
Biological Profile of Synthetic and Natural Indole Derivatives: Paving New Paths in Cancer Treatment



Abstract

1. Introduction
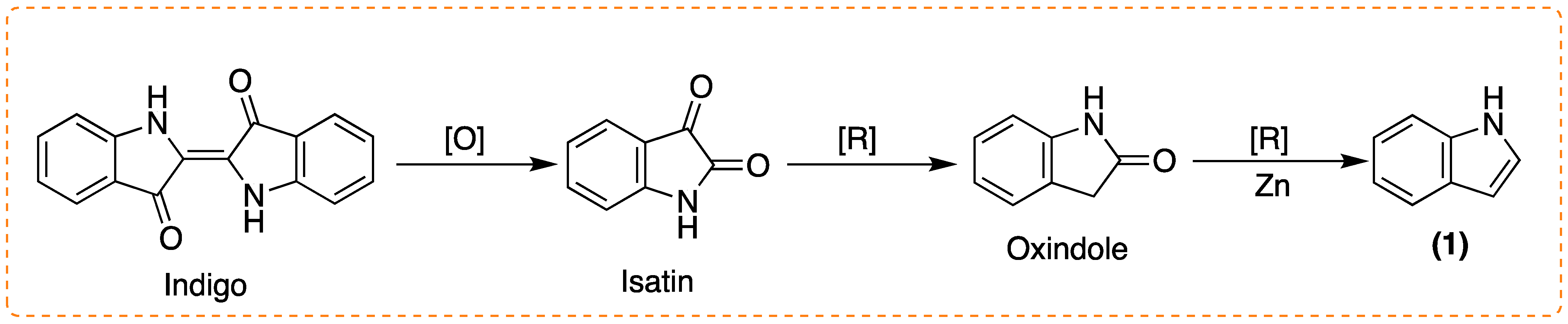
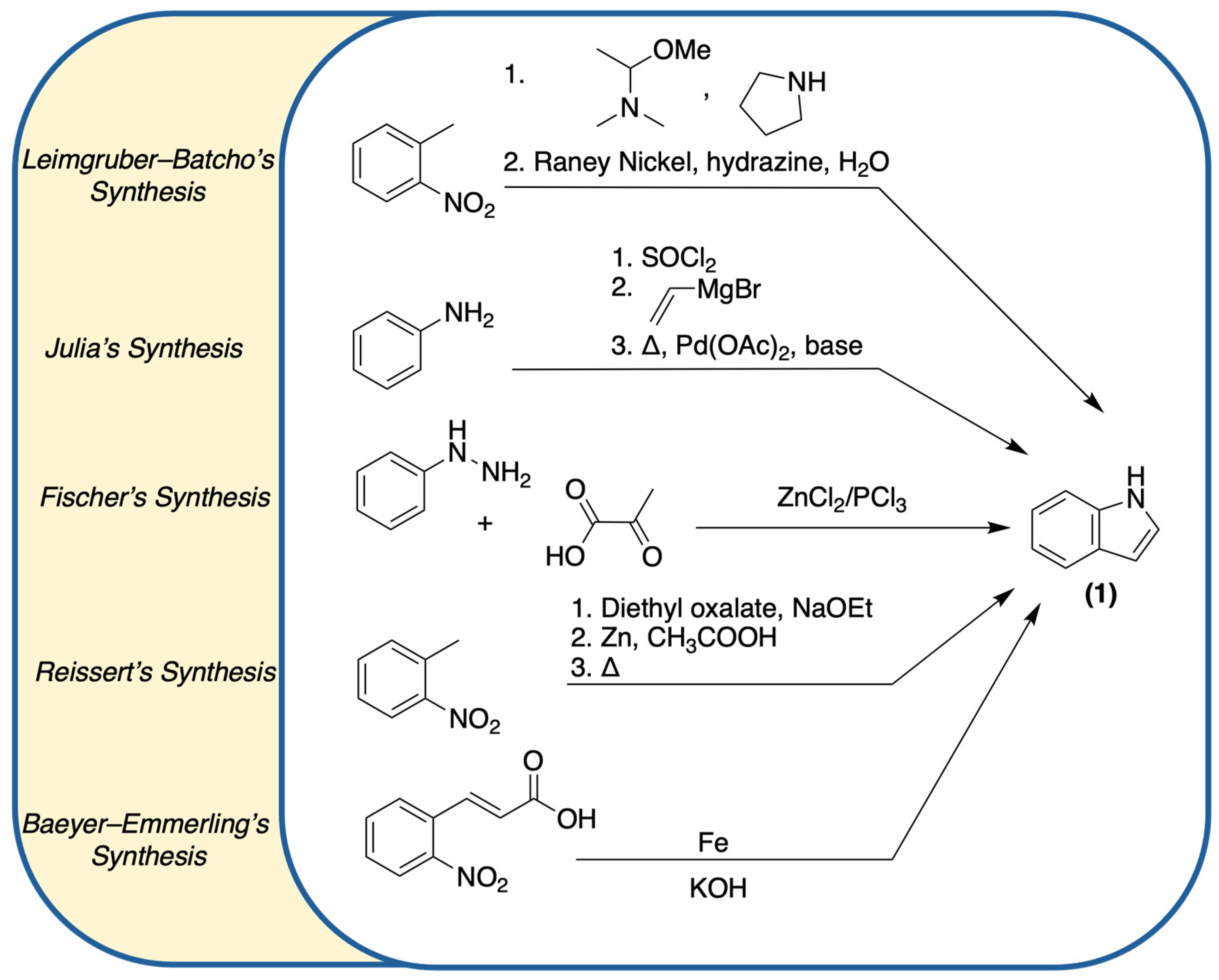
1.1. Accessing (1) and Derivatives: Synthetic Processes
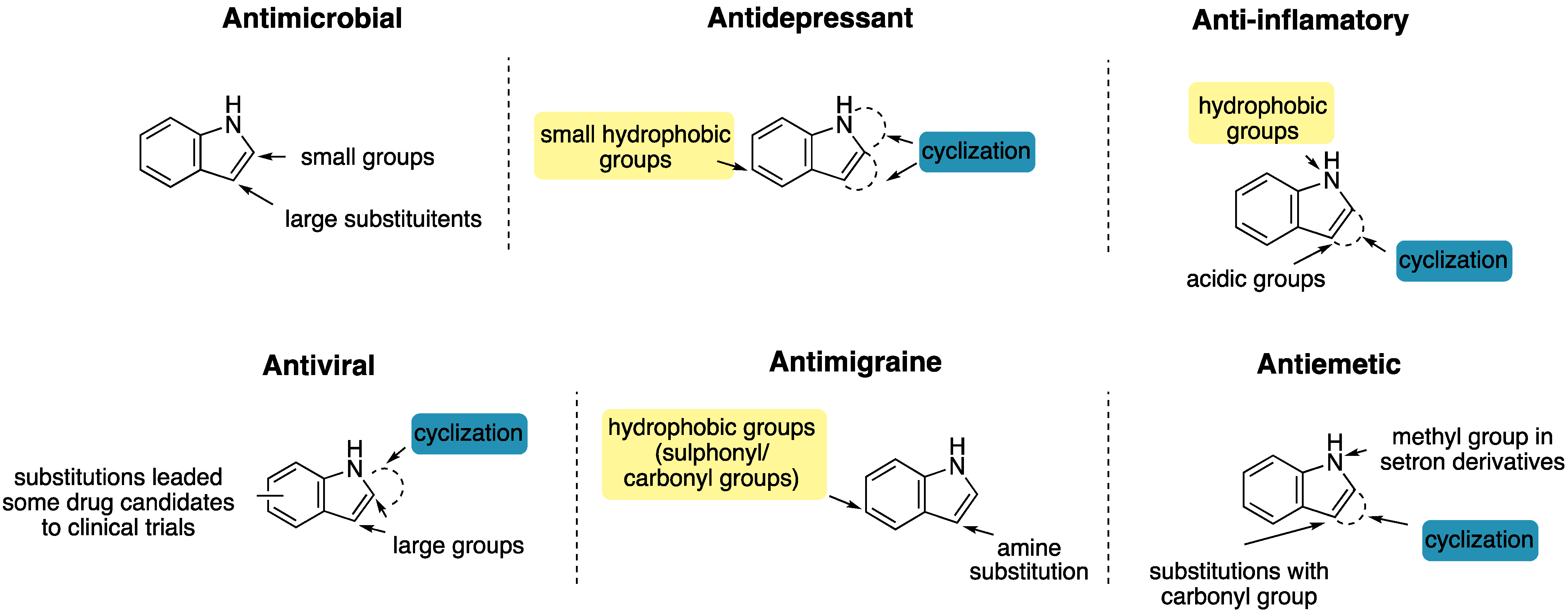
1.2. Biological Profile of the Scaffold
1.2.1. Antimicrobial Activity
1.2.2. Antidepressant Activity
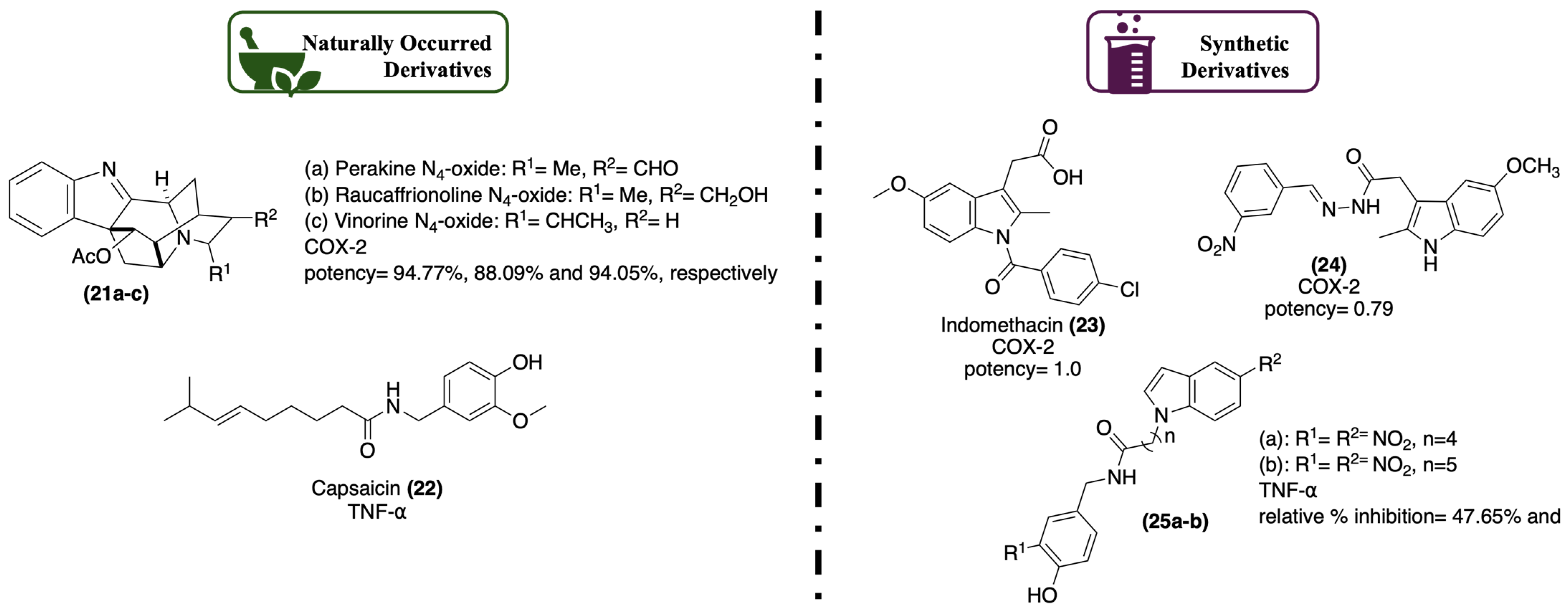
1.2.3. Anti-Inflammatory Activity
1.2.4. Antiviral Activity
1.2.5. Antimigraine Activity
1.2.6. Antiemetic Activity
2. Indole in Cancer
2.1. Main Targets of Indole Derivatives in Cancer
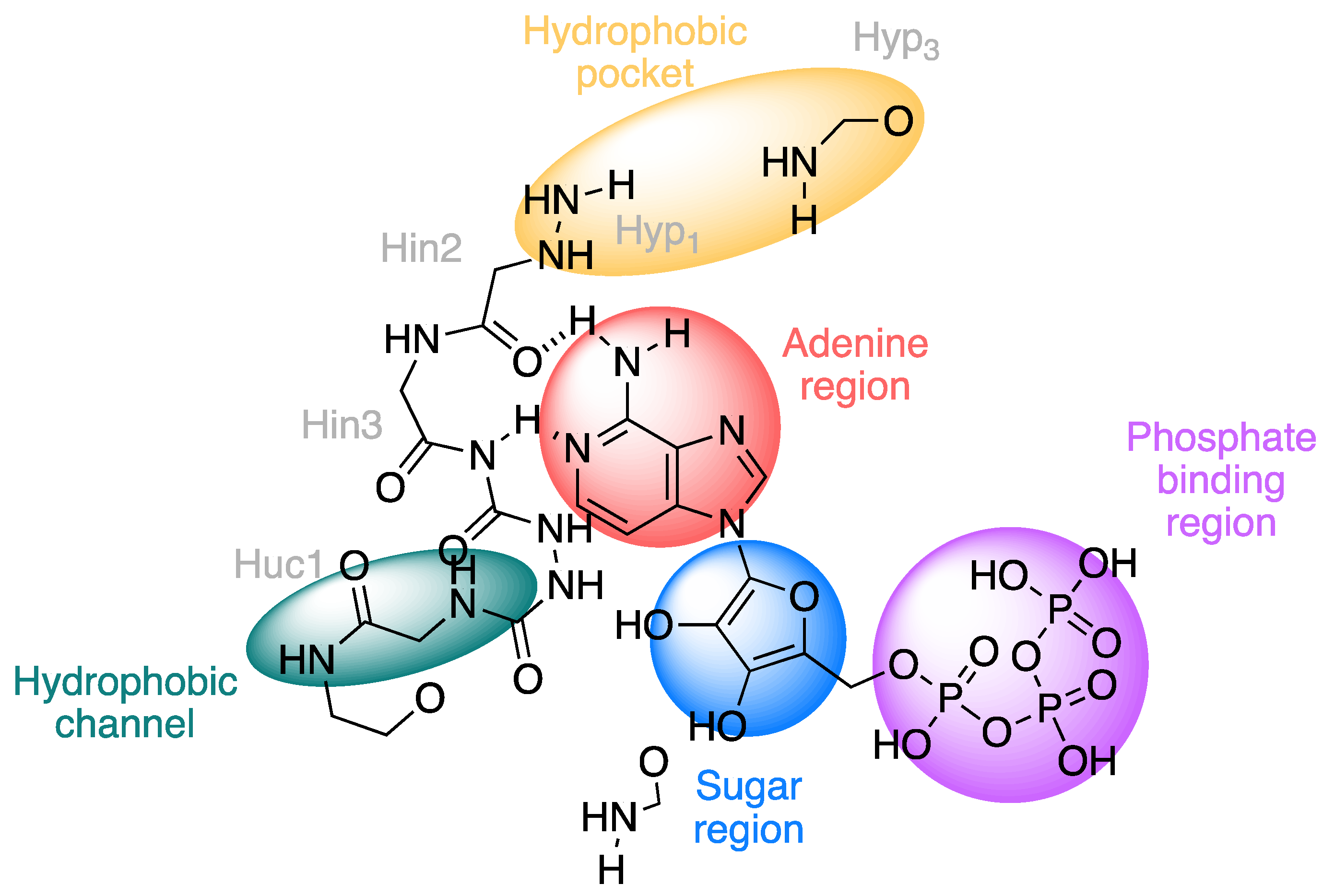
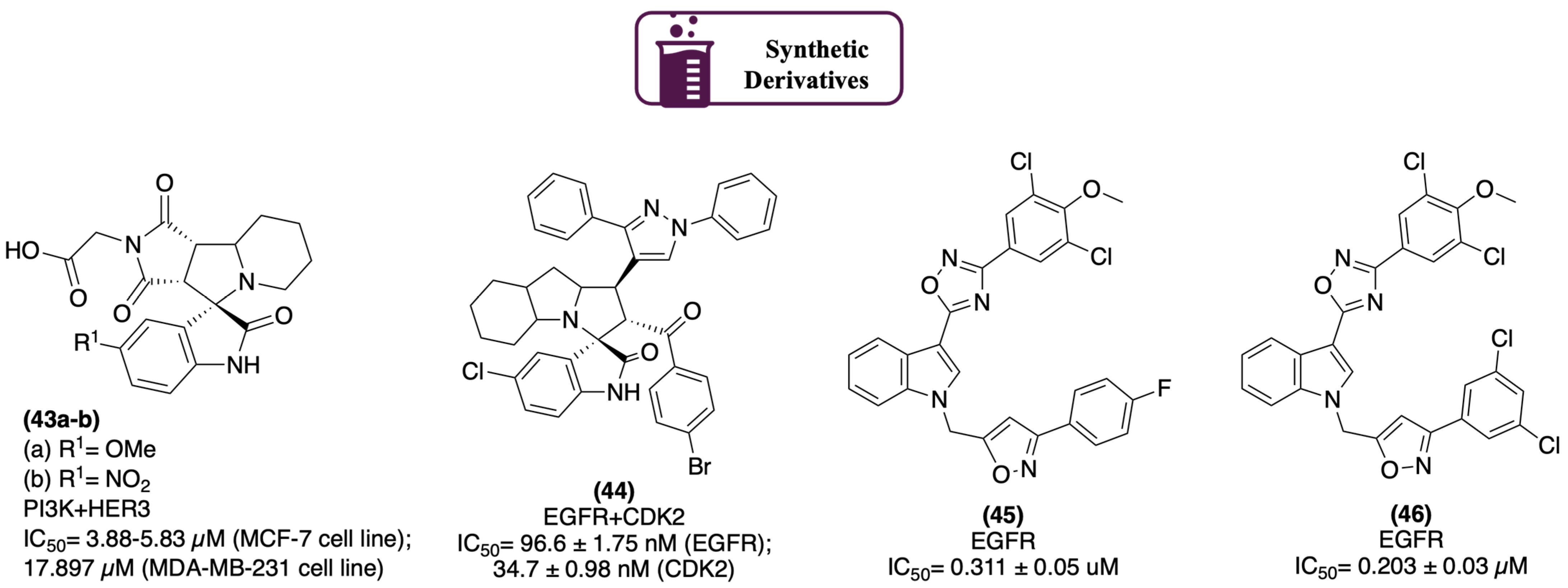
2.1.1. Protein Kinases
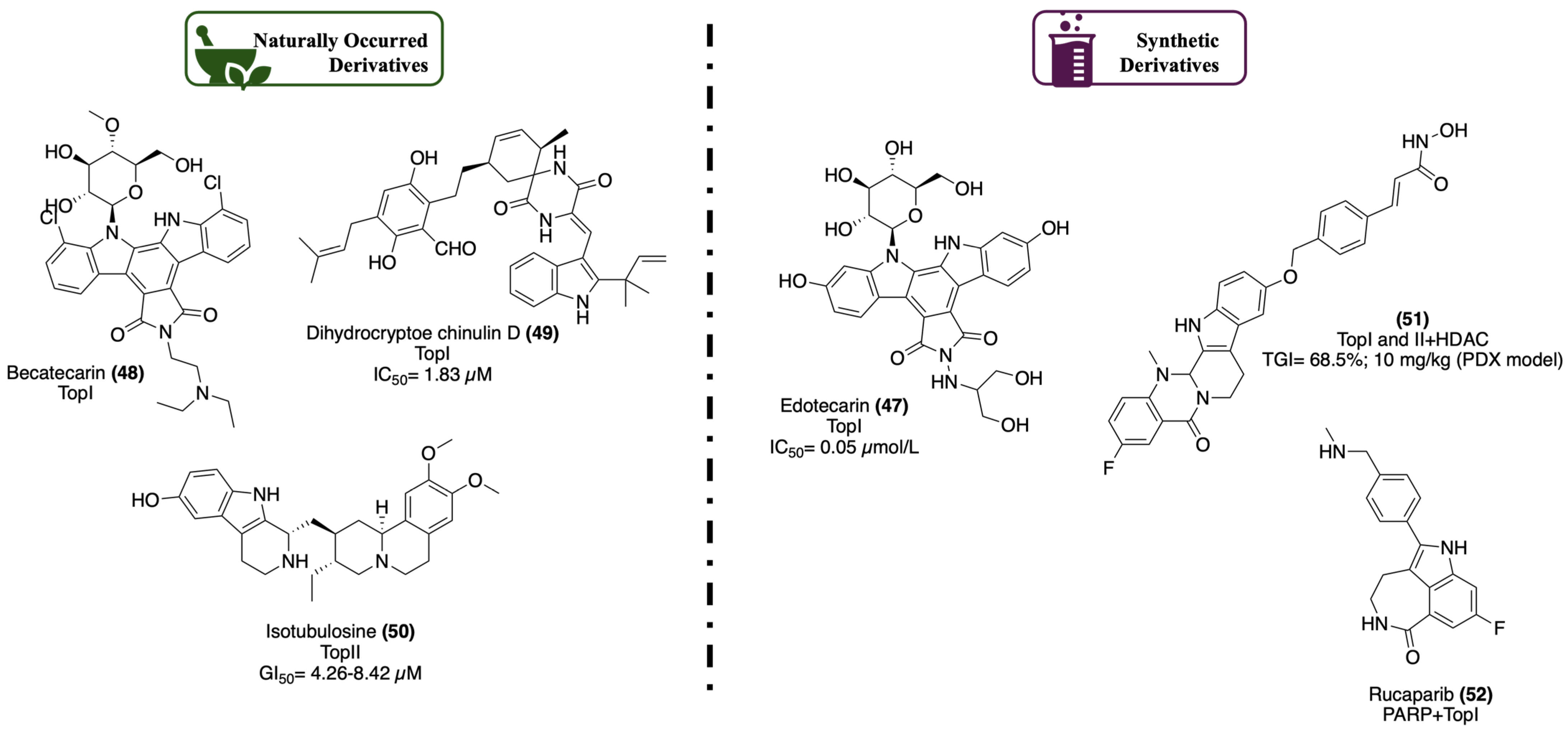
2.1.2. DNA Topoisomerase
2.1.3. Tubulin Polymerization
2.1.4. P-Glycoprotein
2.1.5. Other Interesting Targets
3. Conclusions and Future Perspectives
Funding
Conflicts of Interest
Abbreviations
| Abl | abelson leukemia gene |
| ACh | acetylcholine |
| AChE | acetylcholinesterase |
| ADV | adenovirus |
| Akf | protein kinase B |
| AML | acute myeloid leukemia |
| APC | adenomatous polyposis coli |
| API(s) | active pharmaceutical ingredient(s) |
| ATP | adenosine triphosphate |
| BRCA1 | associated ring domain 1 |
| BRCA1 | breast cancer gene 1 |
| BRCA2 | breast cancer gene 2 |
| CDK | cyclin-dependent kinase |
| COX-2 | cyclooxygenase-2 |
| DRAK1 | kinase-related apoptosis-inducing protein kinase 1 |
| EGFR | epithelial growth factor receptor |
| FLT3 | fms-like tyrosine kinase 3 |
| GSK-β | glycogene synthase kinase beta |
| GTP | guanosine triphosphate |
| HCV | hepatitis C virus |
| HDAC | topoisomerase-histone deacetylase |
| HER2 | human epidermal growth factor receptor 2 |
| HER3 | human epidermal growth factor receptor 3 |
| HSV | herpes simplex virus |
| HVB | hepatitis B virus |
| KIT | proto-oncogene c-kit |
| L-Gln | glutamine |
| MAO | monoamine oxidase |
| MDM2 | mouse double minute 2 homolog |
| MLH1 | mutL homolog 1 |
| mRNA | messenger ribonucleic acid |
| NNRTI | nonnucleoside reverse transcriptase inhibitor |
| NRTK | non-receptor tyrosine kinase |
| P | phosphate |
| p53 | tumor protein 53 |
| PAR | parental chemosensitive cells |
| PARP | poly ADP-ribose polymerase |
| PDGFR | platelet-derived growth factor receptor alpha |
| PI3K | phosphoinositide 3-kinase |
| PIM1 | serine/threonine kinase |
| PK(s) | protein kinase(s) |
| PTEN | phosphatase and tensin homolog |
| PTKI | protein tyrosine kinase inhibitor |
| Rab | ras-associated binding gene |
| Raf | rapidly accelerated fibrosarcoma gene |
| Ras | rat sarcoma gene |
| Rho | rhodopsin gene |
| ROS | reactive oxygen species |
| RTK | receptor tyrosine kinase |
| SAR | structure-activity relationship |
| Src | sarcoma gene |
| TK | tyrosine kinase |
| TopI | topoisomerase I |
| TopII | topoisomerase II |
| TPA | 12-tetradecanoylphorbol-13-acetate |
| TSG | tumor-suppressor gene |
| VEGR | vascular endothelial growth factor |
| WT | Wilms’ tumor gene |
References
- Dhuguru, J.; Skouta, R. Role of Indole Scaffolds as Pharmacophores in the Development of Anti-Lung Cancer Agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi Ziarani, G.; Moradi, R.; Ahmadi, T.; Lashgari, N. Recent advances in the application of indoles in multicomponent reactions. RSC Adv. 2018, 8, 12069–12103. [Google Scholar] [CrossRef] [PubMed]
- Bandini, M. Electrophilicity: The “dark-side” of indole chemistry. Org. Biomol. Chem. 2013, 11, 5206. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, A.; Sharma, R.; Singh, R.K. Target-based anticancer indole derivatives and insight into structure–activity relationship: A mechanistic review update (2018–2021). Acta Pharm. Sin. B 2022, 12, 3006–3027. [Google Scholar] [CrossRef] [PubMed]
- Bajad, N.G.; Singh, S.K.; Singh, S.K.; Singh, T.D.; Singh, M. Indole: A promising scaffold for the discovery and development of potential anti-tubercular agents. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100119. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Dehbia, Z.; Zehravi, M.; Das, R.; Sivakumar, M.; Krishnan, K.; Billah, A.A.M.; Bose, B.; Ghosh, A.; Paul, S.; et al. Indole alkaloids from marine resources: Understandings from therapeutic point of view to treat cancers. Chem. Biol. Interact. 2023, 383, 110682. [Google Scholar] [CrossRef] [PubMed]
- Piechowska, P.; Zawirska-Wojtasiak, R.; Mildner-Szkudlarz, S. Bioactive β-Carbolines in Food: A Review. Nutrients 2019, 11, 814. [Google Scholar] [CrossRef]
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Politis, M.; Niccolini, F. Serotonin in Parkinson’s disease. Behav. Brain Res. 2015, 277, 136–145. [Google Scholar] [CrossRef]
- Tian, J.; Du, E.; Guo, L. Mitochondrial Interaction with Serotonin in Neurobiology and Its Implication in Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2023, 7, 1165–1177. [Google Scholar] [CrossRef]
- Szabó, T.; Volk, B.; Milen, M. Recent Advances in the Synthesis of β-Carboline Alkaloids. Molecules 2021, 26, 663. [Google Scholar] [CrossRef]
- Paul, A.; Acharya, K.; Chakraborty, N. Biosynthesis, extraction, detection and pharmacological attributes of vinblastine and vincristine, two important chemotherapeutic alkaloids of Catharanthus roseus (L.) G. Don: A review. S. Afr. J. Bot. 2023, 161, 365–376. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Neto, J.S.S.; Zeni, G. Recent advances in the synthesis of indoles from alkynes and nitrogen sources. Org. Chem. Front. 2020, 7, 155–210. [Google Scholar] [CrossRef]
- Ayesha; Bilal, M.; Rasool, N.; Khan, S.G.; Rashid, U.; Altaf, H.; Ali, I. Synthesis of Indoles via Intermolecular and Intramolecular Cyclization by Using Palladium-Based Catalysts. Catalysts 2021, 11, 1018. [Google Scholar] [CrossRef]
- Kumari, A.; Singh, R.K. Medicinal chemistry of indole derivatives: Current to future therapeutic prospectives. Bioorg. Chem. 2019, 89, 103021. [Google Scholar] [CrossRef] [PubMed]
- Leimgruber, B.W. Leimgruber–Batcho Indole Synthesis. In Indole Ring Synthesis; Wiley: New York, NY, USA, 2016; pp. 338–348. [Google Scholar] [CrossRef]
- Baudin, J.B.; Julia, S.A. Synthesis of indoles from N-aryl-1-alkenylsulphinamides. Tetrahedron Lett. 1986, 27, 837–840. [Google Scholar] [CrossRef]
- Fischer, E.; Jourdan, F. Ueber die Hydrazine der Brenztraubensäure. Berichte Dtsch. Chem. Ges. 1883, 16, 2241–2245. [Google Scholar] [CrossRef]
- Reissert, A. Einwirkung von Oxalester und Natriumäthylat auf Nitrotoluole. Synthese nitrirter Phenylbrenztraubensäuren. Berichte Dtsch. Chem. Ges. 1897, 30, 1030–1053. [Google Scholar] [CrossRef]
- Baeyer, A.; Emmerling, A. Synthese des Indols. Berichte Dtsch. Chem. Ges. 1869, 2, 679–682. [Google Scholar] [CrossRef]
- Sanz, R.; Guilarte, V.; Pérez, A. Straightforward selective preparation of nitro- or amino-indoles from 2-halonitroanilines and alkynes. First synthesis of 7-amino-5-nitroindoles. Tetrahedron Lett. 2009, 50, 4423–4426. [Google Scholar] [CrossRef]
- Ponpandian, T.; Muthusubramanian, S. Copper catalysed domino decarboxylative cross coupling-cyclisation reactions: Synthesis of 2-arylindoles. Tetrahedron Lett. 2012, 53, 4248–4252. [Google Scholar] [CrossRef]
- Liang, S.; Hammond, L.; Xu, B.; Hammond, G.B. Commercial Supported Gold Nanoparticles Catalyzed Alkyne Hydroamination and Indole Synthesis. Adv. Synth. Catal. 2016, 358, 3313–3318. [Google Scholar] [CrossRef]
- Madelung, W. Über eine neue Darstellungsweise für substituierte Indole. I. Berichte Dtsch. Chem. Ges. 1912, 45, 1128–1134. [Google Scholar] [CrossRef]
- Bartoli, G.; Palmieri, G.; Bosco, M.; Dalpozzo, R. The reaction of vinyl grignard reagents with 2-substituted nitroarenes: A new approach to the synthesis of 7-substituted indoles. Tetrahedron Lett. 1989, 30, 2129–2132. [Google Scholar] [CrossRef]
- Larock, R.C.; Yum, E.K. Synthesis of indoles via palladium-catalyzed heteroannulation of internal alkynes. J. Am. Chem. Soc. 1991, 113, 6689–6690. [Google Scholar] [CrossRef]
- Fukuyama, T.; Chen, X.; Peng, G. A Novel Tin-Mediated Indole Synthesis. J. Am. Chem. Soc. 1994, 116, 3127–3128. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2023 update. Pharmacol. Res. 2023, 187, 106552. [Google Scholar] [CrossRef]
- Sravanthi, T.V.; Manju, S.L. Indoles—A promising scaffold for drug development. Eur. J. Pharm. Sci. 2016, 91, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Gopalakrishnan, G. In vitro pharmacological characterization of SPN-810M (molindone). J. Exp. Pharmacol. 2018, 10, 65–73. [Google Scholar] [CrossRef]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.L.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank Knowledgebase for 2024. Nucleic Acids Res. 2024, 52, D1265–D1275. [Google Scholar] [CrossRef]
- Omar, F.; Tareq, A.M.; Alqahtani, A.M.; Dhama, K.; Sayeed, M.A.; Emran, T.B.; Simal-Gandara, J. Plant-Based Indole Alkaloids: A Comprehensive Overview from a Pharmacological Perspective. Molecules 2021, 26, 2297. [Google Scholar] [CrossRef] [PubMed]
- Chadha, N.; Silakari, O. Indoles as therapeutics of interest in medicinal chemistry: Bird’s eye view. Eur. J. Med. Chem. 2017, 134, 159–184. [Google Scholar] [CrossRef] [PubMed]
- Choppara, P.; Bethu, M.S.; Prasad, Y.V.; Rao, J.V.; Ranjan, T.J.U.; Prasad, G.V.S.; Doradla, R.; Murthy, Y.L.N. Synthesis, characterization and cytotoxic investigations of novel bis(indole) analogues besides antimicrobial study. Arab. J. Chem. 2019, 12, 2721–2731. [Google Scholar] [CrossRef]
- Mielczarek, M.; Thomas, R.V.; Ma, C.; Kandemir, H.; Yang, X.; Bhadbhade, M.; Black, D.S.; Griffith, R.; Lewis, P.J.; Kumar, N. Synthesis and biological activity of novel mono-indole and mono-benzofuran inhibitors of bacterial transcription initiation complex formation. Bioorg. Med. Chem. 2015, 23, 1763–1775. [Google Scholar] [CrossRef] [PubMed]
- Siramshetty, V.B.; Grishagin, I.; Nguyễn, Ð.T.; Peryea, T.; Skovpen, Y.; Stroganov, O.; Katzel, D.; Sheils, T.; Jadhav, A.; Mathé, E.A.; et al. NCATS Inxight Drugs: A comprehensive and curated portal for translational research. Nucleic Acids Res. 2022, 50, D1307–D1316. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.A.; Al-Omar, M.A.; Raish, M.; Ansari, M.A.; Abuelizz, H.A.; Bakheit, A.H.; Naglah, A.M. Indole Derivatives as Cyclooxygenase Inhibitors: Synthesis, Biological Evaluation and Docking Studies. Molecules 2018, 23, 1250. [Google Scholar] [CrossRef] [PubMed]
- Mukthung, C.; Chancharunee, S.; Kielar, F.; Pongcharoen, S.; Wichai, U. Capsaicin Derivatives Containing Indole and Nitroindole for Improved Anti-Inflammatory Activity. Naresuan Univ. J. Sci. Technology. 2018, 26, 157–169. [Google Scholar] [CrossRef]
- Girgis, A.S.; Panda, S.S.; Kariuki, B.M.; Bekheit, M.S.; Barghash, R.F.; Aboshouk, D.R. Indole-Based Compounds as Potential Drug Candidates for SARS-CoV-2. Molecules 2023, 28, 6603. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Amend, S.R.; Austin, R.H.; Gatenby, R.A.; Hammarlund, E.U.; Pienta, K.J. Updating the Definition of Cancer. Mol. Cancer Res. 2023, 21, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Hanselmann, R.G.; Welter, C. Origin of Cancer: An Information, Energy, and Matter Disease. Front. Cell Dev. Biol. 2016, 4, 121. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.F.; Frei, E.; Kude, D.W. Multistage Carcinogenes. In Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Gansler, T.S., Holland, J.F., Frei, E., Eds.; BC Decker Inc.: Hamilton, ON, Canada, 2003. [Google Scholar]
- Weinberg, R.A. Multi-step Tumorigenesis. In The Biology of CANCER, 2nd ed.; Bochicchio, A., Zayatz, E., Mickey, R.K., Harik, L., Ej Publishing Services, Eds.; Garland Science, Taylor & Francis Group, LLC: Abingdon, UK, 2014. [Google Scholar]
- Baba, A.I.; Câtoi, C. Tumor Cell Morphology. In Comparative Oncology; The Publishing House of the Romanian Academy: Bucuresti, Romania, 2007. [Google Scholar]
- Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation in Cancer. In Advances in Experimental Medicine and Biology; Springer New York LLC: New York, NY, USA, 2016; Volume 945, pp. 151–172. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal Transduction in Cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed]
- Khetmalis, Y.M.; Shivani, M.; Murugesan, S.; Chandra Sekhar, K.V.G. Oxindole and its derivatives: A review on recent progress in biological activities. Biomed. Pharmacother. 2021, 141, 111842. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Shokat, K.M. The Evolution of Protein Kinase Inhibitors from Antagonists to Agonists of Cellular Signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Salerno, S.; Barresi, E.; Baglini, E.; Poggetti, V.; Da Settimo, F.; Taliani, S. Target-Based Anticancer Indole Derivatives for the Development of Anti-Glioblastoma Agents. Molecules 2023, 28, 2587. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Xie, Y.; Zhang, J.S.; Wang, J.Q.; Dai, S.J.; He, W.F.; Li, S.Y.; Ashby, C.R., Jr.; Chen, Z.S.; He, Q. Sunitinib resistance in renal cell carcinoma: From molecular mechanisms to predictive biomarkers. Drug Resist. Updates 2023, 67, 100929. [Google Scholar] [CrossRef]
- Hong, Y.; Zhu, Y.Y.; He, Q.; Gu, S.X. Indole derivatives as tubulin polymerization inhibitors for the development of promising anticancer agents. Bioorg. Med. Chem. 2022, 55, 116597. [Google Scholar] [CrossRef]
- Sherer, C.; Snape, T.J. Heterocyclic scaffolds as promising anticancer agents against tumours of the central nervous system: Exploring the scope of indole and carbazole derivatives. Eur. J. Med. Chem. 2015, 97, 552–560. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Lazarus, H.M.; Cooper, B.W. Midostaurin: A novel therapeutic agent for patients with FLT3-mutated acute myeloid leukemia and systemic mastocytosis. Ther. Adv. Hematol. 2017, 8, 245–261. [Google Scholar] [CrossRef]
- Jia, Y.; Wen, X.; Gong, Y.; Wang, X. Current scenario of indole derivatives with potential anti-drug-resistant cancer activity. Eur. J. Med. Chem. 2020, 200, 112359. [Google Scholar] [CrossRef]
- Christodoulou, M.S.; Nicoletti, F.; Mangano, K.; Chiacchio, M.A.; Facchetti, G.; Rimoldi, I.; Beccalli, E.M.; Giofrè, S. Novel 3,3-disubstituted oxindole derivatives. Synthesis and evaluation of the anti-proliferative activity. Bioorg. Med. Chem. Lett. 2020, 30, 126845. [Google Scholar] [CrossRef]
- Gompel, M.; Leost, M.; De Kier Joffe, E.B.; Puricelli, L.; Franco, L.H.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the Ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef]
- Barakat, A.; Abu-Serie, M.M.; Ali, M.; Al-Majid, A.M.; Ashraf, S.; Zia, K.; Ul-Haq, Z.; Al-Dhfyan, A.; Abdel-Aziz, H.A.; El-Faham, A.; et al. Synthesis, In Vitro and in Cell Study of a New Spirooxindoles-Based N-Alkylated Maleimides Targeting HER2/3 Signaling Pathway. Polycycl. Aromat. Compd. 2023, 43, 5251–5275. [Google Scholar] [CrossRef]
- Al-Jassas, R.M.; Islam, M.S.; Al-Majid, A.M.; Nafie, M.S.; Haukka, M.; Rahman, A.F.M.M.; Alayyaf, A.M.A.; Barakat, A. Synthesis and SARs study of novel spiro-oxindoles as potent antiproliferative agents with CDK-2 inhibitory activities. Arch. Pharm. 2023, 356, e202300185. [Google Scholar] [CrossRef]
- Dubba, A.; Kumar Koppula, S. Synthesis of Indole-Oxadiazole coupled isoxazole hybrids as potent EGFR targeting anticancer agents. Chem. Biol. Lett. 2024, 11, 651. [Google Scholar] [CrossRef]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA Topoisomerases. EcoSal Plus 2015, 6. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- Wu, S.; Huang, Y.; Wang, T.; Li, K.; Lu, J.; Huang, M.; Dong, G.; Sheng, C. Evodiamine-Inspired Topoisomerase-Histone Deacetylase Dual Inhibitors: Novel Orally Active Antitumor Agents for Leukemia Therapy. J. Med. Chem. 2022, 65, 4818–4831. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Y.; Zhang, Z.; Zhao, J.; Jia, W.; Xia, C.; Wang, F.; Liu, T. Multi-therapies Based on PARP Inhibition: Potential Therapeutic Approaches for Cancer Treatment. J. Med. Chem. 2022, 65, 16099–16127. [Google Scholar] [CrossRef]
- Dockery, L.; Gunderson, C.; Moore, K. Rucaparib: The past, present, and future of a newly approved PARP inhibitor for ovarian cancer. Onco Targets Ther. 2017, 10, 3029–3037. [Google Scholar] [CrossRef]
- Cheng, Z.; Lu, X.; Feng, B. A review of research progress of antitumor drugs based on tubulin targets. Transl. Cancer Res. 2020, 9, 4020–4027. [Google Scholar] [CrossRef]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and Their Role in Cellular Stress in Cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef]
- Sept, D. Microtubule Polymerization: One Step at a Time. Curr. Biol. 2007, 17, R764–R766. [Google Scholar] [CrossRef]
- Dhyani, P.; Quispe, C.; Sharma, E.; Bahukhandi, A.; Sati, P.; Attri, D.C.; Szopa, A.; Sharifi-Rad, J.; Docea, A.O.; Mardare, I.; et al. Anticancer potential of alkaloids: A key emphasis to colchicine, vinblastine, vincristine, vindesine, vinorelbine and vincamine. Cancer Cell Int. 2022, 22, 206. [Google Scholar] [CrossRef]
- Qin, R.; You, F.M.; Zhao, Q.; Xie, X.; Peng, C.; Zhan, G.; Han, B. Naturally derived indole alkaloids targeting regulated cell death (RCD) for cancer therapy: From molecular mechanisms to potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 133. [Google Scholar] [CrossRef]
- Yan, J.; Chen, J.; Zhang, S.; Hu, J.; Huang, L.; Li, X. Synthesis, Evaluation, and Mechanism Study of Novel Indole-Chalcone Derivatives Exerting Effective Antitumor Activity Through Microtubule Destabilization in Vitro and in Vivo. J. Med. Chem. 2016, 59, 5264–5283. [Google Scholar] [CrossRef]
- Romagnoli, R.; Prencipe, F.; Oliva, P.; Kimatrai Salvador, M.; Brancale, A.; Ferla, S.; Hamel, E.; Viola, G.; Bortolozzi, R.; Persoons, L.; et al. Design, synthesis and biological evaluation of 2-alkoxycarbonyl-3-anilinoindoles as a new class of potent inhibitors of tubulin polymerization. Bioorg. Chem. 2020, 97, 103665. [Google Scholar] [CrossRef]
- Pilotto Heming, C.; Muriithi, W.; Wanjiku Macharia, L.; Niemeyer Filho, P.; Moura-Neto, V.; Aran, V. P-glycoprotein and cancer: What do we currently know? Heliyon 2022, 8, e11171. [Google Scholar] [CrossRef]
- Ahmed Juvale, I.I.; Abdul Hamid, A.A.; Abd Halim, K.B.; Che Has, A.T. P-glycoprotein: New insights into structure, physiological function, regulation and alterations in disease. Heliyon 2022, 8, e09777. [Google Scholar] [CrossRef]
- Paterna, A.; Kincses, A.; Spengler, G.; Mulhovo, S.; Molnár, J.; Ferreira, M.J.U. Dregamine and tabernaemontanine derivatives as ABCB1 modulators on resistant cancer cells. Eur. J. Med. Chem. 2017, 128, 247–257. [Google Scholar] [CrossRef]
- Raimundo, L.; Paterna, A.; Calheiros, J.; Ribeiro, J.; Cardoso, D.S.P.; Piga, I.; Neto, S.J.; Hegan, D.; Glazer, P.M.; Indraccolo, S.; et al. BBIT20 inhibits homologous DNA repair with disruption of the BRCA1–BARD1 interaction in breast and ovarian cancer. Br. J. Pharmacol. 2021, 178, 3627–3647. [Google Scholar] [CrossRef]
- Cardoso, D.S.P.; Kincses, A.; Nové, M.; Spengler, G.; Mulhovo, S.; Aires-de-Sousa, J.; Dos Santos, D.J.V.A.; Ferreira, M.U. Alkylated monoterpene indole alkaloid derivatives as potent P-glycoprotein inhibitors in resistant cancer cells. Eur. J. Med. Chem. 2021, 210, 112985. [Google Scholar] [CrossRef]
- Dhokne, P.; Sakla, A.P.; Shankaraiah, N. Structural insights of oxindole based kinase inhibitors as anticancer agents: Recent advances. Eur. J. Med. Chem. 2021, 216, 113334. [Google Scholar] [CrossRef] [PubMed]
- Mehra, A.; Sharma, V.; Verma, A.; Venugopal, S.; Mittal, A.; Singh, G.; Kaur, B. Indole Derived Anticancer Agents. ChemistrySelect 2022, 7, e202202361. [Google Scholar] [CrossRef]
- Yu, B.; Yu, D.Q.; Liu, H.M. Spirooxindoles: Promising scaffolds for anticancer agents. Eur. J. Med. Chem. 2015, 97, 673–698. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Zhang, X.; Srivenugopal, K.S.; Wang, M.H.; Wang, W.; Zhang, R. Targeting MDM2-p53 Interaction for Cancer Therapy: Are We There Yet? Curr. Med. Chem. 2014, 21, 553–574. [Google Scholar] [CrossRef]
- Panathur, N.; Gokhale, N.; Dalimba, U.; Koushik, P.V.; Yogeeswari, P.; Sriram, D. New indole–isoxazolone derivatives: Synthesis, characterisation and in vitro SIRT1 inhibition studies. Bioorg. Med. Chem. Lett. 2015, 25, 2768–2772. [Google Scholar] [CrossRef]
- Solomon, J.M.; Pasupuleti, R.; Xu, L.; McDonagh, T.; Curtis, R.; DiStefano, P.S.; Huber, L.J. Inhibition of SIRT1 Catalytic Activity Increases p53 Acetylation but Does Not Alter Cell Survival following DNA Damage. Mol. Cell. Biol. 2006, 26, 28–38. [Google Scholar] [CrossRef]
- Friberg, A.; Vigil, D.; Zhao, B.; Daniels, R.N.; Burke, J.P.; Garcia-Barrantes, P.M.; Camper, D.; Chauder, B.A.; Lee, T.; Olejniczak, E.T.; et al. Discovery of Potent Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem. 2013, 56, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Niso, M.; Abate, C.; Contino, M.; Ferorelli, S.; Azzariti, A.; Perrone, R.; Colabufo, N.A.; Berardi, F. Sigma-2 Receptor Agonists as Possible Antitumor Agents in Resistant Tumors: Hints for Collateral Sensitivity. ChemMedChem 2013, 8, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Hu, J.; Tian, J.L.; Yan, H.; Zheng, C.G.; Hu, W.L. Novel hybrids from N-hydroxyarylamide and indole ring through click chemistry as histone deacetylase inhibitors with potent antitumor activities. Chin. Chem. Lett. 2015, 26, 675–680. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indole Derivative | Target | Bioactivity | Ref. |
|---|---|---|---|
 | Aurora A | - | [81] |
 | PDK1/Akt and Aurora inhibition | IC50 = 416 nM and 35 nM, respectively | [81] |
 | Inhibition of ROS-mediated MAPK pathway; G1/S cycle arrest; Apoptosis | IC50 = 0.054 µM (PC3) and 1.439 µM (DU-145) | [82] |
 | TopI inhibitor; DNA cross-linking | IC50 = 0.41–3.5 µM | [58] |
 | p53-MDM2 interaction disruption | Ki = 18 nM | [83] |
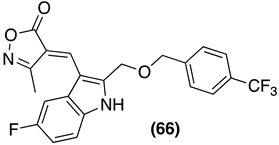 | SIRT | IC50 = 38 nM | [85] |
 | High selectivity over Bcl-2 and Bcl-xl | 16- and 270-fold | [87] |
| Mcl-1 | Ki = 55 nM | ||
 | σ2 receptor | Ki = 5.34 nM | [88] |
 | HDAC | IC50 = 78–380 nM | [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janeiro, A.M.; Marques, C.S. Biological Profile of Synthetic and Natural Indole Derivatives: Paving New Paths in Cancer Treatment. Drugs Drug Candidates 2024, 3, 488-511. https://doi.org/10.3390/ddc3030029
Janeiro AM, Marques CS. Biological Profile of Synthetic and Natural Indole Derivatives: Paving New Paths in Cancer Treatment. Drugs and Drug Candidates. 2024; 3(3):488-511. https://doi.org/10.3390/ddc3030029
Chicago/Turabian StyleJaneiro, Ana Margarida, and Carolina S. Marques. 2024. "Biological Profile of Synthetic and Natural Indole Derivatives: Paving New Paths in Cancer Treatment" Drugs and Drug Candidates 3, no. 3: 488-511. https://doi.org/10.3390/ddc3030029
APA StyleJaneiro, A. M., & Marques, C. S. (2024). Biological Profile of Synthetic and Natural Indole Derivatives: Paving New Paths in Cancer Treatment. Drugs and Drug Candidates, 3(3), 488-511. https://doi.org/10.3390/ddc3030029