
Targeted but Troubling: CYP450 Inhibition by Kinase and PARP Inhibitors and Its Clinical Implications



Abstract
1. Introduction
2. Cytochrome P450 Enzymes and Mechanisms of Inhibition
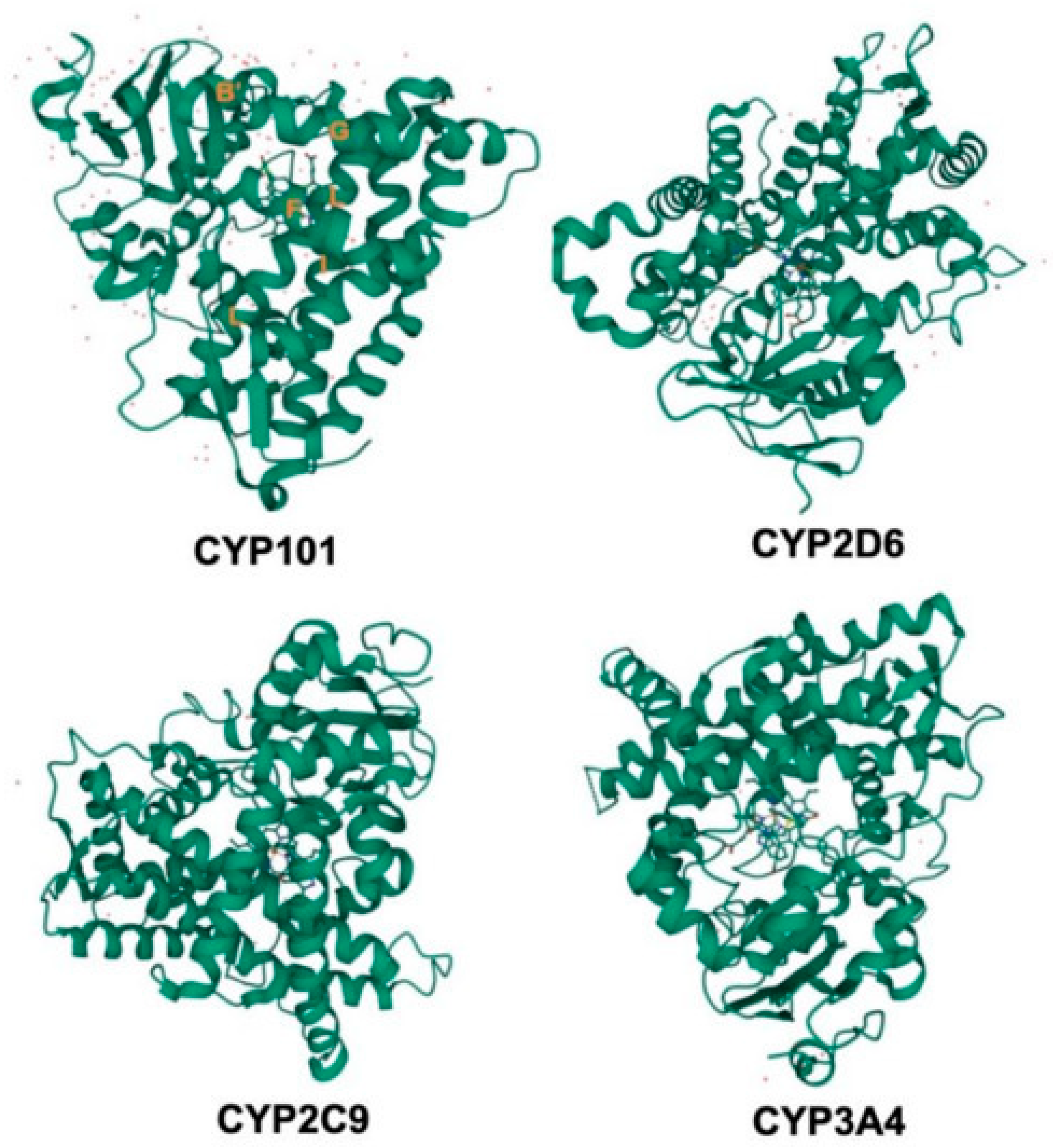
2.1. Structure and Function of CYP450 Enzymes
2.2. Types of CYP Inhibition
2.2.1. Reversible Inhibition
2.2.2. Irreversible Inhibition
2.2.3. Pseudo-Irreversible Inhibition
2.2.4. Mechanism-Based Inhibition
2.2.5. Time-Dependent Inhibition
2.2.6. Direct (Non-Metabolic) Inhibition
2.3. CYP3A4 as a Major Target in Oncology
3. KIs as Perpetrators of CYP Inhibition
4. PARP Inhibitors as Perpetrators of CYP Inhibition
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AUC | Area Under the Curve |
| BTK | Bruton’s Tyrosine Kinase |
| CYP450 | Cytochrome P450 |
| DDI | Drug–Drug Interaction |
| EMA | European Medicines Agency |
| EGFR | Epidermal Growth Factor Receptor |
| FDA | Food and Drug Administration |
| FMN | Flavin Mononucleotide |
| FAD | Flavin Adenine Dinucleotide |
| HR+ | Hormone Receptor Positive |
| KI | Kinase Inhibitor |
| MBI | Mechanism-Based Inhibition |
| NSCLC | Non-Small Cell Lung Cancer |
| PARP | Poly-Adenosine Diphosphate Ribose Polymerase |
| PBPK | Physiologically Based Pharmacokinetic (modeling) |
| ROS | Reactive Oxygen Species |
| TDI | Time-Dependent Inhibition |
| TDM | Therapeutic Drug Monitoring |
References
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef] [PubMed]
- Kato, M. Intestinal first-pass metabolism of CYP3A4 substrates. Drug Metab. Pharmacokinet. 2008, 23, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Gougis, P.; Hilmi, M.; Geraud, A.; Mir, O.; Funck-Brentano, C. Potential cytochrome P450-mediated pharmacokinetic interactions between herbs, food, and dietary supplements and cancer treatments. Crit. Rev. Oncol. Hematol. 2021, 166, 103342. [Google Scholar] [CrossRef] [PubMed]
- Zurth, C.; Koskinen, M.; Fricke, R.; Prien, O.; Korjamo, T.; Graudenz, K.; Denner, K.; Bairlein, M.; von Bühler, C.J.; Wilkinson, G.; et al. Drug-Drug Interaction Potential of Darolutamide: In Vitro and Clinical Studies. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 747–759. [Google Scholar] [CrossRef]
- Shirasaka, Y.; Sager, J.E.; Lutz, J.D.; Davis, C.; Isoherranen, N. Inhibition of CYP2C19 and CYP3A4 by omeprazole metabolites and their contribution to drug-drug interactions. Drug Metab. Dispos. 2013, 41, 1414–1424. [Google Scholar] [CrossRef]
- Wang, X.; Dowty, M.E.; Tripathy, S.; Le, V.H.; Huh, Y.; Curto, M.; Winton, J.A.; O’Gorman, M.T.; Chan, G.; Malhotra, B.K. Assessment of the Effects of Abrocitinib on the Pharmacokinetics of Probe Substrates of Cytochrome P450 1A2, 2B6 and 2C19 Enzymes and Hormonal Oral Contraceptives in Healthy Individuals. Eur. J. Drug Metab. Pharmacokinet. 2024, 49, 367–381. [Google Scholar] [CrossRef]
- Sharma, M.; Loh, K.P.; Nightingale, G.; Mohile, S.G.; Holmes, H.M. Polypharmacy and potentially inappropriate medication use in geriatric oncology. J. Geriatr. Oncol. 2016, 7, 346–353. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Current Approaches for Investigating and Predicting Cytochrome P450 3A4-Ligand Interactions. Adv. Exp. Med. Biol. 2015, 851, 83–105. [Google Scholar]
- Ye, F.; Dewanjee, S.; Li, Y.; Jha, N.K.; Chen, Z.S.; Kumar, A.; Vishakha; Behl, T.; Jha, S.K.; Tang, H. Advancements in clinical aspects of targeted therapy and immunotherapy in breast cancer. Mol. Cancer 2023, 22, 105. [Google Scholar] [CrossRef]
- Wendl, T.; Frechen, S.; Gerisch, M.; Heinig, R.; Eissing, T. Physiologically-based pharmacokinetic modeling to predict CYP3A4-mediated drug-drug interactions of finerenone. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 199–211. [Google Scholar] [CrossRef]
- Stodtmann, S.; Nuthalapati, S.; Eckert, D.; Kasichayanula, S.; Joshi, R.; Bach, B.A.; Mensing, S.; Menon, R.; Xiong, H. A Population Pharmacokinetic Meta-Analysis of Veliparib, a PARP Inhibitor, Across Phase 1/2/3 Trials in Cancer Patients. J. Clin. Pharmacol. 2021, 61, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Stanisławiak-Rudowicz, J.; Karbownik, A.; Szkutnik-Fiedler, D.; Otto, F.; Grabowski, T.; Wolc, A.; Grześkowiak, E.; Szałek, E. Bidirectional pharmacokinetic drug interactions between olaparib and metformin. Cancer Chemother. Pharmacol. 2024, 93, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Beunk, L.; Nijenhuis, M.; Soree, B.; de Boer-Veger, N.J.; Buunk, A.M.; Guchelaar, H.J.; Houwink, E.J.F.; Risselada, A.; Rongen, G.A.P.J.M.; van Schaik, R.H.N.; et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction between CYP2D6, CYP3A4 and CYP1A2 and antipsychotics. Eur. J. Hum. Genet. 2024, 32, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.; Crosby, I.; Yip, V.; Maguire, P.; Pirmohamed, M.; Turner, R.M. A Review of the Important Role of CYP2D6 in Pharmacogenomics. Genes 2020, 11, 1295. [Google Scholar] [CrossRef]
- Bojić, M. Predklinička ispitivanja inhibicijskog i interakcijskog potencijala novih lijekova na razini citokroma P450. Farm. Glas. 2015, 71, 229–242. [Google Scholar]
- Stading, R.; Couroucli, X.; Lingappan, K.; Moorthy, B. The role of cytochrome P450 (CYP) enzymes in hyperoxic lung injury. Expert Opin. Drug Metab. Toxicol. 2021, 17, 171–178. [Google Scholar] [CrossRef]
- Rendić, S. Summary of information on human CYP enzymes: Human P450 metabolism data. Drug Metab. Rev. 2002, 34, 83–448. [Google Scholar]
- Laursen, T.; Jensen, K.; Møller, B.L. Conformational changes of the NADPH-dependent cytochrome P450 reductase in the course of electron transfer to cytochromes P450. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 132–138. [Google Scholar] [CrossRef]
- Williams, P.A.; Cosme, J.; Vinkovic, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef]
- Drug Metabolism—The Importance of Cytochrome P450 3A4. Available online: https://www.medsafe.govt.nz/profs/puarticles/march2014drugmetabolismcytochromep4503a4.htm (accessed on 29 March 2025).
- Mar, P.L.; Gopinathannair, R.; Gengler, B.E.; Chung, M.K.; Perez, A.; Dukes, J.; Ezekowitz, M.D.; Lakkireddy, D.; Lip, G.Y.H.; Miletello, M.; et al. Drug Interactions Affecting Oral Anticoagulant Use. Circ. Arrhythm. Electrophysiol. 2022, 15, e007956. [Google Scholar] [CrossRef]
- Coates, S.; Lazarus, P. Hydrocodone, Oxycodone, and Morphine Metabolism and Drug—Drug Interactions. J. Pharmacol. Exp. Ther. 2023, 387, 150–169. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cytochrome P450 reductase (POR) as a ferroptosis fuel. Protein Cell 2021, 12, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, J.; Danek, P.J.; Basińska-Ziobroń, A.; Pukło, R.; Daniel, W.A. In vitro inhibition of human cytochrome P450 enzymes by the novel atypical antipsychotic drug asenapine: A prediction of possible drug-drug interactions. Pharmacol. Rep. 2020, 72, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Kondža, M. Mehanizam Inhibicijskoga Učinka na Citokrom P450 3A4. Ph.D. Thesis, Faculty of Pharmacy and Biochemistry, University of Zagreb, Zagreb, Croatia, 2022. [Google Scholar]
- Rendić, S.; Medić-Šarić, M. Metabolizam Lijekova i Odabranih Ksenobiotika, 1st ed.; Medicinska Naklada: Zagreb, Croatia, 2013. [Google Scholar]
- Zhao, B.; Waterman, M.R. Moonlighting cytochrome P450 monooxygenases. IUBMB Life 2011, 63, 473–477. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef]
- Kondža, M.; Brizić, I.; Jokić, S. Flavonoids as CYP3A4 inhibitors in vitro. Biomedicines 2024, 12, 644. [Google Scholar] [CrossRef]
- Bjelaković, G.; Stojanović, I.; Bjelaković, G.B.; Pavlović, D.; Kocić, G.; Daković-Milić, A. Competitive inhibitors of enzymes and their therapeutic application. Med. Biol. 2002, 9, 201–206. [Google Scholar]
- Usmani, K.A.; Tang, J. Human cytochrome P450: Metabolism of testosterone by CYP3A4 and inhibition by ketoconazole. Curr. Protoc. Toxicol. 2004, 20, 4–13. [Google Scholar] [CrossRef]
- Deodhar, M.; Rihani, S.B.A.; Darakjian, L.; Turgeon, J.; Michaud, V. Assessing the Mechanism of Fluoxetine-Mediated CYP2D6 Inhibition. Pharmaceutics 2021, 13, 148. [Google Scholar] [CrossRef]
- Williams, J. Enzyme inhibition and induction. Anaesth. Intensive Care Med. 2008, 9, 165–166. [Google Scholar] [CrossRef]
- Wang, X.; Dowty, M.E.; Wouters, A.; Tatulych, S.; Connell, C.A.; Le, V.H.; Tripathy, S.; O’Gorman, M.T.; Winton, J.A.; Yin, N.; et al. Assessment of the Effects of Inhibition or Induction of CYP2C19 and CYP2C9 Enzymes, or Inhibition of OAT3, on the Pharmacokinetics of Abrocitinib and Its Metabolites in Healthy Individuals. Eur. J. Drug Metab. Pharmacokinet. 2021, 47, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Pai, M.P.; Graci, D.M.; Amsden, G.W. Macrolide drug interactions: An update. Ann. Pharmacother. 2000, 34, 495–513. [Google Scholar] [CrossRef] [PubMed]
- Westphal, J.F. Macrolide-induced clinically relevant drug interactions with cytochrome P-450 (CYP) 3A4: An update focused on clarithromycin, azithromycin and dirithromycin. Br. J. Clin. Pharmacol. 2000, 50, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Fairman, D.A.; Collins, C.; Chapple, S. Progress curve analysis of CYP1A2 inhibition: A more informative approach to the assessment of mechanism-based inactivation? Drug Metab. Dispos. 2007, 35, 2159–2165. [Google Scholar] [CrossRef]
- Loos, N.H.C.; Beijnen, J.H.; Schinkel, A.H. The Mechanism-Based Inactivation of CYP3A4 by Ritonavir: What Mechanism? Int. J. Mol. Sci. 2022, 23, 9866. [Google Scholar] [CrossRef]
- Wang, Y.H.; Jones, D.R.; Hall, S.D. Prediction of cytochrome P450 3A inhibition by verapamil enantiomers and their metabolites. Drug Metab. Dispos. 2004, 32, 259–266. [Google Scholar] [CrossRef]
- Blat, Y. Non-competitive inhibition by active site binders. Chem. Biol. Drug Des. 2010, 75, 535–540. [Google Scholar] [CrossRef]
- Boulton, S.; Selvaratnam, R.; Blondeau, J.P.; Lezoualc’h, F.; Melacini, G. Mechanism of selective enzyme inhibition through uncompetitive regulation of an allosteric agonist. J. Am. Chem. Soc. 2018, 140, 9624–9637. [Google Scholar] [CrossRef]
- Johnson, D.S.; Weerapana, E.; Cravatt, B.F. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010, 2, 949–964. [Google Scholar] [CrossRef]
- Kim, H.G.; Lee, H.S.; Jeon, J.S.; Choi, Y.J.; Choi, Y.J.; Yoo, S.Y.; Kim, S.K. Quasi-irreversible inhibition of CYP2D6 by berberine. Pharmaceutics 2020, 12, 916. [Google Scholar] [CrossRef]
- Orlando, R.; De Martin, S.; Pegoraro, P.; Quintieri, L.; Palatini, P. Irreversible CYP3A inhibition accompanied by plasma protein–binding displacement: A comparative analysis in subjects with normal and impaired liver function. Clin. Pharmacol. Ther. 2009, 85, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Chan, S.Y.; Goh, B.C.; Chan, E.; Duan, W.; Huang, M.; McLeod, H.L. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokinet. 2005, 44, 279–304. [Google Scholar] [CrossRef] [PubMed]
- Bojić, M.; Kondža, M.; Rimac, H.; Benković, G.; Maleš, Ž. The effect of flavonoid aglycones on the CYP1A2, CYP2A6, CYP2C8 and CYP2D6 enzymes activity. Molecules 2019, 24, 3174. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.G.; Dresser, G.K.; Bend, J.R. Bergamottin, lime juice, and red wine as inhibitors of cytochrome P450 3A4 activity: Comparison with grapefruit juice. Clin. Pharmacol. Ther. 2003, 73, 529–537. [Google Scholar] [CrossRef]
- Kondža, M.; Mandić, M.; Ivančić, I.; Vladimir-Knežević, S.; Brizić, I. Artemisia annua L. Extracts Irreversibly Inhibit the Activity of CYP2B6 and CYP3A4 Enzymes. Biomedicines 2023, 11, 232. [Google Scholar] [CrossRef]
- Food and Drug Administration. ICH M12 Drug Interaction Final Guidance—In Vitro DDI Assessments. Available online: https://www.fda.gov/media/184437/download (accessed on 26 April 2025).
- European Medicines Agency. Guideline on the investigation of drug interactions. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf (accessed on 26 April 2025).
- Ochs, R.S. Understanding enzyme inhibition. J. Chem. Educ. 2000, 77, 1453. [Google Scholar] [CrossRef]
- Van Erp, N.P.; Gelderblom, H.; Karlsson, M.O.; Li, J.; Zhao, M.; Ouwerkerk, J.; Sparreboom, A. Influence of CYP3A4 inhibition on the steady-state pharmacokinetics of imatinib. Clin. Cancer Res. 2007, 13, 7394–7400. [Google Scholar] [CrossRef]
- Hamilton, M.; Wolf, J.L.; Drolet, D.W.; Fettner, S.H.; Rakhit, A.K.; Witt, K.; Lum, B.L. The effect of rifampicin, a prototypical CYP3A4 inducer, on erlotinib pharmacokinetics in healthy subjects. Cancer Chemother. Pharmacol. 2014, 73, 613–621. [Google Scholar] [CrossRef]
- Gao, N.; Zhang, X.; Hu, X.; Kong, Q.; Cai, J.; Hu, G.; Qian, J. The influence of CYP3A4 genetic polymorphism and proton pump inhibitors on osimertinib metabolism. Front. Pharmacol. 2022, 13, 794931. [Google Scholar] [CrossRef]
- Teo, Y.L.; Saetaew, M.; Chanthawong, S.; Yap, Y.S.; Chan, E.C.Y.; Ho, H.K.; Chan, A. Effect of CYP3A4 inducer dexamethasone on hepatotoxicity of lapatinib: Clinical and in vitro evidence. Breast Cancer Res. Treat. 2012, 133, 703–711. [Google Scholar] [CrossRef]
- De Zwart, L.; Snoeys, J.; De Jong, J.; Sukbuntherng, J.; Mannaert, E.; Monshouwer, M. Ibrutinib dosing strategies based on interaction potential of CYP3A4 perpetrators using physiologically based pharmacokinetic modeling. Clin. Pharmacol. Ther. 2016, 100, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Posada, M.M.; Morse, B.L.; Turner, P.K.; Kulanthaivel, P.; Hall, S.D.; Dickinson, G.L. Predicting clinical effects of CYP3A4 modulators on abemaciclib and active metabolites exposure using physiologically based pharmacokinetic modeling. J. Clin. Pharmacol. 2020, 60, 915–930. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, A.; Kumondai, M.; Kobayashi, D.; Kikuchi, M.; Ueki, Y.; Sato, Y.; Mano, N. Plasma venetoclax concentrations in patients with acute myeloid leukemia treated with CYP3A4 inhibitors. Yakugaku Zasshi 2024, 144, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.M.; Li, Y.H.; Lu, X.R.; Wang, R.; Pang, N.H.; Xu, R.A.; Hu, G.X. Characterization of genetic variation in CYP3A4 on the metabolism of cabozantinib in vitro. Chem. Res. Toxicol. 2019, 32, 1583–1590. [Google Scholar] [CrossRef]
- Dutreix, C.; Munarini, F.; Lorenzo, S.; Roesel, J.; Wang, Y. Investigation into CYP3A4-mediated drug–drug interactions on midostaurin in healthy volunteers. Cancer Chemother. Pharmacol. 2013, 72, 1223–1234. [Google Scholar] [CrossRef]
- Gibbons, J.A.; de Vries, M.; Krauwinkel, W.; Ohtsu, Y.; Noukens, J.; van der Walt, J.S.; Ouatas, T. Pharmacokinetic drug interaction studies with enzalutamide. Clin. Pharmacokinet. 2015, 54, 1057–1069. [Google Scholar] [CrossRef]
- Mikus, G.; Foerster, K.I. Role of CYP3A4 in kinase inhibitor metabolism and assessment of CYP3A4 activity. Transl. Cancer Res. 2017, 6 (Suppl. S10), S1592–S1599. [Google Scholar] [CrossRef]
- Guilhaumou, R.; Solas, C.; Bourgarel-Rey, V.; Quaranta, S.; Rome, A.; Simon, N.; Andre, N. Impact of plasma and intracellular exposure and CYP3A4, CYP3A5, and ABCB1 genetic polymorphisms on vincristine-induced neurotoxicity. Cancer Chemother. Pharmacol. 2011, 68, 1633–1638. [Google Scholar] [CrossRef]
- Gertz, M.; Cartwright, C.M.; Hobbs, M.J.; Kenworthy, K.E.; Rowland, M.; Houston, J.B.; Galetin, A. Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: Application of PBPK modeling in the assessment of drug—Drug interaction potential. Pharm. Res. 2013, 30, 761–780. [Google Scholar] [CrossRef]
- Kato, M.; Chiba, K.; Ito, T.; Koue, T.; Sugiyama, Y. Prediction of interindividual variability in pharmacokinetics for CYP3A4 substrates in humans. Drug Metab. Pharmacokinet. 2010, 25, 367–378. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, R.W.; van Gelder, T.; Mathijssen, R.H.; Jansman, F.G. Drug–drug interactions with tyrosine-kinase inhibitors: A clinical perspective. Lancet Oncol. 2014, 15, e315–e326. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Haap, M.; Kopp, H.G.; Lipp, H.P. Tyrosine kinase inhibitors—A review on pharmacology, metabolism and side effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef]
- Gabora, K.; Piciu, A.; Badulescu, I.C.; Larg, M.I.; Stoian, A.I.; Piciu, D. Current evidence on thyroid related adverse events in patients treated with protein tyrosine kinase inhibitors. Drug Metab. Rev. 2019, 51, 562–569. [Google Scholar] [CrossRef]
- Xu, X.Y.; Chen, J.; Chen, Z.X.; Zhang, Z.Y.; Jin, L.H.; Luo, J.C.; Qian, J.C. CYP3A4 activity variations can lead to stratified metabolism of abemaciclib. Int. J. Biol. Macromol. 2025, 304, 140836. [Google Scholar] [CrossRef]
- Eisenmann, E.D.; Fu, Q.; Muhowski, E.M.; Jin, Y.; Uddin, M.E.; Garrison, D.A.; Baker, S.D. Intentional modulation of ibrutinib pharmacokinetics through CYP3A inhibition. Cancer Res. Commun. 2021, 1, 79–89. [Google Scholar] [CrossRef]
- MacLeod, A.K.; Lin, D.; Huang, J.T.J.; McLaughlin, L.A.; Henderson, C.J.; Wolf, C.R. Identification of novel pathways of osimertinib disposition and potential implications for the outcome of lung cancer therapy. Clin. Cancer Res. 2018, 24, 2138–2147. [Google Scholar] [CrossRef]
- Teng, W.C.; Oh, J.W.; New, L.S.; Wahlin, M.D.; Nelson, S.D.; Ho, H.K.; Chan, E.C.Y. Mechanism-based inactivation of cytochrome P450 3A4 by lapatinib. Mol. Pharmacol. 2010, 78, 693–703. [Google Scholar] [CrossRef]
- McCormick, A.; Swaisland, H.; Reddy, V.P.; Learoyd, M.; Scarfe, G. In vitro evaluation of the inhibition and induction potential of olaparib, a potent poly (ADP-ribose) polymerase inhibitor, on cytochrome P450. Xenobiotica 2018, 48, 555–564. [Google Scholar] [CrossRef]
- Liao, M.; Beltman, J.; Giordano, H.; Harding, T.C.; Maloney, L.; Simmons, A.D.; Xiao, J.J. Clinical pharmacokinetics and pharmacodynamics of rucaparib. Clin. Pharmacokinet. 2022, 61, 1477–1493. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Long, X.; Wang, J. Metabolism-related pharmacokinetic drug–drug interactions with poly (ADP-ribose) polymerase inhibitors. Oncol. Rep. 2022, 47, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, R.J.; Verheggen, R.; Boerrigter, E.; Stanzani, M.; Verweij, P.E.; Blijlevens, N.M.; Lewis, R.E. Management of drug—Drug interactions of targeted therapies for haematological malignancies and triazole antifungal drugs. Lancet Haematol. 2022, 9, e58–e72. [Google Scholar] [CrossRef] [PubMed]
- Dziadkowiec, K.N.; Gąsiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP inhibitors: Review of mechanisms of action and BRCA1/2 mutation targeting. Menopause Rev. 2016, 15, 215–219. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; De Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Dirix, L.; Swaisland, H.; Verheul, H.M.; Rottey, S.; Leunen, K.; Jerusalem, G.; Plummer, R. Effect of itraconazole and rifampin on the pharmacokinetics of olaparib in patients with advanced solid tumors: Results of two phase I open-label studies. Clin. Ther. 2016, 38, 2286–2299. [Google Scholar] [CrossRef]
- Bruin, M.; Sonke, G.S.; Beijnen, J.H.; Huitema, A.D.R. Pharmacokinetics and pharmacodynamics of PARP inhibitors in oncology. Clin. Pharmacokinet. 2022, 61, 13. [Google Scholar] [CrossRef]
- Liao, M.; Jaw-Tsai, S.; Beltman, J.; Simmons, A.D.; Harding, T.C.; Xiao, J.J. Evaluation of in vitro absorption, distribution, metabolism, and excretion and assessment of drug-drug interaction of rucaparib, an orally potent poly (ADP-ribose) polymerase inhibitor. Xenobiotica 2020, 50, 1032–1042. [Google Scholar] [CrossRef]
- Deshpande, P.P.; Perazella, M.A.; Jhaveri, K.D. PARP inhibitors and the kidney. J. Onco-Nephrol. 2021, 5, 42–47. [Google Scholar] [CrossRef]
- Graziani, G.; Szabó, C. Clinical perspectives of PARP inhibitors. Pharmacol. Res. 2005, 52, 109–118. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Type of Inhibition | Effect on Km/Vmax | Clinical Examples | Relevance in Oncology | References |
|---|---|---|---|---|
| competitive | ↑ Km Vmax unchanged | ketoconazole (CYP3A4) fluoxetine (CYP2D6) | high (e.g., oral anticancer agents) | [31,32] |
| non-competitive | ↓ Vmax Km unchanged | ketoconazole (CYP3A4) | rare in clinical practice | [33] |
| uncompetitive | ↓ Km ↓ Vmax | theoretical; rare in vivo | no practical relevance | [32] |
| direct | depends on subtype | fluconazole (CYP2C9) | relevant in supportive care | [34] |
| pseudo-irreversible | similar to irreversible | erythromycin (CYP3A4) | may prolong cytotoxic exposure | [35,36] |
| irreversible | permanent enzyme inactivation | furafylline (CYP1A2) | may increase toxicity of anticancer drugs | [37] |
| mechanism-based inhibition | enzyme inactivation | ritonavir (CYP3A4) | highly relevant with kinase inhibitors | [38] |
| time-dependent inhibition | inhibition increases over time | verapamil (CYP3A4) | clinically important due to progressive effect | [39] |
| Drug Name | Drug Class/Target | Main Clinical Uses | Notes on CYP3A4 Metabolism | References |
|---|---|---|---|---|
| imatinib | tyrosine kinase inhibitor (BCR-ABL) | chronic myeloid leukemia | primarily metabolized by CYP3A4; exposure can be increased by CYP3A4 inhibitors | [52] |
| erlotinib | EGFR inhibitor | NSCLC | extensively metabolized by CYP3A4 and CYP1A2 | [53] |
| osimertinib | EGFR inhibitor (T790M mutant) | NSCLC (EGFR-mutant) | CYP3A4 is the main enzyme responsible for its metabolism | [54] |
| lapatinib | HER2/EGFR inhibitor | HER2-positive breast cancer | metabolized by CYP3A4; caution with inhibitors/inducers | [55] |
| ibrutinib | BTK inhibitor | chronic lymphocytic leukemia | primarily metabolized by CYP3A4; strong inhibitors increase AUC significantly | [56] |
| abemaciclib | CDK4/6 inhibitor | HR+/HER2- breast cancer | major substrate of CYP3A4; time-dependent inhibition potential | [57] |
| venetoclax | BCL-2 inhibitor | CLL, AML | Metabolized by CYP3A4; risk of tumor lysis syndrome increased with strong inhibitors | [58] |
| cabozantinib | multi-kinase inhibitor | renal cell carcinoma, thyroid cancer | CYP3A4 is the primary metabolic pathway | [59] |
| midostaurin | FLT3 inhibitor | acute myeloid leukemia | CYP3A4 extensively involved in metabolism; dose adjustment recommended with inhibitors | [60] |
| enzalutamide | androgen receptor inhibitor | prostate cancer | CYP3A4 involved in metabolism; also induces CYP3A4 | [61] |
| Drug Name | Class | Type of Inhibition | Mechanism | Clinical Implications | References |
|---|---|---|---|---|---|
| abemaciclib | CDK4/6 inhibitor | TDI | TDI observed in vitro; major substrate and weak inhibitor | increased exposure with CYP3A4 inhibitors; requires dose adjustment | [71] |
| ibrutinib | BTK inhibitor | pseudo-irreversible | forms a strong complex with CYP3A4; also self-inhibits | high risk of toxicity with strong CYP3A4 inhibitors (e.g., azoles) | [72] |
| osimertinib | EGFR inhibitor | weak reversible | minimal inhibition in vitro; mainly a substrate | moderate interaction potential in combinations | [73] |
| lapatinib | HER2/EGFR inhibitor | reversible competitive | substrate and reversible inhibitor; may self-inhibit metabolism | nonlinear PK; requires monitoring in combinations | [74] |
| olaparib | PARP inhibitor | TDI | moderate TDI; metabolism via CYP3A4; inhibition increases with exposure | requires dose adjustment with CYP3A4 inhibitors/inducers | [75] |
| rucaparib | PARP inhibitor | direct time-dependent | moderate inhibitor of multiple CYPs; affects metabolism and transporters | increases exposure to CYP3A4 substrates | [76] |
| niraparib | PARP inhibitor | weak | primarily metabolized by non-CYP enzymes | low DDI risk via CYP450 | [77] |
| talazoparib | PARP inhibitor | weak | minimal CYP involvement; primarily renally excreted | low potential for CYP-mediated DDIs | [78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondža, M.; Bukić, J.; Ćavar, I.; Tubić, B. Targeted but Troubling: CYP450 Inhibition by Kinase and PARP Inhibitors and Its Clinical Implications. Drugs Drug Candidates 2025, 4, 24. https://doi.org/10.3390/ddc4020024
Kondža M, Bukić J, Ćavar I, Tubić B. Targeted but Troubling: CYP450 Inhibition by Kinase and PARP Inhibitors and Its Clinical Implications. Drugs and Drug Candidates. 2025; 4(2):24. https://doi.org/10.3390/ddc4020024
Chicago/Turabian StyleKondža, Martin, Josipa Bukić, Ivan Ćavar, and Biljana Tubić. 2025. "Targeted but Troubling: CYP450 Inhibition by Kinase and PARP Inhibitors and Its Clinical Implications" Drugs and Drug Candidates 4, no. 2: 24. https://doi.org/10.3390/ddc4020024
APA StyleKondža, M., Bukić, J., Ćavar, I., & Tubić, B. (2025). Targeted but Troubling: CYP450 Inhibition by Kinase and PARP Inhibitors and Its Clinical Implications. Drugs and Drug Candidates, 4(2), 24. https://doi.org/10.3390/ddc4020024