and proteomics) and network modeling to elucidate immune signatures and predict vaccine efficacy, enabling high-throughput analysis, mechanism-driven insights, and accelerated identification of effective candidates. However, the computational tools used may exhibit biases, and the reliability of predictions depends heavily on the quality and diversity of the training datasets. For example, one of the top-performing MHC binding prediction tools [334], NetMHCpan [217], has been shown to be affected by biases in its training data [218], resulting in cases where it predicts highly hydrophobic peptides as strong binders, even though biochemical evidence suggests otherwise [219].
7. Conclusions
This review presents a system theoretic perspective on drug discovery and vaccine formulation, grounded in computational techniques that integrate machine learning, control theory, and optimization for modeling networked biological systems. It is written to equip researchers with conceptual understanding and practical insight into how these methods enhance each stage of the therapeutic pipeline, including target discovery, virtual screening, lead optimization, dosing, and vaccine antigen design. We highlight current state-of-the-art tools such as graph-based models, deep generative networks, and omics-informed vaccine predictors, which offer faster, cost-effective, and mechanistically grounded alternatives to traditional experimental methods. Challenges remain in terms of data quality, model interpretability, and capturing biological complexity. Nonetheless, system theoretic methods continue to show growing potential in advancing precision therapeutics. This review aims to serve as both a knowledge resource and a foundation for developing skills in applying system theoretical methods across drug and vaccine development.
Author Contributions
Conceptualization, A.S. and A.D.; methodology, A.S., A.D. and Y.-C.H.; formal analysis, A.S., A.D. and Y.-C.H.; investigation, A.S., A.D. and Y.-C.H.; writing—original draft preparation, A.S. and A.D.; writing—review and editing, A.S., A.D. and Y.-C.H.; visualization, A.S., A.D. and Y.-C.H.; supervision, A.D.; project administration, A.D.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
During the preparation of this work, the author(s) used ChatGPT (OpenAI, GPT-4) to assist with language refinement, grammar correction, clarity improvements, and preliminary literature search. After using this tool, the author(s) reviewed and edited the content as needed, taking full responsibility for the publication’s content.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACC | Auto Cross Covariance |
| ADMET | Absorption, Distribution, Metabolism, Excretion, Toxicity |
| AGL-Score | Algebraic Graph Learning Score |
| ARACNe | Algorithm for the Reconstruction of Accurate Cellular Networks |
| BNN | Bayesian Neural Network |
| BPAs | Bacterial Protective Antigens |
| CADD | Computer-Aided Drug Design |
| CNN | Convolutional Neural Network |
| CPI | Compound Protein Interaction |
| DG-GL | Differential Geometry-based Geometric Learning |
| DL | Deep Learning |
| DNA | Deoxyribonucleic Acid |
| DNC | Differentiable Neural Compute |
| DNNs | Deep Neural Networks |
| DR-A | Dimensionality Reduction with Adversarial variational autoencode |
| EN | Electron Microscopy |
| ESPH | Element-Specific Persistent Homology |
| ESTD | Element Specific Topological Descriptor |
| EV | Epitope-based Vaccines |
| FFT-BP | Feature Functional Theory–Binding Predictor |
| GAN | Generative Adversarial Network |
| GBDT | Gradient Boosting Decision Tree |
| GCN | Graph Convolutional Network |
| GENTRL | Generative Tensorial Reinforcement Learning |
| GNN | Graph Neural Network |
| HEFalMp | Human Experimental/Functional Mapper |
| KNN | K-Nearest Neighbors |
| LINCS | Library of Integrated Network-based Cellular Signature |
| LINE | Large-Scale Information Embedding |
| LSTM | Long Short Term Memory |
| MD | Molecular Dynamics |
| MDeePred | Multi-channel Deep Proteochemometric Predictor for Binding Affinity |
| MEBPV | Multi-Epitope-Based Peptide Vaccine |
| MHC | Major Histocompatibility Complex |
| MINDy | Modulator Inference by Network Dynamics |
| ML | Machine learning |
| NERVE | New Enhanced Reverse Vaccinology Environment |
| NLP | Natural Language Processing |
| NMR | Nucleic Magnetic Resonance |
| ORGAN | Objective-Reinforced Generative Adversarial Networks |
| ORGANIC | Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry |
| PASS | Prediction of Activity Spectra for Substances |
| PDB | Protein Data Bank |
| PDD | Phenotypic Drug Discovery |
| PLSR | Partial Least Squares Regression |
| PVC | Protein Vaccine Candidate |
| QSAR | Quantitative Structure–Activity Relationships |
| RANC | Reinforced Adversarial Neural Computer |
| ReLeaSE | Reinforcement Learning for Structural Evolution |
| RF | Random Forest |
| RL | Reinforcement Learning |
| RNA | Ribonucleic Acid |
| RNN | Recurrent Neural Networks |
| RVFV | Rift Valley Fever Virus |
| SDAP | Structural Database of Allergenic Proteins |
| SF | Scoring Function |
| SMOTE | Synthetic Minority Over-sampling Technique |
| SQL | Structured Query Language |
| SVM | Support Vector Machine |
| TDD | Target Drug Discover |
| TF | Transcription Factor |
| TPM | Target Prediction Mode |
References
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural products for drug discovery in the 21st century: Innovations for novel drug discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef]
- Chan, H.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing drug discovery via artificial intelligence. Trends Pharmacol. Sci. 2019, 40, 592–604. [Google Scholar] [CrossRef]
- Gupta, R.; Srivastava, D.; Sahu, M.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Artificial intelligence to deep learning: Machine intelligence approach for drug discovery. Mol. Divers. 2021, 25, 1315–1360. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhao, S.; Gilvary, C.; Elemento, O.; Zhou, J.; Wang, F. Graph convolutional networks for computational drug development and discovery. Briefings Bioinform. 2020, 21, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Murugan, N.A.; Podobas, A.; Gadioli, D.; Vitali, E.; Palermo, G.; Markidis, S. A review on parallel virtual screening softwares for high-performance computers. Pharmaceuticals 2022, 15, 63. [Google Scholar] [CrossRef]
- Frazier, K.C. Biopharmaceutical Research & Development: The Process Behind New Medicines; PhRMA: Washington, DC, USA, 2015. [Google Scholar]
- Pardridge, W.M. Drug transport across the blood–brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Silva, G.A. Nanotechnology applications and approaches for neuroregeneration and drug delivery to the central nervous system. Ann. N. Y. Acad. Sci. 2010, 1199, 221–230. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Zhang, Z.; Ren, J.; Peluffo, A.E.; Zhang, W.; Zhao, Y.; Wu, J.; Yan, K.; Cohen, D.; et al. Network bioinformatics analysis provides insight into drug repurposing for COVID-19. Med. Drug Discov. 2021, 10, 100090. [Google Scholar] [CrossRef] [PubMed]
- Isgut, M.; Rao, M.; Yang, C.; Subrahmanyam, V.; Rida, P.C.; Aneja, R. Application of combination high-throughput phenotypic screening and target identification methods for the discovery of natural product-based combination drugs. Med. Res. Rev. 2018, 38, 504–524. [Google Scholar] [CrossRef]
- Swinney, D.C.; Lee, J.A. Recent advances in phenotypic drug discovery. F1000Research 2020, 9, F1000 Faculty Rev-944. [Google Scholar] [CrossRef]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef]
- Wang, Y.; Lupala, C.S.; Liu, H.; Lin, X. Identification of drug binding sites and action mechanisms with molecular dynamics simulations. Curr. Top. Med. Chem. 2018, 18, 2268–2277. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Searls, D.B. Data integration: Challenges for drug discovery. Nat. Rev. Drug Discov. 2005, 4, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, F.; Tang, J.; Nussinov, R.; Cheng, F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit. Health 2020, 2, e667–e676. [Google Scholar] [CrossRef]
- Chatterjee, R.; Ghosh, M.; Sahoo, S.; Padhi, S.; Misra, N.; Raina, V.; Suar, M.; Son, Y.O. Next-generation bioinformatics approaches and resources for coronavirus vaccine discovery and development—A perspective review. Vaccines 2021, 9, 812. [Google Scholar] [CrossRef]
- Trovato, M.; Krebs, S.J.; Haigwood, N.L.; De Berardinis, P. Delivery strategies for novel vaccine formulations. World J. Virol. 2012, 1, 4. [Google Scholar] [CrossRef]
- Donnelly, R.F. Vaccine delivery systems. Hum. Vaccines Immunother. 2017, 13, 17–18. [Google Scholar] [CrossRef]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and challenges in the delivery of mRNA-based vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef]
- Pepini, T.; Pulichino, A.M.; Carsillo, T.; Carlson, A.L.; Sari-Sarraf, F.; Ramsauer, K.; Debasitis, J.C.; Maruggi, G.; Otten, G.R.; Geall, A.J.; et al. Induction of an IFN-mediated antiviral response by a self-amplifying RNA vaccine: Implications for vaccine design. J. Immunol. 2017, 198, 4012–4024. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Nieto-Torres, J.L.; DeDiego, M.L.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Perlman, S.; Enjuanes, L. Identification of the mechanisms causing reversion to virulence in an attenuated SARS-CoV for the design of a genetically stable vaccine. PLoS Pathog. 2015, 11, e1005215. [Google Scholar] [CrossRef]
- Te Yeh, M.; Bujaki, E.; Dolan, P.T.; Smith, M.; Wahid, R.; Konz, J.; Weiner, A.J.; Bandyopadhyay, A.S.; Van Damme, P.; De Coster, I.; et al. Engineering the live-attenuated polio vaccine to prevent reversion to virulence. Cell Host Microbe 2020, 27, 736–751. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. The advantages and disadvantages of different types of vaccines: DNA vaccine, mRNA vaccine, and inactivated vaccine. Theor. Nat. Sci. 2023, 6, 120–126. [Google Scholar] [CrossRef]
- Liljeroos, L.; Malito, E.; Ferlenghi, I.; Bottomley, M.J. Structural and computational biology in the design of immunogenic vaccine antigens. J. Immunol. Res. 2015, 2015, 156241. [Google Scholar] [CrossRef]
- Nakaya, H.I.; Li, S.; Pulendran, B. Systems vaccinology: Learning to compute the behavior of vaccine induced immunity. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 193–205. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Grandi, G.; Rappuoli, R. Structural vaccinology starts to deliver. Nat. Rev. Microbiol. 2012, 10, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Kulp, D.W.; Schief, W.R. Advances in structure-based vaccine design. Curr. Opin. Virol. 2013, 3, 322–331. [Google Scholar] [CrossRef]
- Sharma, A.; Hsiao, Y.C.; Dutta, A. System Theoretic Methods in Drug Discovery and Vaccine Formulation: Review and Perspectives. SSRN Electron. J. 2023. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A deep learning approach to antibiotic discovery. Cell 2020, 180, 688–702. [Google Scholar] [CrossRef]
- Liu, G.; Catacutan, D.B.; Rathod, K.; Swanson, K.; Jin, W.; Mohammed, J.C.; Chiappino-Pepe, A.; Syed, S.A.; Fragis, M.; Rachwalski, K.; et al. Deep learning-guided discovery of an antibiotic targeting Acinetobacter baumannii. Nat. Chem. Biol. 2023, 19, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Itoh, A.; Murayama, S.; Suzue, S.; Irikura, T. Structure-activity relationships of antibacterial 6, 7-and 7, 8-disubstituted 1-alkyl-1, 4-dihydro-4-oxoquinoline-3-carboxylic acids. J. Med. Chem. 1980, 23, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal–epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H. Role of pharmacokinetics in the discovery and development of indinavir. Adv. Drug Deliv. Rev. 1999, 39, 33–49. [Google Scholar] [CrossRef]
- Janssen, P.A.; Lewi, P.J.; Arnold, E.; Daeyaert, F.; De Jonge, M.; Heeres, J.; Koymans, L.; Vinkers, M.; Guillemont, J.; Pasquier, E.; et al. In search of a novel anti-HIV drug: Multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1 E)-2-cyanoethenyl]-2, 6-dimethylphenyl] amino]-2-pyrimidinyl] amino] benzonitrile (R278474, rilpivirine). J. Med. Chem. 2005, 48, 1901–1909. [Google Scholar] [CrossRef]
- Zhang, P.; Huang, W.; Wang, L.; Bao, L.; Jia, Z.J.; Bauer, S.M.; Goldman, E.A.; Probst, G.D.; Song, Y.; Su, T.; et al. Discovery of betrixaban (PRT054021), N-(5-chloropyridin-2-yl)-2-(4-(N, N-dimethylcarbamimidoyl) benzamido)-5-methoxybenzamide, a highly potent, selective, and orally efficacious factor Xa inhibitor. Bioorganic Med. Chem. Lett. 2009, 19, 2179–2185. [Google Scholar] [CrossRef]
- Wood, J.M.; Maibaum, J.; Rahuel, J.; Grütter, M.G.; Cohen, N.C.; Rasetti, V.; Rüger, H.; Göschke, R.; Stutz, S.; Fuhrer, W.; et al. Structure-based design of aliskiren, a novel orally effective renin inhibitor. Biochem. Biophys. Res. Commun. 2003, 308, 698–705. [Google Scholar] [CrossRef]
- Huang, W.S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of brigatinib (AP26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 2004, 3, 215–225. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Phoebe Chen, Y.P.; Chen, F. Identifying targets for drug discovery using bioinformatics. Expert Opin. Ther. Targets 2008, 12, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Ratti, E.; Trist, D. Continuing evolution of the drug discovery process in the pharmaceutical industry. Pure Appl. Chem. 2001, 73, 67–75. [Google Scholar] [CrossRef]
- Kumble, K.D. An update on using protein microarrays in drug discovery. Expert Opin. Drug Discov. 2007, 2, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Russ, A.; Grosse, J. Mouse genetics in drug target discovery and validation: No simple answers to complex problems. Expert Opin. Drug Discov. 2007, 2, 1379–1387. [Google Scholar] [CrossRef]
- Blundell, T.L.; Sibanda, B.L.; Montalvão, R.W.; Brewerton, S.; Chelliah, V.; Worth, C.L.; Harmer, N.J.; Davies, O.; Burke, D. Structural biology and bioinformatics in drug design: Opportunities and challenges for target identification and lead discovery. Philos. Trans. R. Soc. Biol. Sci. 2006, 361, 413–423. [Google Scholar] [CrossRef]
- Katsila, T.; Spyroulias, G.A.; Patrinos, G.P.; Matsoukas, M.T. Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 2016, 14, 177–184. [Google Scholar] [CrossRef]
- You, Y.; Lai, X.; Pan, Y.; Zheng, H.; Vera, J.; Liu, S.; Deng, S.; Zhang, L. Artificial intelligence in cancer target identification and drug discovery. Signal Transduct. Target. Ther. 2022, 7, 1–24. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797. [Google Scholar] [CrossRef]
- Aryaa, H.; Coumarb, M.S. Target identification and validation. In The Design and Development of Novel Drugs and Vaccines: Principles and Protocols; Academic Press: Cambridge, MA, USA, 2021; p. 11. [Google Scholar]
- Yue, X.; Wang, Z.; Huang, J.; Parthasarathy, S.; Moosavinasab, S.; Huang, Y.; Lin, S.M.; Zhang, W.; Zhang, P.; Sun, H. Graph embedding on biomedical networks: Methods, applications and evaluations. Bioinformatics 2020, 36, 1241–1251. [Google Scholar] [CrossRef]
- Gaudelet, T.; Day, B.; Jamasb, A.R.; Soman, J.; Regep, C.; Liu, G.; Hayter, J.B.; Vickers, R.; Roberts, C.; Tang, J.; et al. Utilizing graph machine learning within drug discovery and development. Briefings Bioinform. 2021, 22, bbab159. [Google Scholar] [CrossRef]
- Lee, J.B.; Rossi, R.; Kong, X. Graph classification using structural attention. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, UK, 19–23 August 2018; pp. 1666–1674. [Google Scholar]
- Zhao, T.; Zhang, X.; Wang, S. Graphsmote: Imbalanced node classification on graphs with graph neural networks. In Proceedings of the 14th ACM International Conference on Web Search and Data Mining, Virtual Event, 8–12 March 2021; pp. 833–841. [Google Scholar]
- Tang, J.; Aggarwal, C.; Liu, H. Node classification in signed social networks. In Proceedings of the 2016 SIAM International Conference on Data Mining, Miami, FL, USA, 5–7 May 2016; pp. 54–62. [Google Scholar]
- Kunegis, J.; Lommatzsch, A. Learning spectral graph transformations for link prediction. In Proceedings of the 26th Annual International Conference on Machine Learning, Montreal, QC, Canada, 14–18 June 2009; pp. 561–568. [Google Scholar]
- MacLean, F. Knowledge graphs and their applications in drug discovery. Expert Opin. Drug Discov. 2021, 16, 1057–1069. [Google Scholar] [CrossRef]
- Cao, Y.; Romero, J.; Aspuru-Guzik, A. Potential of quantum computing for drug discovery. IBM J. Res. Dev. 2018, 62, 6:1–6:20. [Google Scholar] [CrossRef]
- Chan, J.N.; Nislow, C.; Emili, A. Recent advances and method development for drug target identification. Trends Pharmacol. Sci. 2010, 31, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Czodrowski, P.; Kriegl, J.M.; Scheuerer, S.; Fox, T. Computational approaches to predict drug metabolism. Expert Opin. Drug Metab. Toxicol. 2009, 5, 15–27. [Google Scholar] [CrossRef]
- Bredel, M.; Jacoby, E. Chemogenomics: An emerging strategy for rapid target and drug discovery. Nat. Rev. Genet. 2004, 5, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Spring, D.R. Chemical genetics to chemical genomics: Small molecules offer big insights. Chem. Soc. Rev. 2005, 34, 472–482. [Google Scholar] [CrossRef]
- Campillos, M.; Kuhn, M.; Gavin, A.C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef]
- Lim, S.; Lu, Y.; Cho, C.Y.; Sung, I.; Kim, J.; Kim, Y.; Park, S.; Kim, S. A review on compound-protein interaction prediction methods: Data, format, representation and model. Comput. Struct. Biotechnol. J. 2021, 19, 1541–1556. [Google Scholar] [CrossRef]
- Tsubaki, M.; Tomii, K.; Sese, J. Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 2019, 35, 309–318. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Cetin Atalay, R.; Cansen Kahraman, D.; Doğan, T.; Martin, M.; Atalay, V. MDeePred: Novel multi-channel protein featurization for deep learning-based binding affinity prediction in drug discovery. Bioinformatics 2021, 37, 693–704. [Google Scholar] [CrossRef]
- Kumari, P.; Nath, A.; Chaube, R. Identification of human drug targets using machine-learning algorithms. Comput. Biol. Med. 2015, 56, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Kira, K.; Rendell, L.A. A practical approach to feature selection. In Machine Learning Proceedings 1992; Elsevier: Amsterdam, The Netherlands, 1992; pp. 249–256. [Google Scholar]
- Rodriguez, J.J.; Kuncheva, L.I.; Alonso, C.J. Rotation forest: A new classifier ensemble method. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 1619–1630. [Google Scholar] [CrossRef]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a cancer dependency map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef]
- Brazma, A.; Kapushesky, M.; Parkinson, H.; Sarkans, U.; Shojatalab, M. [20] Data Storage and Analysis in ArrayExpress. Methods Enzymol. 2006, 411, 370–386. [Google Scholar]
- Ayton, G.S.; Noid, W.G.; Voth, G.A. Multiscale modeling of biomolecular systems: In serial and in parallel. Curr. Opin. Struct. Biol. 2007, 17, 192–198. [Google Scholar] [CrossRef]
- Simonetti, L.; Ivarsson, Y. Genetically Encoded Cyclic Peptide Phage Display Libraries. ACS Cent. Sci. 2020, 6, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Iskandar, S.E.; Chiou, L.F.; Leisner, T.M.; Shell, D.J.; Norris-Drouin, J.L.; Vaziri, C.; Pearce, K.H.; Bowers, A.A. Identification of covalent cyclic peptide inhibitors in mRNA display. J. Am. Chem. Soc. 2023, 145, 15065–15070. [Google Scholar] [CrossRef]
- Tavassoli, A. SICLOPPS cyclic peptide libraries in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 30–35. [Google Scholar] [CrossRef]
- Sohrabi, C.; Foster, A.; Tavassoli, A. Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery. Nat. Rev. Chem. 2020, 4, 90–101. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Wei, M.; Wang, L.; Li, Y.; Li, Z.; Zhao, B.; Su, X.; Wei, Y.; You, Z. BioKG-CMI: A multi-source feature fusion model based on biological knowledge graph for predicting circRNA-miRNA interactions. Sci. China Inf. Sci. 2024, 67, 189104. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers), Minneapolis, MN, USA, 2–7 June 2019; Association for Computational Linguistics: Stroudsburg, PA, USA, 2019; pp. 4171–4186. [Google Scholar]
- Liang, S.Z.; Wang, L.; You, Z.H.; Yu, C.Q.; Wei, M.M.; Wei, Y.; Shi, T.L.; Jiang, C. Predicting circRNA–Disease Associations through Multisource Domain-Aware Embeddings and Feature Projection Networks. J. Chem. Inf. Model. 2025, 65, 1666–1676. [Google Scholar] [CrossRef]
- Fan, C.; Lei, X.; Fang, Z.; Jiang, Q.; Wu, F.X. CircR2Disease: A manually curated database for experimentally supported circular RNAs associated with various diseases. Database 2018, 2018, bay044. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Rosales, A.R.; Wahlers, J.; Limé, E.; Meadows, R.E.; Leslie, K.W.; Savin, R.; Bell, F.; Hansen, E.; Helquist, P.; Munday, R.H.; et al. Rapid virtual screening of enantioselective catalysts using CatVS. Nat. Catal. 2019, 2, 41–45. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Ewing, T.J.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput.-Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FLEXX incremental construction algorithm for protein–ligand docking. Proteins Struct. Funct. Bioinform. 1999, 37, 228–241. [Google Scholar] [CrossRef]
- Mcgann, M.R.; Almond, H.R.; Nicholls, A.; Grant, J.A.; Brown, F.K. Gaussian docking functions. Biopolym. Orig. Res. Biomol. 2003, 68, 76–90. [Google Scholar] [CrossRef]
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Kumar, V.; Krishna, S.; Siddiqi, M.I. Virtual screening strategies: Recent advances in the identification and design of anti-cancer agents. Methods 2015, 71, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Pal, U. Interaction of Proteins with Small Molecules and Peptides. Ph.D. Thesis, Jadavpur University, Kolkata, India, 2016. [Google Scholar]
- Kucera, T. Virtual Screening in Drug Design–Overview of Most Frequent Techniques. Mil. Med Sci. Lett. 2016, 85, 75–79. [Google Scholar] [CrossRef]
- Jahn, A.; Hinselmann, G.; Fechner, N.; Zell, A. Optimal assignment methods for ligand-based virtual screening. J. Cheminform. 2009, 1, 14. [Google Scholar] [CrossRef]
- Villoutreix, B.O.; Renault, N.; Lagorce, D.; Sperandio, O.; Montes, M.; Miteva, M.A. Free resources to assist structure-based virtual ligand screening experiments. Curr. Protein Pept. Sci. 2007, 8, 381–411. [Google Scholar] [CrossRef]
- Leach, A.R.; Gillet, V.J. Similarity methods. In An Introduction to Chemoinformatics; Springer Netherlands: Dordrecht, The Netherlands, 2007; pp. 99–117. [Google Scholar]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef]
- Bleicher, K.H.; Böhm, H.J.; Müller, K.; Alanine, A.I. Hit and lead generation: Beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378. [Google Scholar] [CrossRef]
- Leach, A.R.; Gillet, V.J.; Lewis, R.A.; Taylor, R. Three-dimensional pharmacophore methods in drug discovery. J. Med. Chem. 2010, 53, 539–558. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.J. Machine learning for molecular modelling in drug design. Biomolecules 2019, 9, 216. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef]
- Wang, Y.; Suzek, T.; Zhang, J.; Wang, J.; He, S.; Cheng, T.; Shoemaker, B.A.; Gindulyte, A.; Bryant, S.H. PubChem bioassay: 2014 update. Nucleic Acids Res. 2014, 42, D1075–D1082. [Google Scholar] [CrossRef]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure–activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef]
- Norinder, U.; Carlsson, L.; Boyer, S.; Eklund, M. Introducing conformal prediction in predictive modeling. A transparent and flexible alternative to applicability domain determination. J. Chem. Inf. Model. 2014, 54, 1596–1603. [Google Scholar] [CrossRef]
- Wu, H.; Gao, L.; Dong, J.; Yang, X. Detecting overlapping protein complexes by rough-fuzzy clustering in protein-protein interaction networks. PLoS ONE 2014, 9, e91856. [Google Scholar] [CrossRef]
- Ji, B.Y.; You, Z.H.; Jiang, H.J.; Guo, Z.H.; Zheng, K. Prediction of drug-target interactions from multi-molecular network based on LINE network representation method. J. Transl. Med. 2020, 18, 347. [Google Scholar] [CrossRef]
- Costa, P.R.; Acencio, M.L.; Lemke, N. A machine learning approach for genome-wide prediction of morbid and druggable human genes based on systems-level data. BMC Genomics. 2010, 11, S9. [Google Scholar] [CrossRef]
- Bender, A.; Mussa, H.Y.; Glen, R.C.; Reiling, S. Molecular similarity searching using atom environments, information-based feature selection, and a naive Bayesian classifier. J. Chem. Inf. Comput. Sci. 2004, 44, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Poroikov, V.; Filimonov, D.; Borodina, Y.V.; Lagunin, A.; Kos, A. Robustness of biological activity spectra predicting by computer program PASS for noncongeneric sets of chemical compounds. J. Chem. Inf. Comput. Sci. 2000, 40, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.j.; Lee, H. An analysis of disease-gene relationship from Medline abstracts by DigSee. Sci. Rep. 2017, 7, 40154. [Google Scholar] [CrossRef]
- Ryu, S.; Kwon, Y.; Kim, W.Y. A Bayesian graph convolutional network for reliable prediction of molecular properties with uncertainty quantification. Chem. Sci. 2019, 10, 8438–8446. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Li, Q.; Zhou, L. Bayesian neural networks for selection of drug sensitive genes. J. Am. Stat. Assoc. 2018, 113, 955–972. [Google Scholar] [CrossRef]
- Beker, W.; Wołos, A.; Szymkuć, S.; Grzybowski, B.A. Minimal-uncertainty prediction of general drug-likeness based on Bayesian neural networks. Nat. Mach. Intell. 2020, 2, 457–465. [Google Scholar] [CrossRef]
- Yue, R.; Dutta, A. Repurposing Drugs for Infectious Diseases by Graph Convolutional Network with Sensitivity-Based Graph Reduction. Interdiscip. Sci. Comput. Life Sci. 2025, 17, 185–199. [Google Scholar] [CrossRef]
- Dutta, A. Predicting Drug Mechanics by Deep Learning on Gene and Cell Activities. In Proceedings of the 2022 44th Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), Glasgow, UK, 11–15 July 2022; IEEE: New York, NY, USA, 2022; pp. 2916–2919. [Google Scholar]
- Hsiao, Y.C.; Yue, R.; Dutta, A. Derivation of back-propagation for graph convolutional networks using matrix calculus and its application to explainable artificial intelligence. IEEE Trans. Artif. Intell. 2025; in press. 1–11. [Google Scholar] [CrossRef]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Ramsundar, B.; Liu, B.; Wu, Z.; Verras, A.; Tudor, M.; Sheridan, R.P.; Pande, V. Is multitask deep learning practical for pharma? J. Chem. Inf. Model. 2017, 57, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Preuer, K. Deep Learning in Drug Discovery. Ph.D. Thesis, Johannes Kepler University Linz, Linz, Austria, May 2019. [Google Scholar]
- Bravo, À.; Piñero, J.; Queralt-Rosinach, N.; Rautschka, M.; Furlong, L.I. Extraction of relations between genes and diseases from text and large-scale data analysis: Implications for translational research. BMC Bioinform. 2015, 16, 55. [Google Scholar] [CrossRef]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [PubMed]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef]
- Lin, E.; Mukherjee, S.; Kannan, S. A deep adversarial variational autoencoder model for dimensionality reduction in single-cell RNA sequencing analysis. BMC Bioinform. 2020, 21, 64. [Google Scholar] [CrossRef]
- Wu, K.; Wei, G.W. Quantitative toxicity prediction using topology based multitask deep neural networks. J. Chem. Inf. Model. 2018, 58, 520–531. [Google Scholar] [CrossRef]
- Yang, M.; Simm, J.; Lam, C.C.; Zakeri, P.; van Westen, G.J.; Moreau, Y.; Saez-Rodriguez, J. Linking drug target and pathway activation for effective therapy using multi-task learning. Sci. Rep. 2018, 8, 8322. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Outeiral, C.; Guimaraes, G.L.; Aspuru-Guzik, A. Optimizing distributions over molecular space. An objective-reinforced generative adversarial network for inverse-design chemistry (ORGANIC). ChemRxiv 2017. [Google Scholar] [CrossRef]
- Putin, E.; Asadulaev, A.; Ivanenkov, Y.; Aladinskiy, V.; Sanchez-Lengeling, B.; Aspuru-Guzik, A.; Zhavoronkov, A. Reinforced adversarial neural computer for de novo molecular design. J. Chem. Inf. Model. 2018, 58, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminform. 2019, 11, 74. [Google Scholar] [CrossRef]
- Theodoris, C.V.; Xiao, L.; Chopra, A.; Chaffin, M.D.; Al Sayed, Z.R.; Hill, M.C.; Mantineo, H.; Brydon, E.M.; Zeng, Z.; Liu, X.S.; et al. Transfer learning enables predictions in network biology. Nature 2023, 618, 616–624. [Google Scholar] [CrossRef]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Koh, H.Y.; Yang, M.; Li, L.; May, L.T.; Webb, G.I.; Pan, S.; Church, G. Large language models in drug discovery and development: From disease mechanisms to clinical trials. arXiv 2024, arXiv:2409.04481. [Google Scholar]
- Hu, W.; Ohue, M. SpatialPPIv2: Enhancing protein–protein interaction prediction through graph neural networks with protein language models. Comput. Struct. Biotechnol. J. 2025, 27, 508–518. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Towards Cracking the Language of Life’s Code Through Self-Supervised Deep Learning and High Performance Computing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. biorxiv 2022. [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 2022, 13, 1265. [Google Scholar] [CrossRef]
- Hu, W.; Ohue, M. SpatialPPI: Three-dimensional space protein-protein interaction prediction with AlphaFold Multimer. Comput. Struct. Biotechnol. J. 2024, 23, 1214–1225. [Google Scholar] [CrossRef]
- Peng, Y.; Wu, J.; Sun, Y.; Zhang, Y.; Wang, Q.; Shao, S. Contrastive-learning of language embedding and biological features for cross modality encoding and effector prediction. Nat. Commun. 2025, 16, 1299. [Google Scholar] [CrossRef]
- Vincoff, S.; Goel, S.; Kholina, K.; Pulugurta, R.; Vure, P.; Chatterjee, P. FusOn-pLM: A fusion oncoprotein-specific language model via adjusted rate masking. Nat. Commun. 2025, 16, 1436. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, T.I.; Oyedele, A.Q.K.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Idris, M.O.; Olaoba, O.T.; Adedotun, I.O.; Kolawole, O.E.; et al. Molecular modeling in drug discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- DesJarlais, R.L.; Sheridan, R.P.; Seibel, G.L.; Dixon, J.S.; Kuntz, I.D.; Venkataraghavan, R. Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure. J. Med. Chem. 1988, 31, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; Joshi, H.V.; Shah, U.A.; Patel, J.K. A Review on Computational Software Tools for Drug Design and Discovery. Indo Glob. J. Pharm. Sci. 2022, 12, 53–81. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Altae-Tran, H.; Ramsundar, B.; Pappu, A.S.; Pande, V. Low data drug discovery with one-shot learning. ACS Cent. Sci. 2017, 3, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Gao, M.; Skolnick, J. Comprehensive prediction of drug-protein interactions and side effects for the human proteome. Sci. Rep. 2015, 5, 11090. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular docking with deep learning. J. Cheminform. 2021, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2010, 39, D1035–D1041. [Google Scholar] [CrossRef]
- Kuhn, M.; von Mering, C.; Campillos, M.; Jensen, L.J.; Bork, P. STITCH: Interaction networks of chemicals and proteins. Nucleic Acids Res. 2007, 36, D684–D688. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, G.; Hu, S.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph neural networks: A review of methods and applications. AI Open 2020, 1, 57–81. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Canese, K.; Weis, S. PubMed: The bibliographic database. In The NCBI Handbook; NCBI: Bethesda, Maryland, 2013; Volume 2. [Google Scholar]
- Keenan, A.B.; Jenkins, S.L.; Jagodnik, K.M.; He, E.; Torre, D.; Wang, Z.; Dohlman, A.B.; Silverstein, M.C.; Lachmann, A.; Kuleshov, M.V.; et al. The library of integrated network-based cellular signatures NIH program: System-level cataloging of human cells response to perturbations. Cell Syst. 2018, 6, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Reid, S.P.; Clark, N.R.; Wang, Z.; Fernandez, N.F.; Rouillard, A.D.; Readhead, B.; Tritsch, S.R.; Hodos, R.; Hafner, M.; et al. L1000CDS2: LINCS L1000 characteristic direction signatures search engine. NPJ Syst. Biol. Appl. 2016, 2, 16015. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016, 4, gkw1000. [Google Scholar]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Pan, X.; Lin, X.; Cao, D.; Zeng, X.; Yu, P.S.; He, L.; Nussinov, R.; Cheng, F. Deep learning for drug repurposing: Methods, databases, and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1597. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Chen, J.H.; Linstead, E.; Swamidass, S.J.; Wang, D.; Baldi, P. ChemDB update—Full-text search and virtual chemical space. Bioinformatics 2007, 23, 2348–2351. [Google Scholar] [CrossRef]
- Duran-Frigola, M.; Pauls, E.; Guitart-Pla, O.; Bertoni, M.; Alcalde, V.; Amat, D.; Juan-Blanco, T.; Aloy, P. Extending the small-molecule similarity principle to all levels of biology with the Chemical Checker. Nat. Biotechnol. 2020, 38, 1087–1096. [Google Scholar] [CrossRef]
- Ursu, O.; Holmes, J.; Bologa, C.G.; Yang, J.J.; Mathias, S.L.; Stathias, V.; Nguyen, D.T.; Schürer, S.; Oprea, T. DrugCentral 2018: An update. Nucleic Acids Res. 2019, 47, D963–D970. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Alexer, S.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; Buneman, O.P.; et al. The concise guide to pharmacology 2017/18: Overview. Br. J. Pharmacol. 2017, 174, S1–S16. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; Von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, F.; Zhou, Y.; Zhang, Y.; Wang, Z.; Zhang, R.; Zhu, J.; Ren, Y.; Tan, Y.; et al. Therapeutic target database 2020: Enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2020, 48, D1031–D1041. [Google Scholar] [CrossRef]
- Martin, R.; Löchel, H.F.; Welzel, M.; Hattab, G.; Hauschild, A.C.; Heider, D. CORDITE: The curated CORona drug InTERactions database for SARS-CoV-2. Iscience 2020, 23, 101297. [Google Scholar] [CrossRef]
- Shrotri, M.; Swinnen, T.; Kampmann, B.; Parker, E.P. An interactive website tracking COVID-19 vaccine development. Lancet Glob. Health 2021, 9, e590–e592. [Google Scholar] [CrossRef]
- Parker, E.P.; Shrotri, M.; Kampmann, B. Keeping track of the SARS-CoV-2 vaccine pipeline. Nat. Rev. Immunol. 2020, 20, 650. [Google Scholar] [CrossRef] [PubMed]
- Douguet, D. e-LEA3D: A computational-aided drug design web server. Nucleic Acids Res. 2010, 38, W615–W621. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, L.; Wang, Y.; Zou, J.; Yang, R.; Luo, X.; Wu, C.; Yang, W.; Tian, C.; Xu, H.; et al. Generative deep learning enables the discovery of a potent and selective RIPK1 inhibitor. Nat. Commun. 2022, 13, 6891. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Park, J.; Han, E.T.; Park, W.S.; Han, J.H.; Chun, W. Drug repositioning via graph neural networks: Identifying novel JAK2 inhibitors from FDA-Approved drugs through molecular docking and biological validation. Molecules 2024, 29, 1363. [Google Scholar] [CrossRef]
- Keserü, G.M.; Makara, G.M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 2009, 8, 203–212. [Google Scholar] [CrossRef]
- Deprez, B.; Deprez-Poulain, R. Trends in hit-to-lead: An update. In Frontiers in Medicinal Chemistry; Bentham Science Publishers: Sharjah, United Arab Emirates, 2006; Volume 653, pp. 653–673. [Google Scholar]
- Wunberg, T.; Hendrix, M.; Hillisch, A.; Lobell, M.; Meier, H.; Schmeck, C.; Wild, H.; Hinzen, B. Improving the hit-to-lead process: Data-driven assessment of drug-like and lead-like screening hits. Drug Discov. Today 2006, 11, 175–180. [Google Scholar] [CrossRef]
- Garbett, N.C.; Chaires, J.B. Thermodynamic studies for drug design and screening. Expert Opin. Drug Discov. 2012, 7, 299–314. [Google Scholar] [CrossRef]
- Siddharthan, N.; Prabu, M.R.; Sivasankari, B. Bioinformatics in Drug Discovery a Review. Int. J. Res. Arts Sci. 2016, 2, 11–13. [Google Scholar] [CrossRef]
- Cox, P.B.; Gupta, R. Contemporary Computational Applications and Tools in Drug Discovery. ACS Med. Chem. Lett. 2022, 13, 1016–1029. [Google Scholar] [CrossRef] [PubMed]
- Berg, E.L. The future of phenotypic drug discovery. Cell Chem. Biol. 2021, 28, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Miyao, T.; Kaneko, H.; Funatsu, K. Inverse QSPR/QSAR analysis for chemical structure generation (from y to x). J. Chem. Inf. Model. 2016, 56, 286–299. [Google Scholar] [CrossRef]
- Wong, W.W.; Burkowski, F.J. A constructive approach for discovering new drug leads: Using a kernel methodology for the inverse-QSAR problem. J. Cheminform. 2009, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Ramsundar, B.; Kearnes, S.; Riley, P.; Webster, D.; Konerding, D.; Pande, V. Massively multitask networks for drug discovery. arXiv 2015, arXiv:1502.02072. [Google Scholar]
- Jiménez, J.; Skalic, M.; Martinez-Rosell, G.; De Fabritiis, G. K deep: Protein–ligand absolute binding affinity prediction via 3d-convolutional neural networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef]
- Cang, Z.; Wei, G.W. TopologyNet: Topology based deep convolutional and multi-task neural networks for biomolecular property predictions. PLoS Comput. Biol. 2017, 13, e1005690. [Google Scholar] [CrossRef]
- Feinberg, E.N.; Sur, D.; Wu, Z.; Husic, B.E.; Mai, H.; Li, Y.; Sun, S.; Yang, J.; Ramsundar, B.; Pande, V.S. PotentialNet for molecular property prediction. ACS Cent. Sci. 2018, 4, 1520–1530. [Google Scholar] [CrossRef]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef]
- Li, B.; Shin, H.; Gulbekyan, G.; Pustovalova, O.; Nikolsky, Y.; Hope, A.; Bessarabova, M.; Schu, M.; Kolpakova-Hart, E.; Merberg, D.; et al. Development of a drug-response modeling framework to identify cell line derived translational biomarkers that can predict treatment outcome to erlotinib or sorafenib. PLoS ONE 2015, 10, e0130700. [Google Scholar] [CrossRef]
- Ashtawy, H.M.; Mahapatra, N.R. A comparative assessment of predictive accuracies of conventional and machine learning scoring functions for protein-ligand binding affinity prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 12, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.J.; Mitchell, J.B. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, Z.; Nguyen, D.D.; Wei, G.W. Feature functional theory–binding predictor (FFT–BP) for the blind prediction of binding free energies. Theor. Chem. Accounts 2017, 136, 1–22. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Wei, G.W. AGL-score: Algebraic graph learning score for protein–ligand binding scoring, ranking, docking, and screening. J. Chem. Inf. Model. 2019, 59, 3291–3304. [Google Scholar] [CrossRef]
- Cang, Z.; Mu, L.; Wei, G.W. Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening. PLoS Comput. Biol. 2018, 14, e1005929. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Wei, G.W. DG-GL: Differential geometry-based geometric learning of molecular datasets. Int. J. Numer. Methods Biomed. Eng. 2019, 35, e3179. [Google Scholar] [CrossRef]
- Boyles, F.; Deane, C.M.; Morris, G.M. Learning from the ligand: Using ligand-based features to improve binding affinity prediction. Bioinformatics 2020, 36, 758–764. [Google Scholar] [CrossRef]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef]
- Sautto, G.A.; Kirchenbaum, G.A.; Ecker, J.W.; Bebin-Blackwell, A.G.; Pierce, S.R.; Ross, T.M. Elicitation of broadly protective antibodies following infection with influenza viruses expressing H1N1 computationally optimized broadly reactive hemagglutinin antigens. Immunohorizons 2018, 2, 226–237. [Google Scholar] [CrossRef]
- Clementi, N.; Mancini, N.; Criscuolo, E.; Cappelletti, F.; Clementi, M.; Burioni, R. Epitope mapping by epitope excision, hydrogen/deuterium exchange, and peptide-panning techniques combined with in silico analysis. In Monoclonal Antibodies; Springer: Amsterdam, The Netherlands, 2014; pp. 427–446. [Google Scholar]
- Castelli, M.; Cappelletti, F.; Diotti, R.A.; Sautto, G.; Criscuolo, E.; Dal Peraro, M.; Clementi, N. Peptide-based vaccinology: Experimental and computational approaches to target hypervariable viruses through the fine characterization of protective epitopes recognized by monoclonal antibodies and the identification of T-cell-activating peptides. Clin. Dev. Immunol. 2013, 2013, 521231. [Google Scholar] [CrossRef]
- Criscuolo, E.; Caputo, V.; Diotti, R.A.; Sautto, G.A.; Kirchenbaum, G.A.; Clementi, N. Alternative methods of vaccine delivery: An overview of edible and intradermal vaccines. J. Immunol. Res. 2019, 2019, 8303648. [Google Scholar] [CrossRef]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Atkins, T.K.; Solanki, A.; Vasmatzis, G.; Cornette, J.; Riedel, M. Evaluating NetMHCpan performance on non-European HLA alleles not present in training data. Front. Immunol. 2024, 14, 1288105. [Google Scholar] [CrossRef]
- Solanki, A.; Riedel, M.; Cornette, J.; Udell, J.; Koratkar, I.; Vasmatzis, G. The role of hydrophobicity in peptide-MHC binding. In Proceedings of the International Symposium on Mathematical and Computational Oncology, Virtual Event, 11–13 October 2021; Springer: Berlin/Heidelberg, Germany, 2021; pp. 24–37. [Google Scholar]
- Sunita; Sajid, A.; Singh, Y.; Shukla, P. Computational tools for modern vaccine development. Hum. Vaccines Immunother. 2020, 16, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T.; Ahsan, M.; Ali, A. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 4. [Google Scholar] [CrossRef]
- Goodswen, S.J.; Kennedy, P.J.; Ellis, J.T. Vacceed: A high-throughput in silico vaccine candidate discovery pipeline for eukaryotic pathogens based on reverse vaccinology. Bioinformatics 2014, 30, 2381–2383. [Google Scholar] [CrossRef]
- Yang, B.; Sayers, S.; Xiang, Z.; He, Y. Protegen: A web-based protective antigen database and analysis system. Nucleic Acids Res. 2011, 39, D1073–D1078. [Google Scholar] [CrossRef]
- Patiyal, S.; Kaur, D.; Kaur, H.; Sharma, N.; Dhall, A.; Sahai, S.; Agrawal, P.; Maryam, L.; Arora, C.; Raghava, G.P. A web-based platform on coronavirus disease-19 to maintain predicted diagnostic, drug, and vaccine candidates. Monoclon. Antibodies Immunodiagn. Immunother. 2020, 39, 204–216. [Google Scholar] [CrossRef]
- Ansari, H.R.; Flower, D.R.; Raghava, G. AntigenDB: An immunoinformatics database of pathogen antigens. Nucleic Acids Res. 2010, 38, D847–D853. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Mahapatra, S.R.; Parida, B.K.; Rath, S.; Dehury, B.; Raina, V.; Mohakud, N.K.; Misra, N.; Suar, M. DBCOVP: A database of coronavirus virulent glycoproteins. Comput. Biol. Med. 2021, 129, 104131. [Google Scholar] [CrossRef]
- Mei, L.C.; Jin, Y.; Wang, Z.; Hao, G.F.; Yang, G.F. Web resources facilitate drug discovery in treatment of COVID-19. Drug Discov. Today 2021, 26, 2358–2366. [Google Scholar] [CrossRef]
- Wu, J.; Chen, W.; Zhou, J.; Zhao, W.; Sun, Y.; Zhu, H.; Yao, P.; Chen, S.; Jiang, J.; Zhou, Z. COVIEdb: A database for potential immune epitopes of coronaviruses. Front. Pharmacol. 2020, 11, 572249. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.N.; McAdam, P.R.; Vivona, S.; Zhang, J.X.; Luong, T.; Belew, R.K.; Sahota, H.; Guiney, D.; Valafar, F.; Fierer, J.; et al. Improving reverse vaccinology with a machine learning approach. Vaccine 2011, 29, 8156–8164. [Google Scholar] [CrossRef]
- Ong, E.; Wang, H.; Wong, M.U.; Seetharaman, M.; Valdez, N.; He, Y. Vaxign-ML: Supervised machine learning reverse vaccinology model for improved prediction of bacterial protective antigens. Bioinformatics 2020, 36, 3185–3191. [Google Scholar] [CrossRef] [PubMed]
- Heinson, A.I.; Gunawardana, Y.; Moesker, B.; Denman Hume, C.C.; Vataga, E.; Hall, Y.; Stylianou, E.; McShane, H.; Williams, A.; Niranjan, M.; et al. Enhancing the biological relevance of machine learning classifiers for reverse vaccinology. Int. J. Mol. Sci. 2017, 18, 312. [Google Scholar] [CrossRef]
- Ong, E.; Cooke, M.F.; Huffman, A.; Xiang, Z.; Wong, M.U.; Wang, H.; Seetharaman, M.; Valdez, N.; He, Y. Vaxign2: The second generation of the first Web-based vaccine design program using reverse vaccinology and machine learning. Nucleic Acids Res. 2021, 49, W671–W678. [Google Scholar] [CrossRef]
- Basso, K.; Margolin, A.A.; Stolovitzky, G.; Klein, U.; Dalla-Favera, R.; Califano, A. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 2005, 37, 382–390. [Google Scholar] [CrossRef]
- Dalsass, M.; Brozzi, A.; Medini, D.; Rappuoli, R. Comparison of open-source reverse vaccinology programs for bacterial vaccine antigen discovery. Front. Immunol. 2019, 10, 113. [Google Scholar] [CrossRef]
- Tobuse, A.J.; Ang, C.W.; Yeong, K.Y. Modern vaccine development via reverse vaccinology to combat antimicrobial resistance. Life Sci. 2022, 302, 120660. [Google Scholar] [CrossRef] [PubMed]
- Shahid, F.; Ashraf, S.T.; Ali, A. Reverse vaccinology approach to potential vaccine candidates against Acinetobacter baumannii. In Acinetobacter baumannii: Methods and Protocols; Biswas, I., Rather, P.N., Eds.; Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1946, pp. 329–336. [Google Scholar]
- Yang, Z.; Bogdan, P.; Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci. Rep. 2021, 11, 3238. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xiang, Z.; Mobley, H.L. Vaxign: The first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J. Biomed. Biotechnol. 2010, 2010, 297505. [Google Scholar] [CrossRef] [PubMed]
- Vivona, S.; Bernante, F.; Filippini, F. NERVE: New enhanced reverse vaccinology environment. BMC Biotechnol. 2006, 6, 35. [Google Scholar] [CrossRef]
- Jaiswal, V.; Chanumolu, S.K.; Gupta, A.; Chauhan, R.S.; Rout, C. Jenner-predict server: Prediction of protein vaccine candidates (PVCs) in bacteria based on host-pathogen interactions. BMC Bioinform. 2013, 14, 211. [Google Scholar] [CrossRef]
- Masignani, V.; Pizza, M.; Moxon, E.R. The development of a vaccine against meningococcus B using reverse vaccinology. Front. Immunol. 2019, 10, 751. [Google Scholar] [CrossRef]
- Maiden, M.C.J. The impact of nucleotide sequence analysis on meningococcal vaccine development and assessment. Front. Immunol. 2019, 9, 3151. [Google Scholar] [CrossRef]
- Bianconi, I.; Alcalá-Franco, B.; Scarselli, M.; Dalsass, M.; Buccato, S.; Colaprico, A.; Marchi, S.; Masignani, V.; Bragonzi, A. Genome-based approach delivers vaccine candidates against Pseudomonas aeruginosa. Front. Immunol. 2019, 9, 3021. [Google Scholar] [CrossRef]
- Nagpal, G.; Usmani, S.S.; Raghava, G.P. A web resource for designing subunit vaccine against major pathogenic species of bacteria. Front. Immunol. 2018, 9, 2280. [Google Scholar] [CrossRef]
- Howard, A.; O’Donoghue, M.; Feeney, A.; Sleator, R.D. Acinetobacter baumannii: An emerging opportunistic pathogen. Virulence 2012, 3, 243–250. [Google Scholar] [CrossRef]
- Moriel, D.G.; Beatson, S.A.; Wurpel, D.J.; Lipman, J.; Nimmo, G.R.; Paterson, D.L.; Schembri, M.A. Identification of novel vaccine candidates against multidrug-resistant Acinetobacter baumannii. PLoS ONE 2013, 8, e77631. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. 2017. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 16 June 2025).
- Ahmad, S.; Azam, S.S. A novel approach of virulome based reverse vaccinology for exploring and validating peptide-based vaccine candidates against the most troublesome nosocomial pathogen: Acinetobacter baumannii. J. Mol. Graph. Model. 2018, 83, 1–11. [Google Scholar] [CrossRef]
- Shahid, F.; Zaheer, T.; Ashraf, S.T.; Shehroz, M.; Anwer, F.; Naz, A.; Ali, A. Chimeric vaccine designs against Acinetobacter baumannii using pan genome and reverse vaccinology approaches. Sci. Rep. 2021, 11, 13213. [Google Scholar] [CrossRef]
- Beiranvand, S.; Doosti, A.; Mirzaei, S.A. Putative novel B-cell vaccine candidates identified by reverse vaccinology and genomics approaches to control Acinetobacter baumannii serotypes. Infect. Genet. Evol. 2021, 96, 105138. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, F.; Zhou, Z.; Ma, S.; Zhang, P.; Tan, C.; Luo, Y.; Qin, R.; Chen, J.; Pan, P. A novel mRNA multi-epitope vaccine of Acinetobacter baumannii based on multi-target protein design in immunoinformatic approach. BMC Genom. 2024, 25, 791. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Ulmer, J.B.; Rappuoli, R. Structure-based antigen design: A strategy for next generation vaccines. Trends Biotechnol. 2008, 26, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.V.; Van Regenmortel, M.H. B cell epitope prediction. Struct. Bioinform. 2009, 2, 849–879. [Google Scholar]
- Khan, A.M.; Miotto, O.; Heiny, A.; Salmon, J.; Srinivasan, K.; Nascimento, E.J.; Marques, E.T., Jr.; Brusic, V.; Tan, T.W.; August, J.T. A systematic bioinformatics approach for selection of epitope-based vaccine targets. Cell. Immunol. 2006, 244, 141–147. [Google Scholar] [CrossRef]
- Yoder, J.D.; Dormitzer, P.R. Alternative intermolecular contacts underlie the rotavirus VP5* two-to three-fold rearrangement. EMBO J. 2006, 25, 1559–1568. [Google Scholar] [CrossRef][Green Version]
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc. Natl. Acad. Sci. USA 2021, 118, e2024302118. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.K.; Arul, M.N.; Patel, P.; Verma, S.K.; Luo, W.; Rubahn, H.G.; Mishra, Y.K.; Suar, M.; Ahuja, R. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci. Adv. 2020, 6, eabb8097. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Ivanciuc, O.; Schein, C.H.; Braun, W. SDAP: Database and computational tools for allergenic proteins. Nucleic Acids Res. 2003, 31, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Haste Andersen, P.; Nielsen, M.; Lund, O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006, 15, 2558–2567. [Google Scholar] [CrossRef]
- El-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting flexible length linear B-cell epitopes. In Computational Systems Bioinformatics: (Volume 7); World Scientific: Singapore, 2008; pp. 121–132. [Google Scholar]
- Degoot, A.M.; Chirove, F.; Ndifon, W. Trans-allelic model for prediction of peptide: MHC-II interactions. Front. Immunol. 2018, 9, 1410. [Google Scholar] [CrossRef]
- Mayrose, I.; Penn, O.; Erez, E.; Rubinstein, N.D.; Shlomi, T.; Freund, N.T.; Bublil, E.M.; Ruppin, E.; Sharan, R.; Gershoni, J.M.; et al. Pepitope: Epitope mapping from affinity-selected peptides. Bioinformatics 2007, 23, 3244–3246. [Google Scholar] [CrossRef]
- Mayrose, I.; Shlomi, T.; Rubinstein, N.D.; Gershoni, J.M.; Ruppin, E.; Sharan, R.; Pupko, T. Epitope mapping using combinatorial phage-display libraries: A graph-based algorithm. Nucleic Acids Res. 2007, 35, 69–78. [Google Scholar] [CrossRef]
- Kringelum, J.V.; Nielsen, M.; Padkjær, S.B.; Lund, O. Structural analysis of B-cell epitopes in antibody: Protein complexes. Mol. Immunol. 2013, 53, 24–34. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J.M. Prediction of protein-protein interaction sites using patch analysis. J. Mol. Biol. 1997, 272, 133–143. [Google Scholar] [CrossRef]
- Sela-Culang, I.; Ashkenazi, S.; Peters, B.; Ofran, Y. PEASE: Predicting B-cell epitopes utilizing antibody sequence. Bioinformatics 2015, 31, 1313–1315. [Google Scholar] [CrossRef] [PubMed]
- Sela-Culang, I.; Benhnia, M.R.E.I.; Matho, M.H.; Kaever, T.; Maybeno, M.; Schlossman, A.; Nimrod, G.; Li, S.; Xiang, Y.; Zajonc, D.; et al. Using a combined computational-experimental approach to predict antibody-specific B cell epitopes. Structure 2014, 22, 646–657. [Google Scholar] [CrossRef]
- Simek, M.D.; Rida, W.; Priddy, F.H.; Pung, P.; Carrow, E.; Laufer, D.S.; Lehrman, J.K.; Boaz, M.; Tarragona-Fiol, T.; Miiro, G.; et al. Human immunodeficiency virus type 1 elite neutralizers: Individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J. Virol. 2009, 83, 7337–7348. [Google Scholar] [CrossRef] [PubMed]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient unbound docking of rigid molecules. In Proceedings of the International Workshop on Algorithms in Bioinformatics, Rome, Italy, 17–21 September 2002; Springer: Berlin/Heidelberg, Germany, 2002; pp. 185–200. [Google Scholar]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef]
- Krarup, A.; Truan, D.; Furmanova-Hollenstein, P.; Bogaert, L.; Bouchier, P.; Bisschop, I.J.; Widjojoatmodjo, M.N.; Zahn, R.; Schuitemaker, H.; McLellan, J.S.; et al. A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism. Nat. Commun. 2015, 6, 8143. [Google Scholar] [CrossRef]
- Joyce, M.G.; Zhang, B.; Ou, L.; Chen, M.; Chuang, G.Y.; Druz, A.; Kong, W.P.; Lai, Y.T.; Rundlet, E.J.; Tsybovsky, Y.; et al. Iterative structure-based improvement of a fusion-glycoprotein vaccine against RSV. Nat. Struct. Mol. Biol. 2016, 23, 811–820. [Google Scholar] [CrossRef]
- Moin, A.T.; Ullah, M.A.; Patil, R.B.; Faruqui, N.A.; Araf, Y.; Das, S.; Uddin, K.M.K.; Hossain, M.S.; Miah, M.F.; Moni, M.A.; et al. A computational approach to design a polyvalent vaccine against human respiratory syncytial virus. Sci. Rep. 2023, 13, 9702. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Li, S.; Nakaya, H.I. Systems vaccinology. Immunity 2010, 33, 516–529. [Google Scholar] [CrossRef]
- Ideker, T.; Galitski, T.; Hood, L. A new approach to decoding life: Systems biology. Annu. Rev. Genom. Hum. Genet. 2001, 2, 343–372. [Google Scholar] [CrossRef]
- Kitano, H. Computational systems biology. Nature 2002, 420, 206–210. [Google Scholar] [CrossRef]
- Berry, M.P.; Graham, C.M.; McNab, F.W.; Xu, Z.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977. [Google Scholar] [CrossRef]
- Vahey, M.T.; Wang, Z.; Kester, K.E.; Cummings, J.; Heppner Jr, D.G.; Nau, M.E.; Ofori-Anyinam, O.; Cohen, J.; Coche, T.; Ballou, W.R.; et al. Expression of genes associated with immunoproteasome processing of major histocompatibility complex peptides is indicative of protection with adjuvanted RTS, S malaria vaccine. J. Infect. Dis. 2010, 201, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.P.; Grundy, W.N.; Lin, D.; Cristianini, N.; Sugnet, C.W.; Furey, T.S.; Ares Jr, M.; Haussler, D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl. Acad. Sci. USA 2000, 97, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Uriarte, R.; Alvarez de Andrés, S. Gene selection and classification of microarray data using random forest. BMC Bioinform. 2006, 7, 3. [Google Scholar] [CrossRef]
- Shi, L.; Campbell, G.; Jones, W.D.; Campagne, F.; Wen, Z.; Walker, S.J.; Su, Z.; Chu, T.-M.; Goodsaid, F.M.; Pusztai, L.; et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nature Biotechnol. 2010, 28, 827–838. [Google Scholar]
- Amit, I.; Garber, M.; Chevrier, N.; Leite, A.P.; Donner, Y.; Eisenhaure, T.; Guttman, M.; Grenier, J.K.; Li, W.; Zuk, O.; et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 2009, 326, 257–263. [Google Scholar] [CrossRef]
- Litvak, V.; Ramsey, S.A.; Rust, A.G.; Zak, D.E.; Kennedy, K.A.; Lampano, A.E.; Nykter, M.; Shmulevich, I.; Aderem, A. Function of C/EBPδ in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat. Immunol. 2009, 10, 437–443. [Google Scholar] [CrossRef]
- Gonzalez-Dias, P.; Lee, E.K.; Sorgi, S.; de Lima, D.S.; Urbanski, A.H.; Silveira, E.L.; Nakaya, H.I. Methods for predicting vaccine immunogenicity and reactogenicity. Hum. Vaccines Immunother. 2020, 16, 269–276. [Google Scholar] [CrossRef]
- Creighton, R.; Schuch, V.; Urbanski, A.H.; Giddaluru, J.; Costa-Martins, A.G.; Nakaya, H.I. Network vaccinology. Semin. Immunol. 2020, 50, 101420. [Google Scholar] [CrossRef]
- Wang, K.; Saito, M.; Bisikirska, B.C.; Alvarez, M.J.; Lim, W.K.; Rajbhandari, P.; Shen, Q.; Nemenman, I.; Basso, K.; Margolin, A.A.; et al. Genome-wide identification of post-translational modulators of transcription factor activity in human B cells. Nat. Biotechnol. 2009, 27, 829–837. [Google Scholar] [CrossRef]
- Lynn, D.J.; Winsor, G.L.; Chan, C.; Richard, N.; Laird, M.R.; Barsky, A.; Gardy, J.L.; Roche, F.M.; Chan, T.H.; Shah, N.; et al. InnateDB: Facilitating systems-level analyses of the mammalian innate immune response. Mol. Syst. Biol. 2008, 4, 218. [Google Scholar] [CrossRef] [PubMed]
- Huttenhower, C.; Haley, E.M.; Hibbs, M.A.; Dumeaux, V.; Barrett, D.R.; Coller, H.A.; Troyanskaya, O.G. Exploring the human genome with functional maps. Genome Res. 2009, 19, 1093–1106. [Google Scholar] [CrossRef]
- Dutta, A. COVID-19 waves: Variant dynamics and control. Sci. Rep. 2022, 12, 9332. [Google Scholar] [CrossRef]
- Dutta, A. Optimizing antiviral therapy for COVID-19 with learned pathogenic model. Sci. Rep. 2022, 12, 6873. [Google Scholar] [CrossRef] [PubMed]
- Yue, R.; Dutta, A. Koopman-based impulsive model predictive control of BCG immunotherapy. In Proceedings of the 2023 45th Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), Sydney, Australia, 24–27 July 2023; IEEE: New York, NY, USA, 2023; pp. 1–4. [Google Scholar]
- Toussaint, N.C.; Dönnes, P.; Kohlbacher, O. A mathematical framework for the selection of an optimal set of peptides for epitope-based vaccines. PLoS Comput. Biol. 2008, 4, e1000246. [Google Scholar] [CrossRef]
- Fatima, I.; Ahmad, S.; Abbasi, S.W.; Ashfaq, U.A.; Shahid, F.; ul Qamar, M.T.; Rehman, A.; Allemailem, K.S. Designing of a multi-epitopes-based peptide vaccine against rift valley fever virus and its validation through integrated computational approaches. Comput. Biol. Med. 2022, 141, 105151. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Wei, Y.; Shu, J.; Zheng, W.; Zhang, Y.; Xie, J.; Zhang, D.; Luo, X.; Sun, X.; Wang, X.; et al. Integrated in-silico design and in vivo validation of multi-epitope vaccines for norovirus. Virol. J. 2025, 22, 166. [Google Scholar] [CrossRef]
- Madeira, F.; Madhusoodanan, N.; Lee, J.; Eusebi, A.; Niewielska, A.; Tivey, A.R.; Lopez, R.; Butcher, S. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 2024, 52, W521–W525. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.; Eramian, D.; Shen, M.y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef]
- Qiu, T.; Yang, Y.; Qiu, J.; Huang, Y.; Xu, T.; Xiao, H.; Wu, D.; Zhang, Q.; Zhou, C.; Zhang, X.; et al. CE-BLAST makes it possible to compute antigenic similarity for newly emerging pathogens. Nat. Commun. 2018, 9, 1772. [Google Scholar] [CrossRef]
- Wang, P.; Sidney, J.; Kim, Y.; Sette, A.; Lund, O.; Nielsen, M.; Peters, B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010, 11, 568. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P.S. AlgPred: Prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Naorem, L.D.; Jain, S.; Raghava, G.P. ToxinPred2: An improved method for predicting toxicity of proteins. Briefings Bioinform. 2022, 23, bbac174. [Google Scholar] [CrossRef] [PubMed]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef]
- Walker, J.M. The Proteomics Protocols Handbook; Humana: Totowa, NJ, USA, 2005. [Google Scholar]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Almagro Armenteros, J.J.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. biorxiv 2022. [Google Scholar] [CrossRef]
- Buchan, D.W.; Jones, D.T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Jindal, A.; Ghani, U.; Kotelnikov, S.; Egbert, M.; Hashemi, N.; Vajda, S.; Padhorny, D.; Kozakov, D. Elucidation of protein function using computational docking and hotspot analysis by ClusPro and FTMap. Biol. Crystallogr. 2022, 78, 690–697. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum: Summaries and analyses of PDB structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef]
- Lavecchia, A. Deep learning in drug discovery: Opportunities, challenges and future prospects. Drug Discov. Today 2019, 24, 2017–2032. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Ma, J.; Wang, J.; Ghoraie, L.S.; Men, X.; Liu, L.; Dai, P. Network-based method for drug target discovery at the isoform level. Sci. Rep. 2019, 9, 13868. [Google Scholar] [CrossRef]
- Zhang, Y.; Pu, Y.; Zhang, H.; Cong, Y.; Zhou, J. An extended fractional Kalman filter for inferring gene regulatory networks using time-series data. Chemom. Intell. Lab. Systs. 2014, 138, 57–63. [Google Scholar] [CrossRef]
- Gygi, S.P.; Rochon, Y.; Franza, B.R.; Aebersold, R. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 1999, 19, 1720–1730. [Google Scholar] [CrossRef]
- Stephenson, N.; Shane, E.; Chase, J.; Rowland, J.; Ries, D.; Justice, N.; Zhang, J.; Chan, L.; Cao, R. Survey of machine learning techniques in drug discovery. Curr. Drug Metab. 2019, 20, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.N.; Philot, E.A.; Trossini, G.H.G.; Scott, L.P.B.; Maltarollo, V.G.; Honorio, K.M. Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discov. 2016, 11, 225–239. [Google Scholar] [CrossRef]
- Chaudhury, S.; Duncan, E.H.; Atre, T.; Storme, C.K.; Beck, K.; Kaba, S.A.; Lanar, D.E.; Bergmann-Leitner, E.S. Identification of immune signatures of novel adjuvant formulations using machine learning. Sci. Rep. 2018, 8, 17508. [Google Scholar] [CrossRef]
- Mora, M.; Veggi, D.; Santini, L.; Pizza, M.; Rappuoli, R. Reverse vaccinology. Drug Discov. Today 2003, 8, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Gloanec, N.; Guyard-Nicodème, M.; Chemaly, M.; Dory, D. Reverse vaccinology: A strategy also used for identifying potential vaccine antigens in poultry. Vaccine 2025, 48, 126756. [Google Scholar] [CrossRef]
- Rappuoli, R.; Bottomley, M.J.; D’Oro, U.; Finco, O.; De Gregorio, E. Reverse vaccinology 2.0: Human immunology instructs vaccine antigen design. J. Exp. Med. 2016, 213, 469–481. [Google Scholar] [CrossRef]
- Vishweshwaraiah, Y.L.; Dokholyan, N.V. Toward rational vaccine engineering. Adv. Drug Deliv. Rev. 2022, 183, 114142. [Google Scholar] [CrossRef]
- Pulendran, B. Systems vaccinology: Probing humanity’s diverse immune systems with vaccines. Proc. Natl. Acad. Sci. USA 2014, 111, 12300–12306. [Google Scholar] [CrossRef] [PubMed]
- Sugrue, J.A.; Duffy, D. Systems vaccinology studies–achievements and future potential. Microbes Infect. 2024, 26, 105318. [Google Scholar] [CrossRef]
- Plotkin, S. History of vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 12283–12287. [Google Scholar] [CrossRef]
- Soleymani, S.; Tavassoli, A.; Housaindokht, M.R. An overview of progress from empirical to rational design in modern vaccine development, with an emphasis on computational tools and immunoinformatics approaches. Comput. Biol. Med. 2022, 140, 105057. [Google Scholar] [CrossRef] [PubMed]
- Carri, I.; Schwab, E.; Podaza, E.; Alvarez, H.M.G.; Mordoh, J.; Nielsen, M.; Barrio, M.M. Beyond MHC binding: Immunogenicity prediction tools to refine neoantigen selection in cancer patients. Explor. Immunol. 2023, 3, 82–103. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).