
Beta-Thalassemia: A Pharmacological Drug-Based Treatment

 ,
,  , ,
, ,  ,
,  ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Methodology
2.1. Search Strategy
2.2. Inclusion Criteria
2.3. Exclusion Criteria
3. Pathophysiology of Thalassemia
4. Current Treatment Strategies
4.1. Transfusional Therapy
4.2. Non-Transfusional Therapy
4.3. Drugs Replacing Blood Transfusion Process in Thalassemia
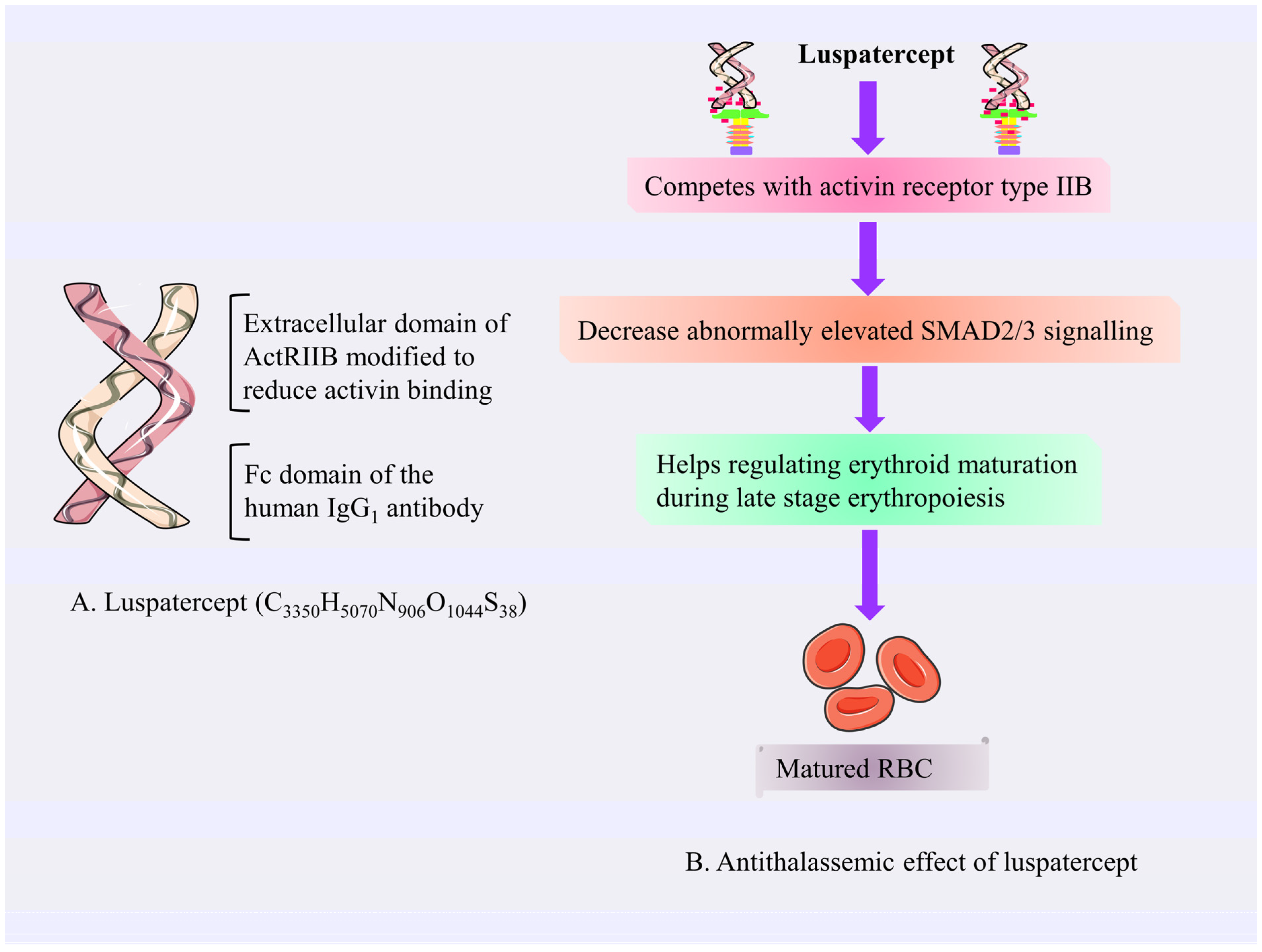
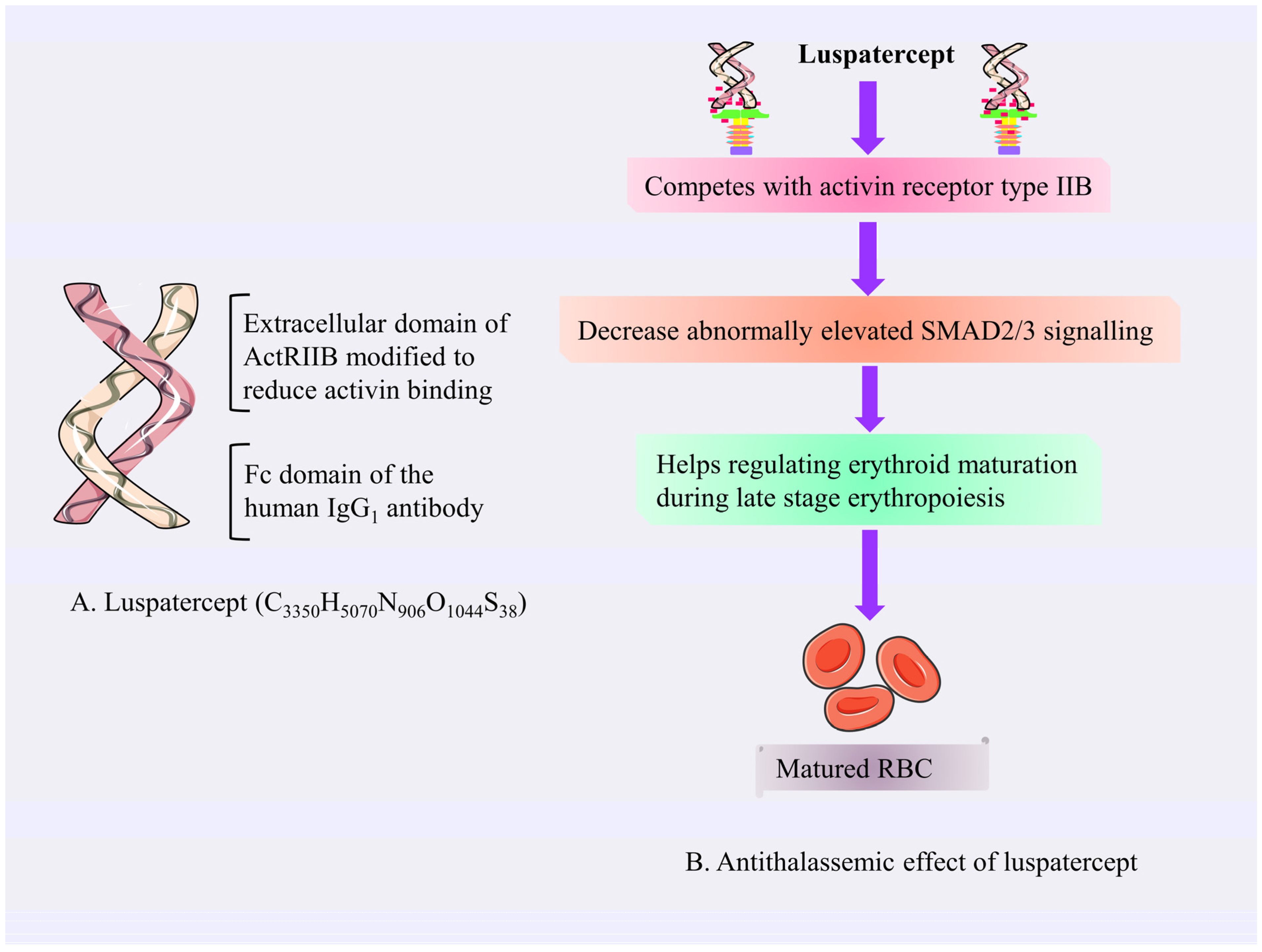
4.3.1. Luspatercept
Mechanism of Action of Luspatercept
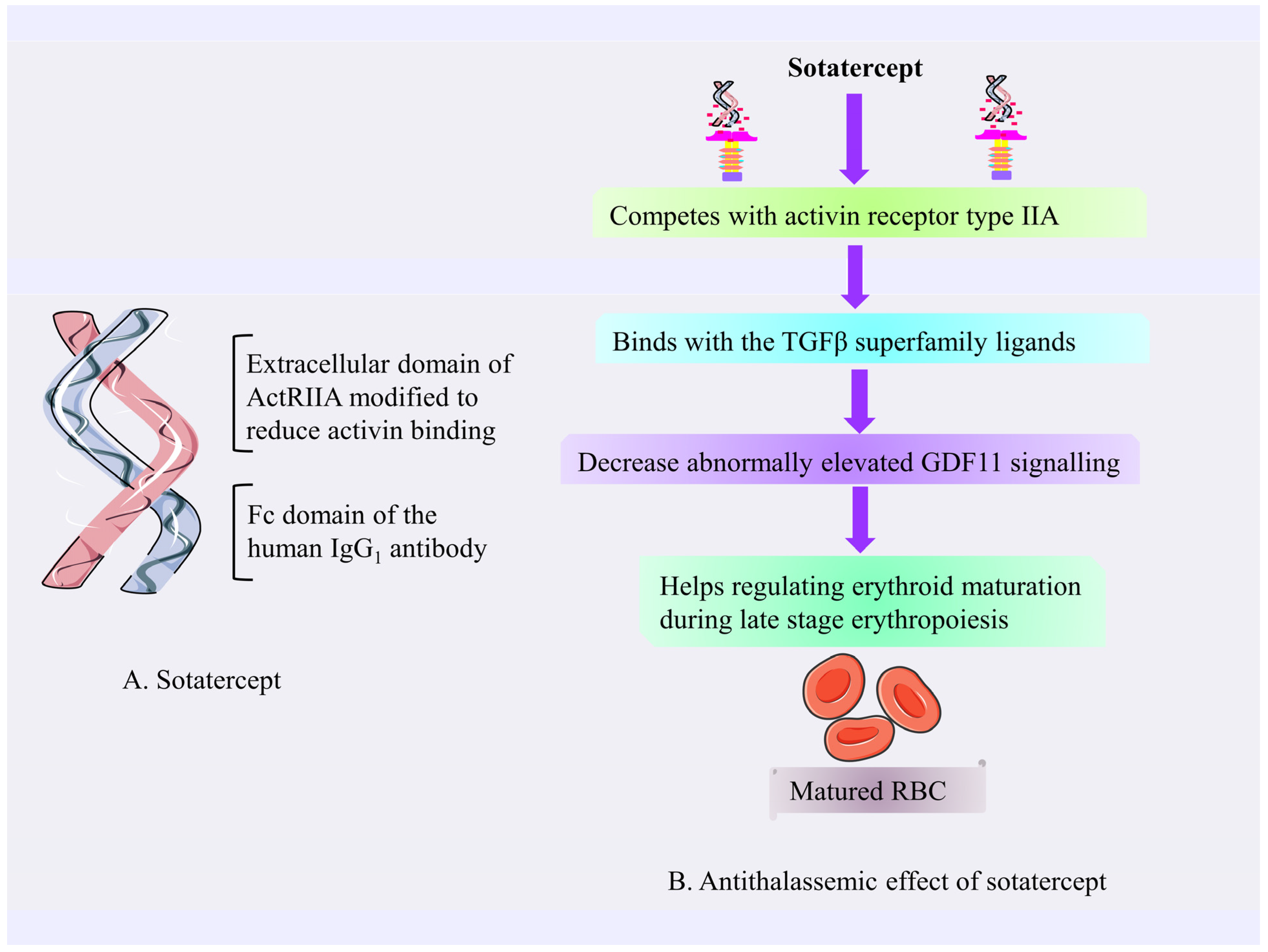
4.3.2. Sotatercept
Mechanism of Action of Sotatercept
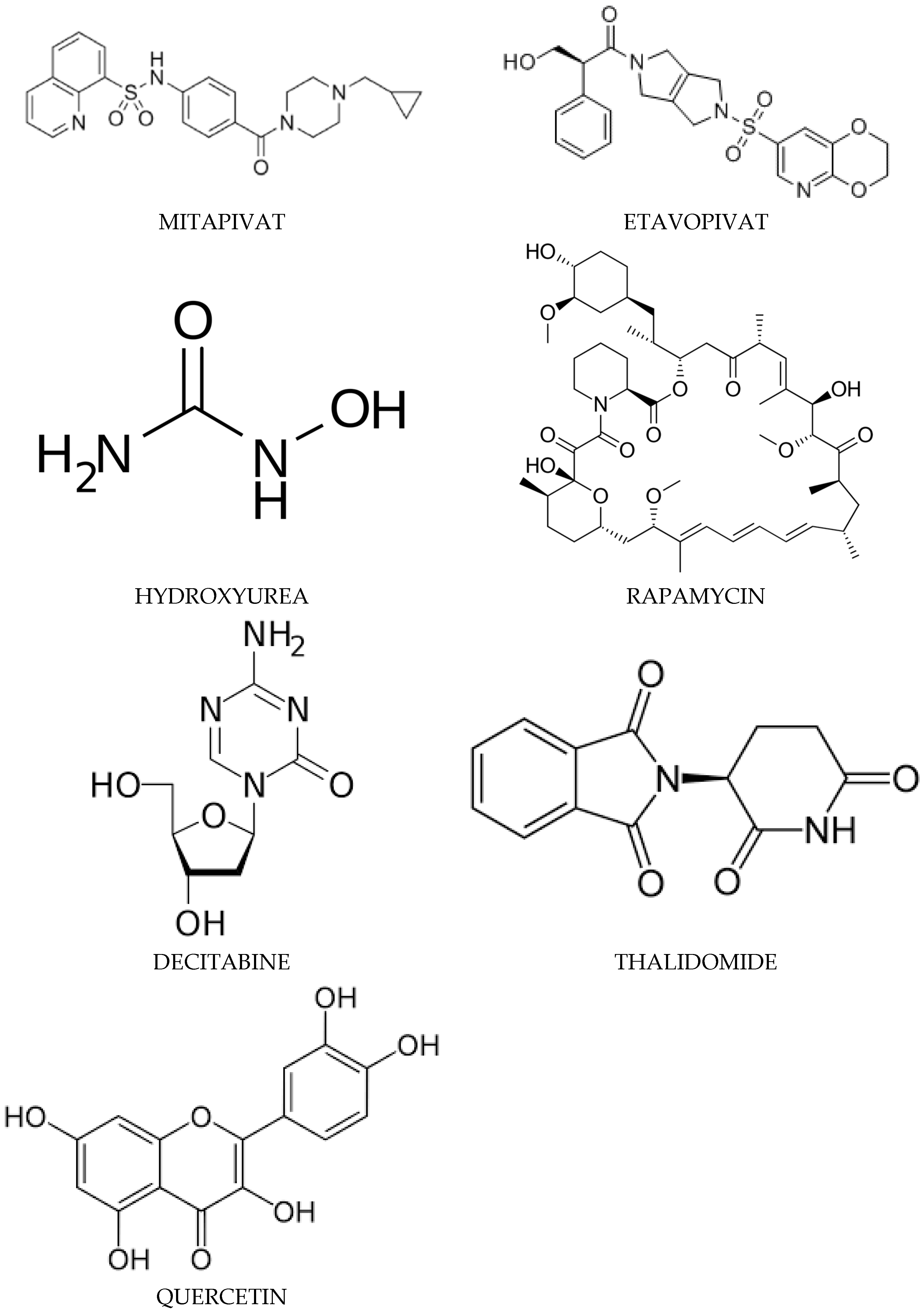
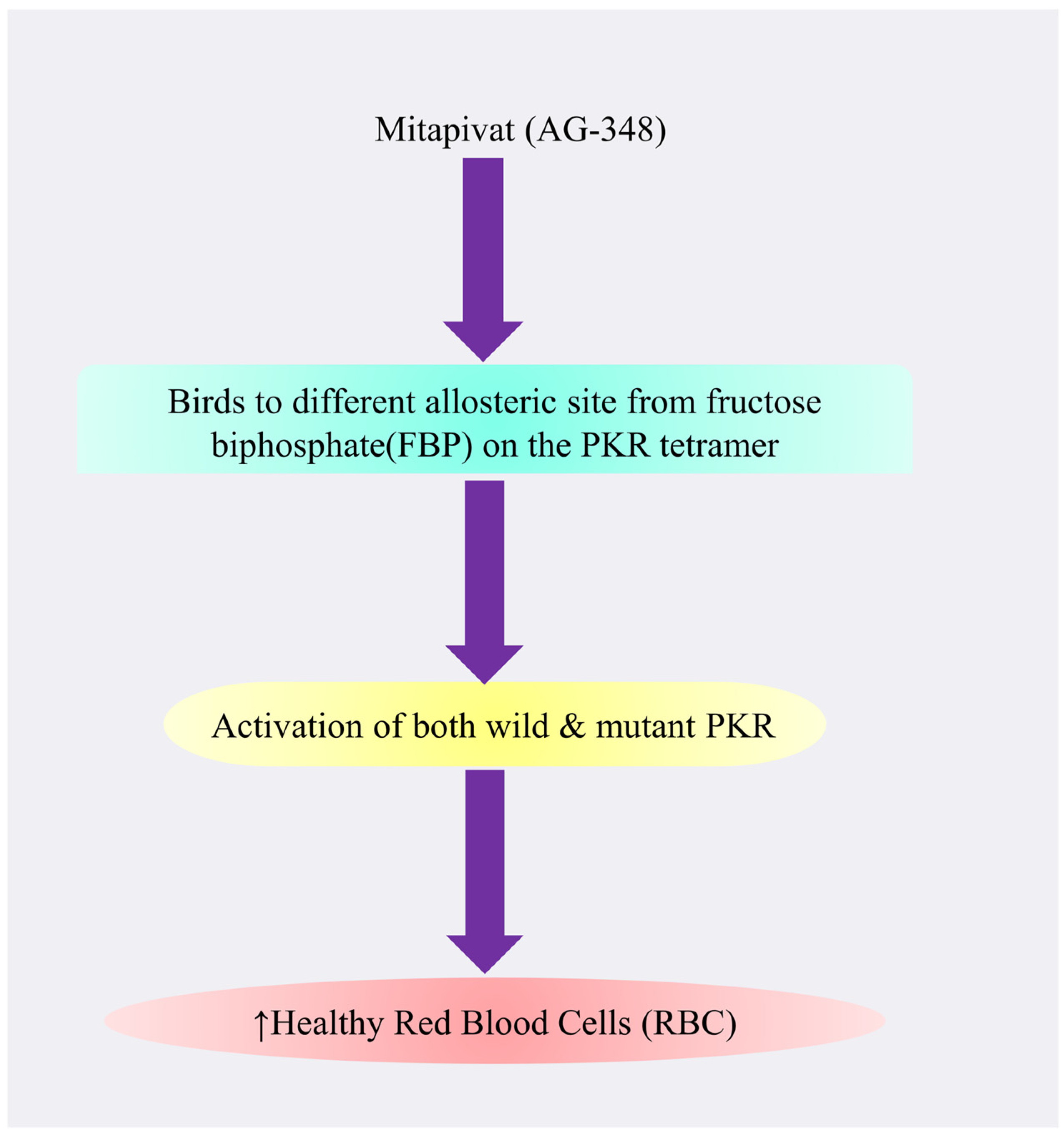
4.3.3. Mitapivat
Mechanism of Action of Mitapivat
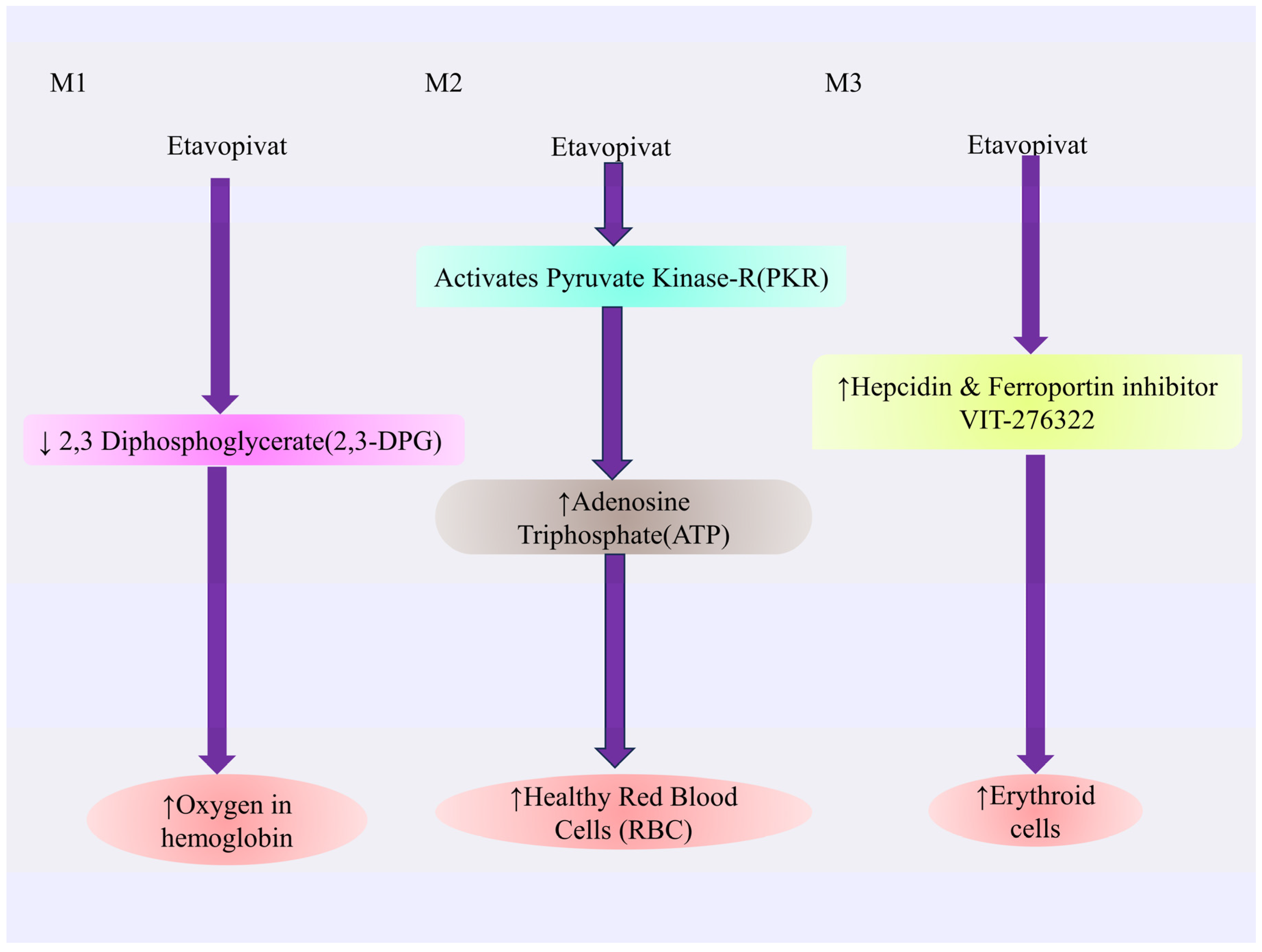
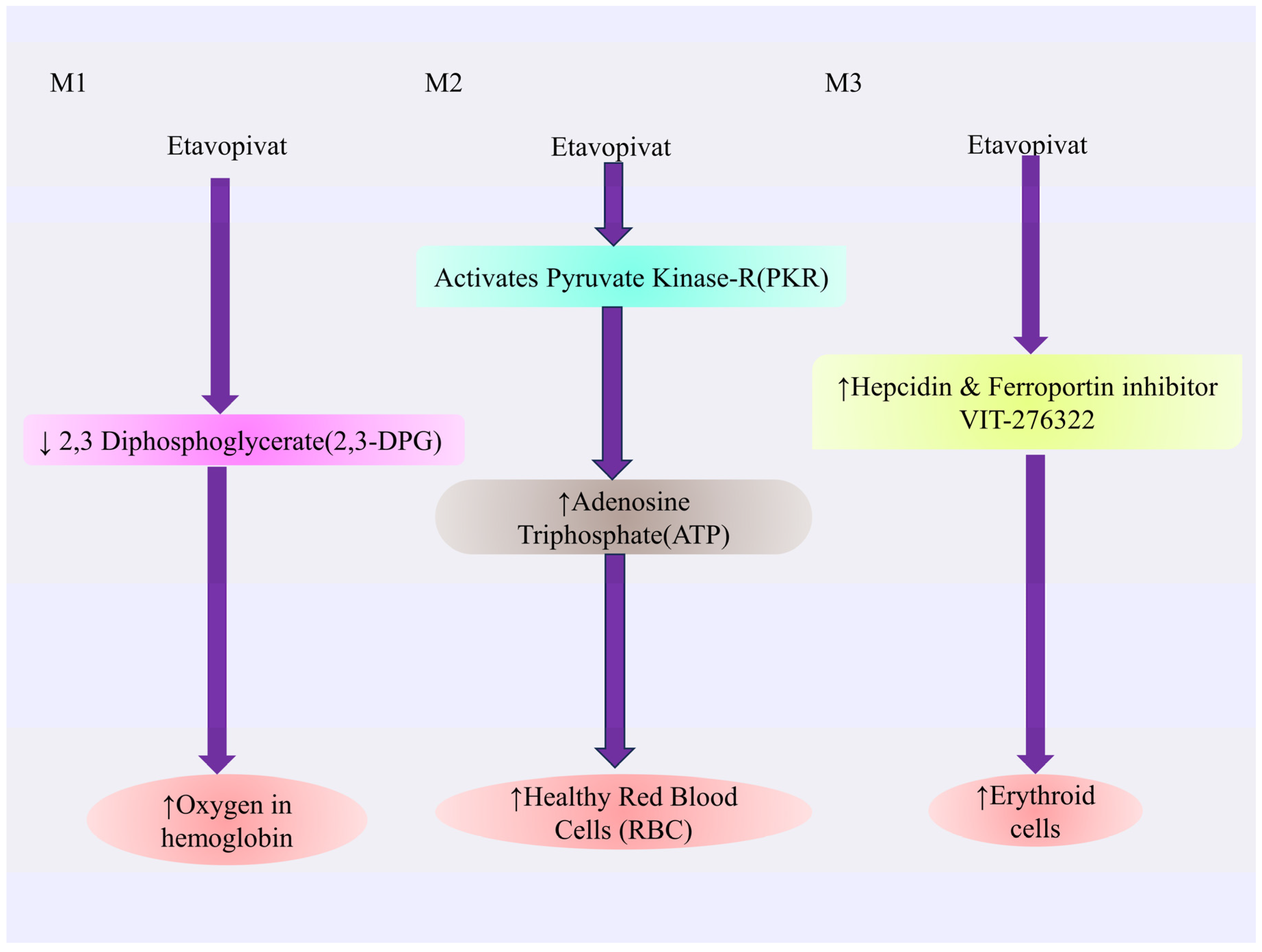
4.3.4. Etavopivat
Mechanism of Action of Etavopivat
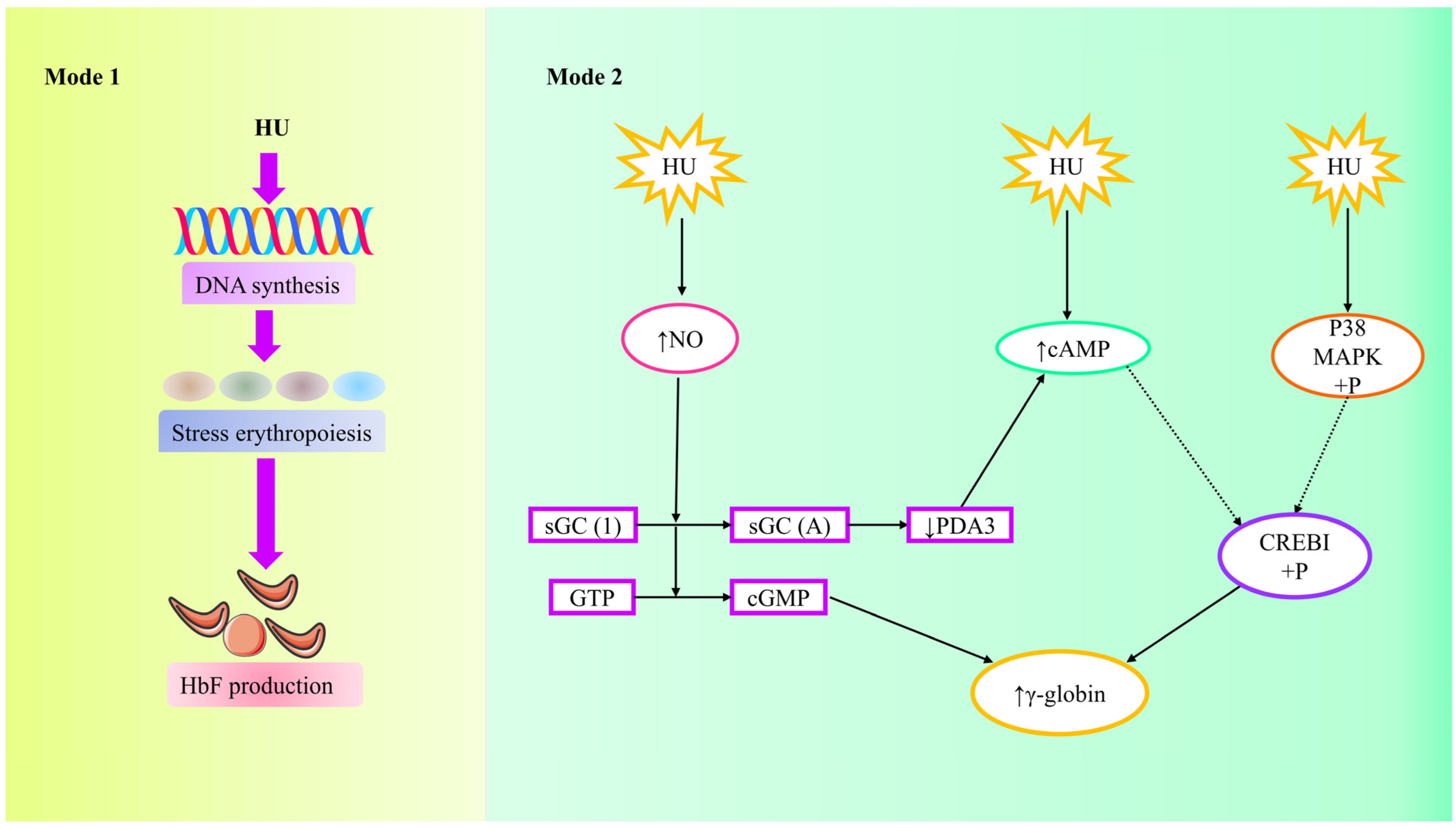
4.3.5. Hydroxyurea
Mechanism of Action of Hydroxyurea (HU)
4.3.6. Rapamycin
Mechanism of Action of Rapamycin
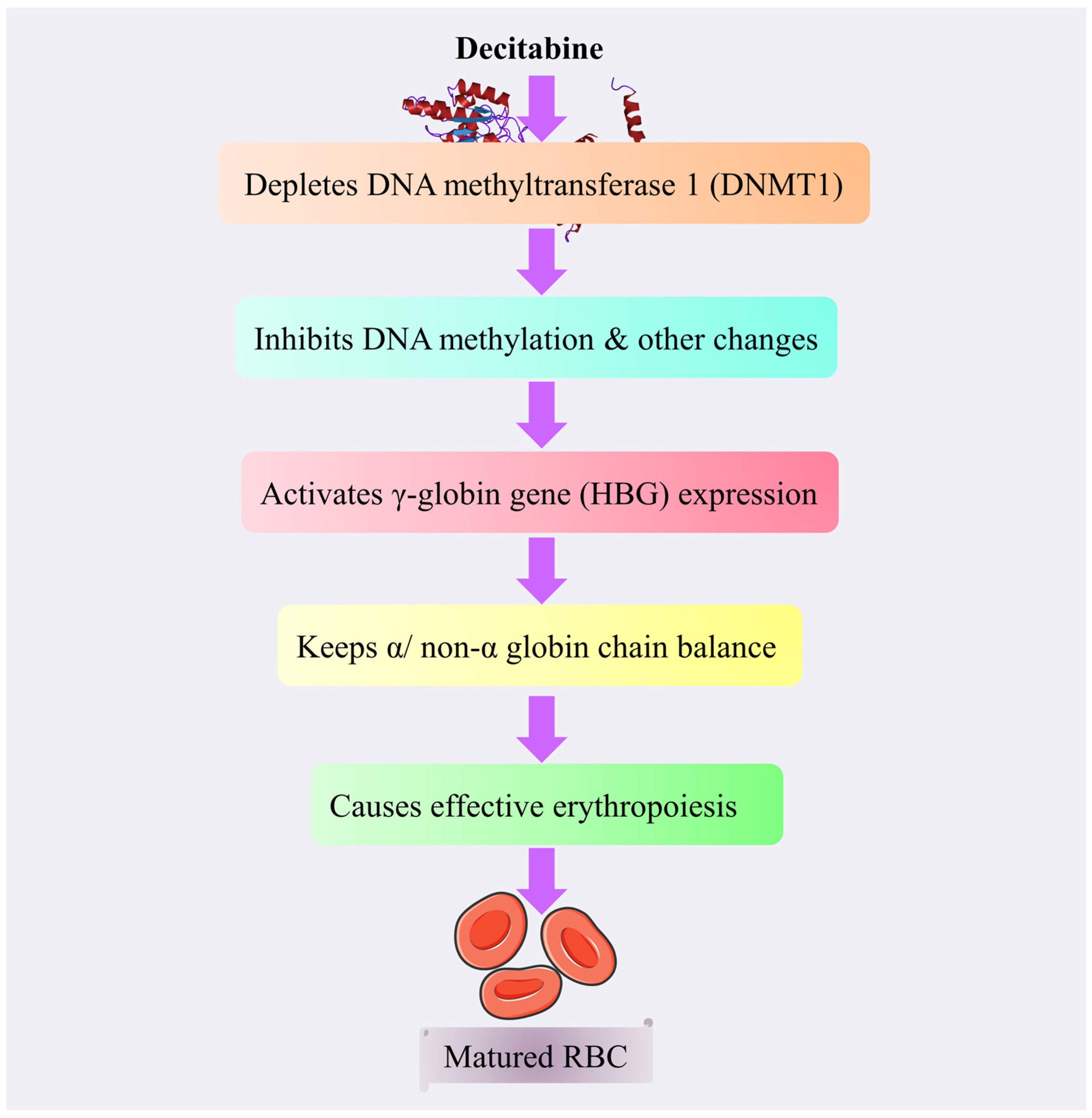
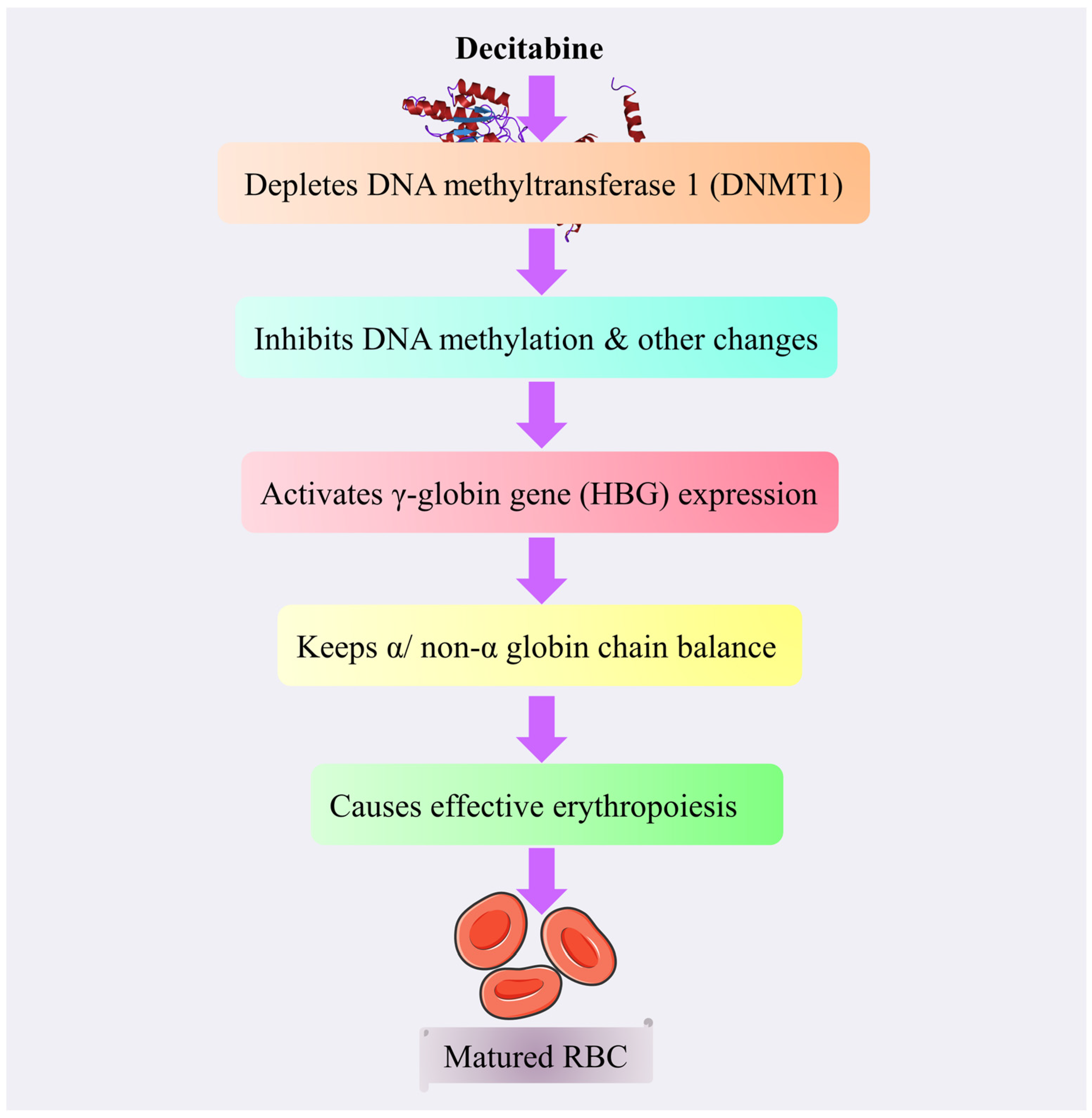
4.3.7. Decitabine
Mechanism of Action of Decitabine
Toxicity
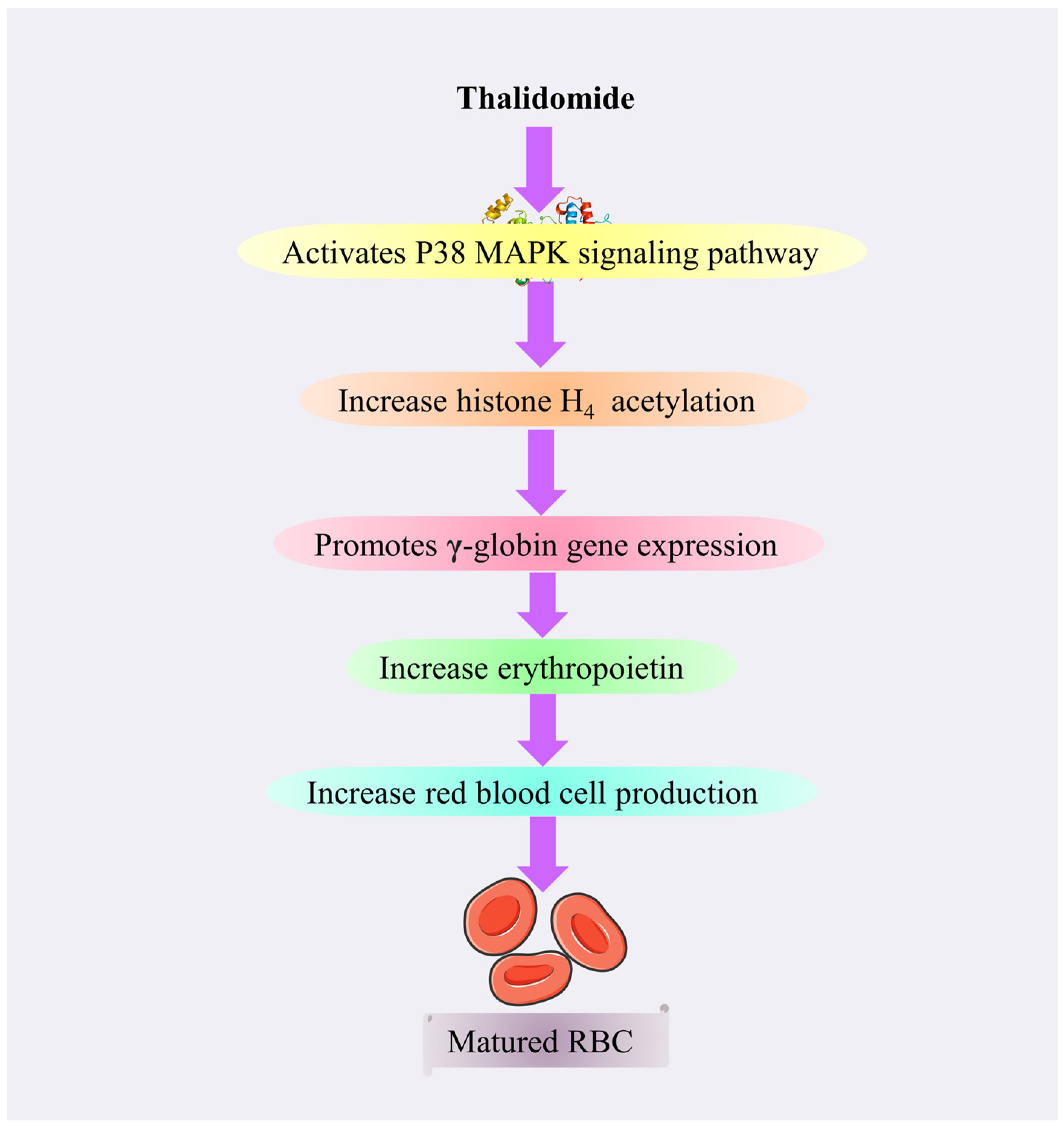
4.3.8. Thalidomide
Mechanism of Action of Thalidomide
Toxicity
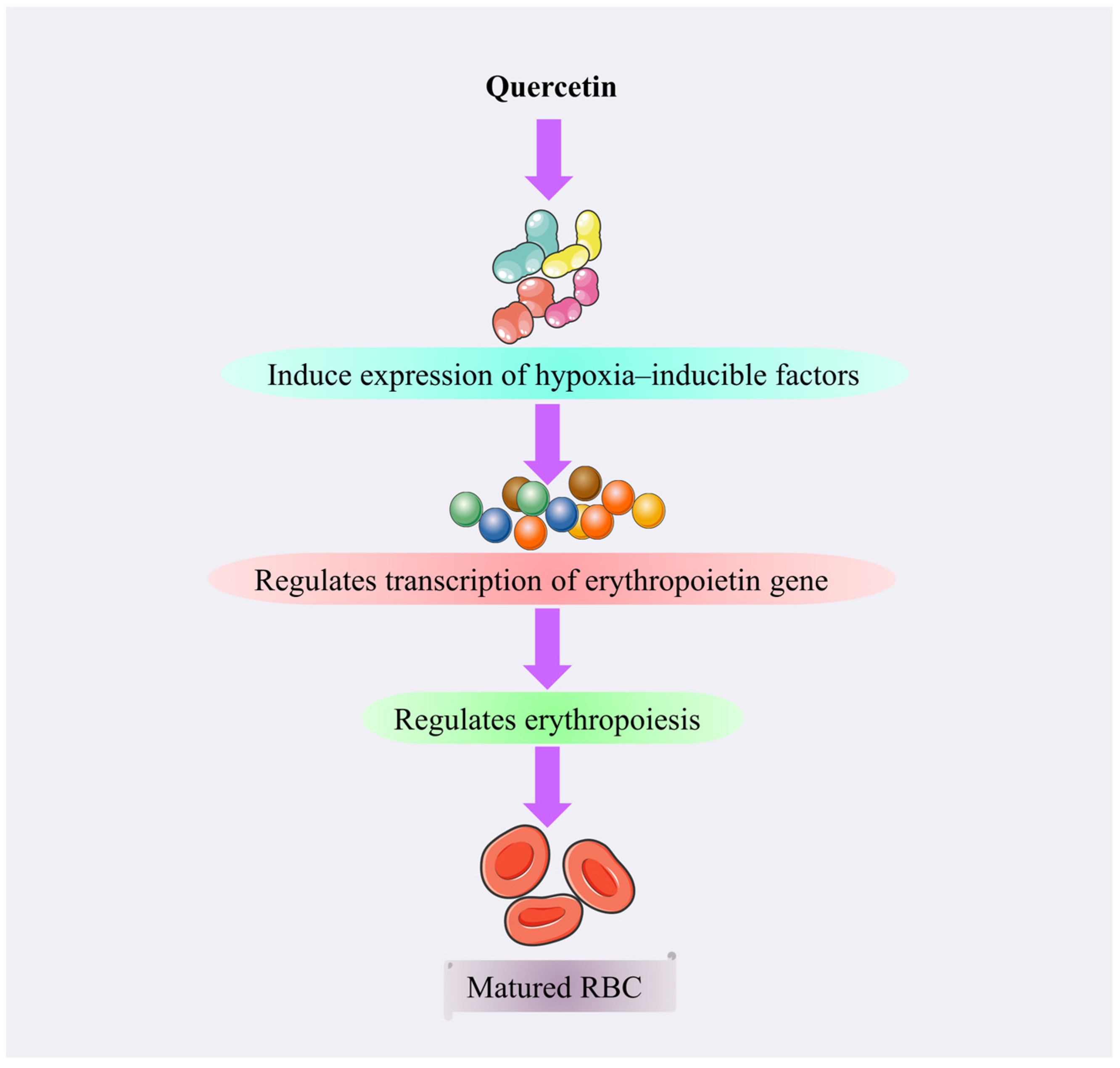
4.3.9. Quercetin
Mechanism of Action of Quercetin
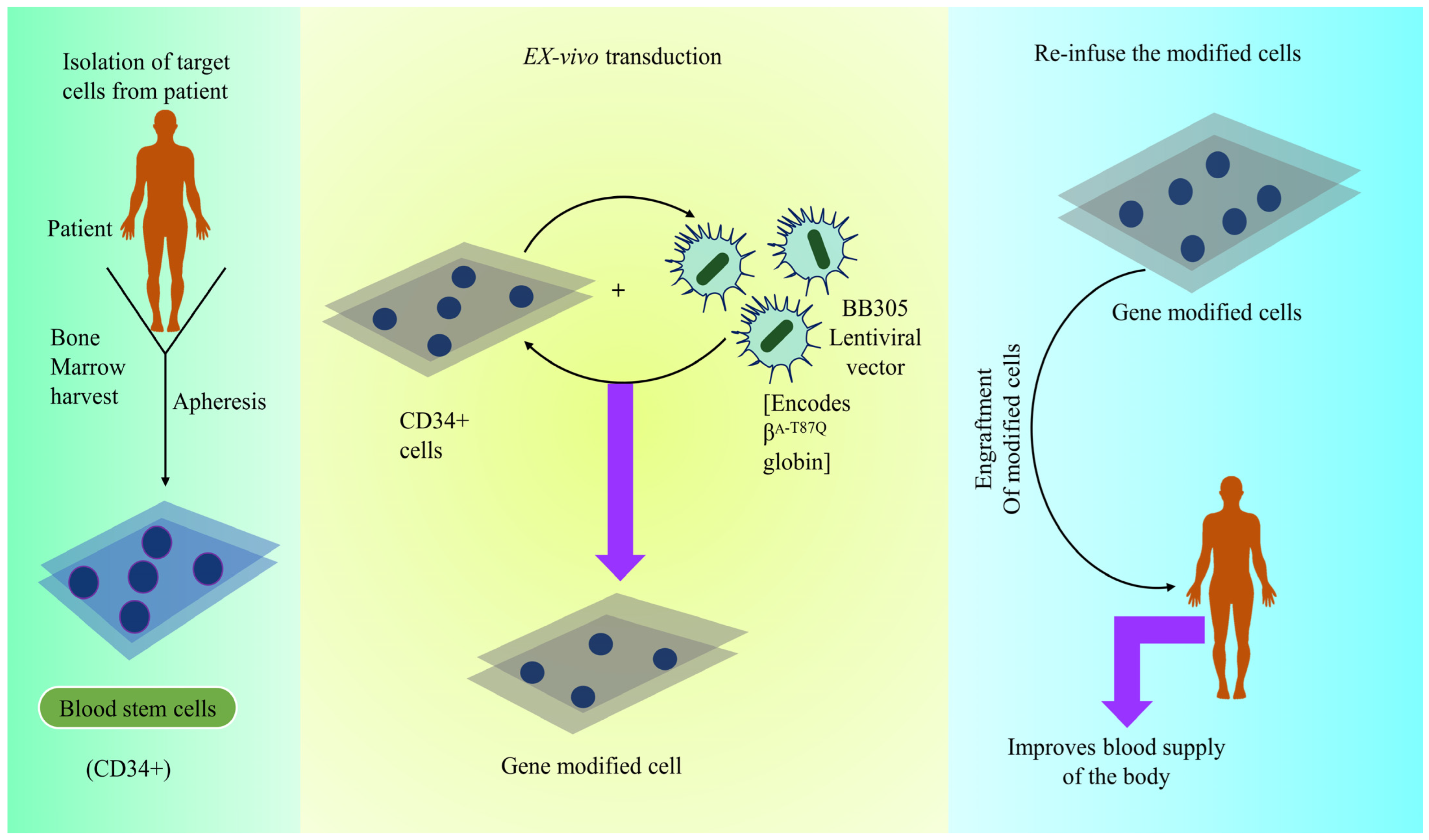
4.3.10. Betibeglogene Autotemcel (Zynteglo)
Mechanism of Action of Betibeglogene Autotemcel (Zynteglo)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Concentration/Dose and Route of Administration | Test system/Model | Results | Reference |
|---|---|---|---|---|
| Luspatercept | 0.2–1.25 mg/kg per body weight; subcutaneously | Clinical trial | A significant proportion of individuals experienced a positive change in their Hb levels or reduced need for blood transfusions after using luspatercept | [52] |
| Luspatercept | 1 mg/kg once every 3 weeks; starting dose (s.c.) enhanced to 1.25 mg/kg in individuals who did not experience a reduction in RBC transfusion problems after ≥2 treatment cycles (6 weeks) | Clinical trial (approved) | Patients may experience a decrease in their need for blood transfusions through treatment with luspatercept | [53] |
| Luspatercept | 0.6–1.25 mg/kg | Phase II clinical trial | Increased Hb level | [54] |
| Luspatercept | 1–1.25 mg/kg | Randomized, phase III, double-blind | Enhanced Hb production | [55] |
| Sotatercept | 0.1, 0.3, 0.5, 0.75, and 1.0 mg/kg used to evaluate the effective and safe dose; subcutaneous injection ≤ 22 months | Clinical trial | In patients with transfusion-dependent β-thalassemia, the result was evaluated as a reduction of at least 20% in the need for blood transfusions that lasted for 24 weeks. For non-transfusion-dependent β-thalassemia patients, the response was evaluated as a sustained increase in the Hb levels of no less than 1.0 g/dL for 12 weeks | [28] |
| HU | 10 mg/kg per day as beginning dose, which was increased by 5 mg/kg (per day) at 4-weekly intervals to a maximum of 20 mg/kg per day or until myelotoxicity appeared | Clinical trial | Out of the total number of patients, which was 26, 70.2% responded positively to the HU therapy. Among the patients, 45.9% presented a significant response, while 24.3% presented a less significant response | [56] |
| HU | 16 mg/kg/day | Clinical trial | The study found that HU was a safe treatment option for patients with α, ꞵ-thalassemia major, and it led to a decrease in the need for blood transfusions | [57] |
| HU | 14 mg/kg/day | Clinical trial | The analysis of gamma-globin expression in the groups under study revealed a notable increase in gamma-globin gene expression | [58] |
| HU | 15 mg/kg | Clinical trial | To obtain a more comprehensive representation of the exact mechanism, a study with a larger number of participants and a longer duration of HU treatment is required | [59] |
| HU | 10–20 mg/kg once; 6 months | Clinical trial | 65.93% responded well | [57] |
| Rapamycin | On the 4th or 5th day of phase II, rapamycin (manufactured by Sigma/Aldrich, Milwaukee, WI, USA) was introduced; the cells were then collected and examined on day 12 | In vitro | The majority of the samples (six out of seven analyzed) showed an increase in the total amount of Hb per cell (measured in pg/cell) | [56] |
| Rapamycin | A concentration of 100 nanomolar rapamycin was administered to the culture | In vitro | Rapamycin enhanced the expression of α-globin mRNA in 60.0% of sickle cell disease samples and 56.0% of b-TI samples examined | [57] |
| Rapamycin | 4 mg/kg once for 30 days | In vivo | Rapamycin decreased by approximately 50% the amount of insoluble α-globin observed in the red blood cells of HbbTh3+ mice (p < 0.001). However, this reduction was not observed in HbbTh3+/Ulk1−/− mice | [36] |
| Rapamycin | 16 and 24 ng/mL | Clinical trial | The main goal was to observe an increase in the fraction of HbF in blood samples taken at day 360 compared to baseline (day 0) | [60] |
| Decitabine (2-deoxy 5-azacytidine) | 0.15–0.30 mg/kg administered on 5 days of the week for 2 weeks | A dose-escalating phase I/II study | Before treatment, the mean ratio was 3.19% with a standard deviation of 1.43%. Following treatment, this ratio increased to 13.66%, with a standard deviation of 4.35% | [39] |
| Decitabine | 0.2 mg/kg administered 2 times each week for 12 weeks (s.c.) | Clinical trial | The total Hb level increased significantly from 7.88 ± 0.88 g/dL to 9.04 ± 0.77 g/dL, and there was also a significant rise in the absolute HbF level from 3.64 ± 1.13 g/dL to 4.29 ± 1.13 g/dL | [40] |
| Decitabine | 0.2 mg/kg of 5-aza-2′-deoxycytidine (decitabine) was administered subcutaneously on two consecutive days each week for a minimum of 12 weeks | Phase II | There was a substantial increase in Hb level, observed as soon as week 2 | [61] |
| Thalidomide | 50 mg each night (p.o.) | Clinical trial | After the treatment period, there was an increase of 2.5 ± 1.8 g/dL in both Hb and HbF levels | [62] |
| Thalidomide | Starting dose of 50 mg/d, or a daily dose of 100 mg/d was administered to individuals requiring blood transfusions at least twice a month (2016–2019) | Clinical trial | Thalidomide appears to be a highly promising cure option for individuals with β-thalassemia, with the potential to substantially enhance Hb level, thereby reducing the need for blood transfusions | [44] |
| Thalidomide | 2–10 mg/kg at any time throughout the therapy; at first, a dose of 3 mg/kg (balanced to closest 50 mg) was utilized | Clinical trial | Thalidomide treatment produced a marked response in over 75% of victims with symptomatic β-thalassemia, regardless of whether they had TDT or NTDT | [23] |
| Thalidomide | 2.5–3.6 mg/kg/d | Clinical trial | Safely enhanced Hb production | [45] |
| Quercetin | 500 mg/day quercetin tablet | Double-blinded, randomized clinical trial | Suppressed inflammation | [63] |
| Quercetin | 500 mg/day | Clinical trial | The use of quercetin may improve iron levels in individuals with thalassemia major, but its impact on inflammation is not clear | [41] |
| Mitapivat | (p.o.) | Clinical trial | Twenty-six patients (50%, mean maximum Hb increase of 3 g/dL, 4 g/dL range, with a good safety profile) had an Hb increase of more than 1 g/dL from baseline | [64] |
| Mitapivat | More often when dosed over 700 mg (p.o.) | Phase I | Positive pharmacokinetics with minimal variability of dose-dependent alterations in blood glycolytic intermediates (increased ATP, decreased 2,3-DPG) consistent with the activation of glycolysis | [65] |
| Mitapivat | 50 mg twice a day | Phase II | An increase in Hb was observed at weeks 12 and 72, with a median (Q1, Q3) of 1.3 g/dL (0.60, 1.90) and 1.2 g/dL (−0.03, 2.2) | [66] |
| Mitapivat | 16 patients received 100 mg of BID mitapivat and one patient received 50 mg | Phase III | Continuous improvements in hemolysis, inefficient erythropoiesis, and hemoglobin | [67] |
| Mitapivat | 50 mg twice a day | Phase III | Hemolysis markers and ineffective erythropoiesis were improved during the core period | [68] |
| Mitapivat | 20 mg twice a day | Phase II | A rise in hemoglobin from the starting point and a corresponding fall in hemolysis-related indicators | [69] |
| Mitapivat | 5–50 mg two times a day | Phase III | Of the 27 patients enrolled, 20 finished the study; with 22% achieving a transfusion-free response and 37% reporting a decrease in their transfusion burden, the primary endpoint was met | [70] |
| Etavopivat | Once daily | Preclinical and phase I clinical trial | Proof of concept without significant adverse events was demonstrated in controls treated for up to 14 days with 7-day follow-up | [71] |
| Etavopivat | Less than or equal to 400 mg once daily | Phase I | Pharmacokinetic/pharmacodynamic analysis results showed that 400 mg taken once daily was the maximal dose at which PKR activation was achieved | [72] |
| Betibeglogene Autotemcel | Patients received autologous CD34+ cells transduced with BB305 lentiviral vector after receiving busulfan, a single-agent, pharmacokinetically adjusted treatment; myeloablation | Phases II and IV | Patients with TDT could maintain and stabilize their response following β-cell gene therapy. Hematologic parameters and the iron burden were improved as a result of sustained levels of HbAT87Q and efficient iron reduction with phlebotomy and/or iron chelation | [73] |
| Betibeglogene autotemcel | In patients with transfusion-dependent β-thalassemia who were adult or pediatric and had a non-β°/β° genotype, Beti-cel was used. Patients received Beti-cel intravenously and underwent myeloablation with busulfan (dosages modified based on pharmacokinetic analysis). A median follow-up of 29.5 months was observed for the 23 patients who were enrolled and received treatment | Phase IV | Higher HbAT87Q levels were obtained in the study compared to the phase I–II studies, which were attributed to improvements in hematopoietic stem-cell transduction. According to these findings, a single Beti-cel infusion may be therapeutic for the majority of patients with transfusion-dependent β-thalassemia and a non-β°/β° genotype, as it can lead to near-normal hemoglobin levels and transfusion independence | [51] |
| Betibeglogene autotemcel | Gene therapy using lentiviral technology and ex vivo autologous hematopoietic stem cells using betibeglogene autotemcel | Phase III | 41 patients received Beti-cel treatment and were cured | [50] |
5. Discussion
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ali, S.; Saffiullah, M.F. Awareness of parents regarding beta thalassemia major disease. Khyber Med. Univ. J. 2015, 7, 72–75. [Google Scholar]
- Rund, D.; Rachmilewitz, E. Beta-thalassemia. N. Engl. J. Med. 2005, 353, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.; Moi, P.; Galanello, R. Recent advances in β-thalassemias. Pediatr. Rep. 2011, 3, e17. [Google Scholar] [CrossRef]
- Williams, T.N.; Weatherall, D.J. World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harb. Perspect. Med. 2012, 2, a011692. [Google Scholar] [CrossRef]
- Fucharoen, S.; Winichagoon, P. Haemoglobinopathies in southeast Asia. Indian J. Med. Res. 2011, 134, 498–506. [Google Scholar] [PubMed]
- Brancaleoni, V.; Di Pierro, E.; Motta, I.; Cappellini, M.D. Laboratory diagnosis of thalassemia. Int. J. Lab. Hematol. 2016, 38 (Suppl. S1), 32–40. [Google Scholar] [CrossRef]
- Muncie, H.L., Jr.; Campbell, J. Alpha and beta thalassemia. Am. Fam. Physician 2009, 80, 339–344. [Google Scholar] [PubMed]
- Galanello, R.; Origa, R. Beta-thalassemia. Orphanet. J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef]
- Pinto, V.M.; Forni, G.L. Management of Iron Overload in Beta-Thalassemia Patients: Clinical Practice Update Based on Case Series. Int. J. Mol. Sci. 2020, 21, 8771. [Google Scholar] [CrossRef]
- Canver, M.C.; Orkin, S.H. Customizing the genome as therapy for the β-hemoglobinopathies. Blood 2016, 127, 2536–2545. [Google Scholar] [CrossRef]
- Farashi, S.; Harteveld, C.L. Molecular basis of α-thalassemia. Blood Cells Mol. Dis. 2018, 70, 43–53. [Google Scholar] [CrossRef]
- Origa, R.; Galanello, R. Pathophysiology of beta thalassaemia. Pediatr. Endocrinol. Rev. 2011, 8 (Suppl. S2), 263–270. [Google Scholar] [PubMed]
- Mettananda, S.; Gibbons, R.J.; Higgs, D.R. α-Globin as a molecular target in the treatment of β-thalassemia. Blood J. Am. Soc. Hematol. 2015, 125, 3694–3701. [Google Scholar] [CrossRef]
- Poggiali, E.; Cassinerio, E.; Zanaboni, L.; Cappellini, M.D. An update on iron chelation therapy. Blood Transfus. 2012, 10, 411–422. [Google Scholar] [CrossRef]
- Kubasch, A.S.; Fenaux, P.; Platzbecker, U. Development of luspatercept to treat ineffective erythropoiesis. Blood Adv. 2021, 5, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Carrancio, S.; Markovics, J.; Wong, P.; Leisten, J.; Castiglioni, P.; Groza, M.C.; Raymon, H.K.; Heise, C.; Daniel, T.; Chopra, R.; et al. An activin receptor IIA ligand trap promotes erythropoiesis resulting in a rapid induction of red blood cells and haemoglobin. Br. J. Haematol. 2014, 165, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H.; van Beers, E.J. Mitapivat, a novel pyruvate kinase activator, for the treatment of hereditary hemolytic anemias. Ther. Adv. Hematol. 2021, 12, 20406207211066070. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, J.; Wang, Z.; Li, J.; Zhang, L.; Hu, X.; Zhang, X.; Tang, J.; Zhang, J.; Li, S. Double-bonded azacycles used as bioisosteric moieties in drug discovery. Tetrahedron Lett. 2023, 129, 154719. [Google Scholar] [CrossRef]
- Larsen, I.K.; Jerslev, B. Crystal and Molecular Structure of Hydroxyurea. Acta Chem. Scand. 1966, 20, 983. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Neupane, Y.R.; Sabir, M.D.; Ahmad, N.; Ali, M.; Kohli, K. Lipid drug conjugate nanoparticle as a novel lipid nanocarrier for the oral delivery of decitabine: Ex vivo gut permeation studies. Nanotechnology 2013, 24, 415102. [Google Scholar] [CrossRef]
- Melchert, M.; List, A. The thalidomide saga. Int. J. Biochem. Cell Biol. 2007, 39, 1489–1499. [Google Scholar] [CrossRef]
- Materska, M. Quercetin and its derivatives: Chemical structure and bioactivity—A review. Pol. J. Food Nutr. Sci. 2008, 58, 407–413. [Google Scholar]
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T.; Georgiev, P.; Kuo, K.H.M.; Coates, T.; Voskaridou, E.; Liew, H.K.; Pazgal-Kobrowski, I.; Forni, G.L.; et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2020, 382, 1219–1231. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Taher, A.T. The use of luspatercept for thalassemia in adults. Blood Adv. 2021, 5, 326–333. [Google Scholar] [CrossRef]
- Abdulkadyrov, K.M.; Salogub, G.N.; Khuazheva, N.K.; Sherman, M.L.; Laadem, A.; Barger, R.; Knight, R.; Srinivasan, S.; Terpos, E. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br. J. Haematol. 2014, 165, 814–823. [Google Scholar] [CrossRef]
- Cappellini, M.-D.; Porter, J.; Origa, R.; Forni, G.L.; Laadem, A.; Galacteros, F.; Miteva, D.; Sung, V.; Chopra, R.; Arlet, J.-B. A phase 2a, open-label, dose-finding study to determine the safety and tolerability of sotatercept (ACE-011) in adults with beta (β)-thalassemia: Interim results. Blood 2013, 122, 3448. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Porter, J.; Origa, R.; Forni, G.L.; Voskaridou, E.; Galactéros, F.; Taher, A.T.; Arlet, J.B.; Ribeil, J.A.; Garbowski, M.; et al. Sotatercept, a novel transforming growth factor β ligand trap, improves anemia in β-thalassemia: A phase II, open-label, dose-finding study. Haematologica 2019, 104, 477–484. [Google Scholar] [CrossRef]
- Iqbal, A.; Habiba, U.; Waseem, R.; Islam, Z. Pyruvate kinase activator: A major breakthrough in the world of Hematology. Ann. Med. Surg. 2022, 82, 104631. [Google Scholar] [CrossRef]
- Schroeder, P.; Fulzele, K.; Forsyth, S.; Ribadeneira, M.D.; Guichard, S.; Wilker, E.; Marshall, C.G.; Drake, A.; Fessler, R.; Konstantinidis, D.G.; et al. Etavopivat, a Pyruvate Kinase Activator in Red Blood Cells, for the Treatment of Sickle Cell Disease. J. Pharmacol. Exp. Ther. 2022, 380, 210–219. [Google Scholar] [CrossRef]
- Musallam, K.M.; Taher, A.T.; Cappellini, M.D.; Hermine, O.; Kuo, K.H.M.; Sheth, S.; Viprakasit, V.; Porter, J.B. Untreated Anemia in Nontransfusion-dependent β-thalassemia: Time to Sound the Alarm. Hemasphere 2022, 6, e806. [Google Scholar] [CrossRef]
- McGann, P.T.; Ware, R.E. Hydroxyurea therapy for sickle cell anemia. Expert Opin. Drug Saf. 2015, 14, 1749–1758. [Google Scholar] [CrossRef]
- Koren, A.; Levin, C.; Dgany, O.; Kransnov, T.; Elhasid, R.; Zalman, L.; Palmor, H.; Tamary, H. Response to hydroxyurea therapy in beta-thalassemia. Am. J. Hematol. 2008, 83, 366–370. [Google Scholar] [CrossRef]
- Banan, M. Hydroxyurea treatment in β-thalassemia patients: To respond or not to respond? Ann. Hematol. 2013, 92, 289–299. [Google Scholar] [CrossRef]
- Abraham, R.T.; Wiederrecht, G.J. Immunopharmacology of rapamycin. Annu. Rev. Immunol. 1996, 14, 483–510. [Google Scholar] [CrossRef]
- Lechauve, C.; Keith, J.; Khandros, E.; Fowler, S.; Mayberry, K.; Freiwan, A.; Thom, C.S.; Delbini, P.; Romero, E.B.; Zhang, J.; et al. The autophagy-activating kinase ULK1 mediates clearance of free α-globin in β-thalassemia. Sci. Transl. Med. 2019, 11, eaav4881. [Google Scholar] [CrossRef]
- Brus, J.; Czernek, J.; Kobera, L.; Urbanova, M.; Abbrent, S.; Husak, M. Predicting the crystal structure of decitabine by powder NMR crystallography: Influence of long-range molecular packing symmetry on NMR parameters. Cryst. Growth Des. 2016, 16, 7102–7111. [Google Scholar] [CrossRef]
- Lal, A.; Vichinsky, E. The role of fetal hemoglobin-enhancing agents in thalassemia. Semin. Hematol. 2004, 41, 17–22. [Google Scholar] [CrossRef]
- Koshy, M.; Dorn, L.; Bressler, L.; Molokie, R.; Lavelle, D.; Talischy, N.; Hoffman, R.; van Overveld, W.; DeSimone, J. 2-deoxy 5-azacytidine and fetal hemoglobin induction in sickle cell anemia. Blood 2000, 96, 2379–2384. [Google Scholar] [CrossRef]
- Olivieri, N.F.; Saunthararajah, Y.; Thayalasuthan, V.; Kwiatkowski, J.; Ware, R.E.; Kuypers, F.A.; Kim, H.Y.; Trachtenberg, F.L.; Vichinsky, E.P. A pilot study of subcutaneous decitabine in β-thalassemia intermedia. Blood 2011, 118, 2708–2711. [Google Scholar] [CrossRef]
- Mehta, P.; Yadav, N.; Soni, P.; Thekuddan, S.F.; Singh, R.; Khushoo, V.; Mirgh, S.P.; Agrawal, N.; Ahmed, R.; Kapoor, J. Experience with Low Dose Thalidomide in Transfusion Dependent Beta Thalassaemia in a Resource Limited Setting. Blood 2019, 134, 963. [Google Scholar] [CrossRef]
- Tseng, S.; Pak, G.; Washenik, K.; Pomeranz, M.K.; Shupack, J.L. Rediscovering thalidomide: A review of its mechanism of action, side effects, and potential uses. J. Am. Acad. Dermatol. 1996, 35, 969–979. [Google Scholar] [CrossRef]
- Yassin, A.K. Promising Response to Thalidomide in Symptomatic β-Thalassemia. Indian J. Hematol. Blood Transfus. 2020, 36, 337–341. [Google Scholar] [CrossRef]
- Yang, K.; Wu, Y.; Zhou, Y.; Long, B.; Lu, Q.; Zhou, T.; Wang, L.; Geng, Z.; Yin, X. Thalidomide for Patients with β-Thalassemia: A Multicenter Experience. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020021. [Google Scholar] [CrossRef]
- Li, X.; Hu, S.; Liu, Y.; Huang, J.; Hong, W.; Xu, L.; Xu, H.; Fang, J. Efficacy of Thalidomide Treatment in Children With Transfusion Dependent β-Thalassemia: A Retrospective Clinical Study. Front. Pharmacol. 2021, 12, 722502. [Google Scholar] [CrossRef]
- Sajadi Hezaveh, Z.; Azarkeivan, A.; Janani, L.; Hosseini, S.; Shidfar, F. The effect of quercetin on iron overload and inflammation in β-thalassemia major patients: A double-blind randomized clinical trial. Complement. Ther. Med. 2019, 46, 24–28. [Google Scholar] [CrossRef]
- Nishimura, K.; Matsumoto, R.; Yonezawa, Y.; Nakagawa, H. Effect of quercetin on cell protection via erythropoietin and cell injury of HepG2 cells. Arch. Biochem. Biophys. 2017, 636, 11–16. [Google Scholar] [CrossRef]
- Asghar, A.A.; Khabir, Y.; Hashmi, M.R. Zynteglo: Betibeglogene autotemcel—An innovative therapy for β- thalassemia patients. Ann. Med. Surg. 2022, 82, 104624. [Google Scholar] [CrossRef]
- Taher, A.T.; Bou-Fakhredin, R.; Kattamis, A.; Viprakasit, V.; Cappellini, M.D. Improving outcomes and quality of life for patients with transfusion-dependent β-thalassemia: Recommendations for best clinical practice and the use of novel treatment strategies. Expert Rev. Hematol. 2021, 14, 897–909. [Google Scholar] [CrossRef]
- Whitney, D.; Shestopalov, I.; Fincker, M.; d’Anjou, M.; Kral, K.; Gayron, M.; Pierciey, F.J.; Colvin, R.A. Drug Product Attributes Predict Clinical Efficacy in betibeglogene autotemcel Gene Therapy for β-thalassemia. Mol. Ther. Methods Clin. Dev. 2023, 31, 101155. [Google Scholar] [CrossRef]
- Locatelli, F.; Thompson, A.A.; Kwiatkowski, J.L.; Porter, J.B.; Thrasher, A.J.; Hongeng, S.; Sauer, M.G.; Thuret, I.; Lal, A.; Algeri, M.; et al. Betibeglogene Autotemcel Gene Therapy for Non-β(0)/β(0) Genotype β-Thalassemia. N. Engl. J. Med. 2022, 386, 415–427. [Google Scholar] [CrossRef]
- Piga, A.; Perrotta, S.; Gamberini, M.R.; Voskaridou, E.; Melpignano, A.; Filosa, A.; Caruso, V.; Pietrangelo, A.; Longo, F.; Tartaglione, I.; et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with β-thalassemia. Blood 2019, 133, 1279–1289. [Google Scholar] [CrossRef]
- Markham, A. Luspatercept: First Approval. Drugs 2020, 80, 85–90. [Google Scholar] [CrossRef]
- Piga, A.; Longo, F.; Gamberini, M.R.; Voskaridou, E.; Ricchi, P.; Caruso, V.; Pietrangelo, A.; Zhang, X.; Shetty, J.K.; Attie, K.M.; et al. Long-term safety and erythroid response with luspatercept treatment in patients with β-thalassemia. Ther. Adv. Hematol. 2022, 13, 20406207221134404. [Google Scholar] [CrossRef] [PubMed]
- Aydinok, Y. Highlights on the Luspatercept Treatment in Thalassemia. Thalass. Rep. 2023, 13, 77–84. [Google Scholar] [CrossRef]
- Dixit, A.; Chatterjee, T.C.; Mishra, P.; Choudhry, D.R.; Mahapatra, M.; Tyagi, S.; Kabra, M.; Saxena, R.; Choudhry, V.P. Hydroxyurea in thalassemia intermedia—A promising therapy. Ann. Hematol. 2005, 84, 441–446. [Google Scholar] [CrossRef]
- Ansari, S.H.; Shamsi, T.S.; Ashraf, M.; Perveen, K.; Farzana, T.; Bohray, M.; Erum, S.; Mehboob, T. Efficacy of hydroxyurea in providing transfusion independence in β-thalassemia. J. Pediatr. Hematol. Oncol. 2011, 33, 339–343. [Google Scholar] [CrossRef]
- Bordbar, M.R.; Silavizadeh, S.; Haghpanah, S.; Kamfiroozi, R.; Bardestani, M.; Karimi, M. Hydroxyurea Treatment in Transfusion-Dependent β-Thalassemia Patients. Iran. Red Crescent Med. J. 2014, 16, e18028. [Google Scholar] [CrossRef]
- Zohaib, M.; Ansari, S.H.; Shamsi, T.S.; Zubarev, R.A.; Zarina, S. Pharmacoproteomics Profiling of Plasma From β-Thalassemia Patients in Response to Hydroxyurea Treatment. J. Clin. Pharmacol. 2019, 59, 98–106. [Google Scholar] [CrossRef]
- Gamberini, M.R.; Prosdocimi, M.; Gambari, R. Sirolimus for treatment of β-thalassemia: From pre-clinical studies to the design of clinical trials. Health Educ. 2021, 4, 3. [Google Scholar]
- Kalantri, S.A.; Ray, R.; Chattopadhyay, A.; Bhattacharjee, S.; Biswas, A.; Bhattacharyya, M. Efficacy of decitabine as hemoglobin F inducer in HbE/β-thalassemia. Ann. Hematol. 2018, 97, 1689–1694. [Google Scholar] [CrossRef]
- Li, Y.; Ren, Q.; Zhou, Y.; Li, P.; Lin, W.; Yin, X. Thalidomide has a significant effect in patients with thalassemia intermedia. Hematology 2018, 23, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, C.; Russo, M.; Bilotto, S.; Tedesco, I.; Laratta, B.; Russo, G.L. Dietary polyphenols in cancer prevention: The example of the flavonoid quercetin in leukemia. Ann. N. Y. Acad. Sci. 2012, 1259, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Fattizzo, B.; Motta, I. Rise of the planet of rare anemias: An update on emerging treatment strategies. Front. Med. 2022, 9, 1097426. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Merica, E.; Chen, Y.; Cohen, M.; Goldwater, R.; Kosinski, P.A.; Kung, C.; Yuan, Z.J.; Silverman, L.; Goldwasser, M.; et al. Phase 1 Single- and Multiple-Ascending-Dose Randomized Studies of the Safety, Pharmacokinetics, and Pharmacodynamics of AG-348, a First-in-Class Allosteric Activator of Pyruvate Kinase R, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2019, 8, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.H.; Layton, D.M.; Lal, A.; Al-Samkari, H.; Kosinski, P.A.; Tong, B.; Estepp, J.H.; Uhlig, K.; Vichinsky, E.P. Mitapivat Improves Markers of Erythropoietic Activity in Long-Term Study of Adults with Alpha-or Beta-Non-Transfusion-Dependent Thalassemia. Blood 2022, 140, 2479–2480. [Google Scholar] [CrossRef]
- Kuo, K.H.; Layton, D.M.; Lal, A.; Al-Samkari, H.; Bhatia, J.; Kosinski, P.A.; Tong, B.; Lynch, M.; Uhlig, K.; Vichinsky, E. Long-term efficacy and safety of the oral pyruvate kinase activator mitapivat in adults with non-transfusion-dependent alpha-or beta-thalassemia. Blood 2021, 138, 576. [Google Scholar] [CrossRef]
- Kuo, K.H.M.; Layton, D.M.; Lal, A.; Al-Samkari, H.; Bhatia, J.; Kosinski, P.A.; Tong, B.; Lynch, M.; Uhlig, K.; Vichinsky, E.P. Safety and efficacy of mitapivat, an oral pyruvate kinase activator, in adults with non-transfusion dependent α-thalassaemia or β-thalassaemia: An open-label, multicentre, phase 2 study. Lancet 2022, 400, 493–501. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, M.J.; Rab, M.A.; Rijneveld, A.W.; Nur, E.; Bartels, M.; Jans, J.J.; van Solinge, W.W.; Schutgens, R.E.; van Wijk, R.; Van Beers, E.J. Safety and efficacy of mitapivat (AG-348), an oral activator of pyruvate kinase R, in subjects with sickle cell disease: A phase 2, open-label study (ESTIMATE). Blood 2021, 138, 2047. [Google Scholar] [CrossRef]
- Luke, N.; Hillier, K.; Al-Samkari, H.; Grace, R.F. Updates and advances in pyruvate kinase deficiency. Trends Mol. Med. 2023, 29, 406–418. [Google Scholar] [CrossRef]
- van Dijk, M.J.; de Wilde, J.R.A.; Bartels, M.; Kuo, K.H.M.; Glenthøj, A.; Rab, M.A.E.; van Beers, E.J.; van Wijk, R. Activation of pyruvate kinase as therapeutic option for rare hemolytic anemias: Shedding new light on an old enzyme. Blood Rev. 2023, 61, 101103. [Google Scholar] [CrossRef] [PubMed]
- Bou-Fakhredin, R.; Kuo, K.H.M.; Taher, A.T. Emerging Therapies in β-Thalassemia. Hematol. Oncol. Clin. N. Am. 2023, 37, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, J.L.; Walters, M.C.; Hongeng, S.; Locatelli, F.; Rasko, J.E.; Cavazzana, M.; Chen, Y.; Colvin, R.A.; Thompson, A.A. Long-term efficacy and safety of betibeglogene autotemcel gene therapy for the treatment of transfusion-dependent β-thalassemia: Results in patients with up to 6 years of follow-up. Blood 2020, 136, 51–52. [Google Scholar] [CrossRef]
- Fibach, E.; Bianchi, N.; Borgatti, M.; Zuccato, C.; Finotti, A.; Lampronti, I.; Prus, E.; Mischiati, C.; Gambari, R. Effects of rapamycin on accumulation of alpha-, beta- and gamma-globin mRNAs in erythroid precursor cells from beta-thalassaemia patients. Eur. J. Haematol. 2006, 77, 437–441. [Google Scholar] [CrossRef]
- Pecoraro, A.; Troia, A.; Calzolari, R.; Scazzone, C.; Rigano, P.; Martorana, A.; Sacco, M.; Maggio, A.; Di Marzo, R. Efficacy of Rapamycin as Inducer of Hb F in Primary Erythroid Cultures from Sickle Cell Disease and β-Thalassemia Patients. Hemoglobin 2015, 39, 225–229. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biswas, S.; Smrity, S.Z.; Bhuia, M.S.; Sonia, F.A.; Aktar, M.A.; Chowdhury, R.; Islam, T.; Islam, M.T.; Gonçalves Alencar, G.; Paulo, C.L.R.; et al. Beta-Thalassemia: A Pharmacological Drug-Based Treatment. Drugs Drug Candidates 2024, 3, 126-147. https://doi.org/10.3390/ddc3010008
Biswas S, Smrity SZ, Bhuia MS, Sonia FA, Aktar MA, Chowdhury R, Islam T, Islam MT, Gonçalves Alencar G, Paulo CLR, et al. Beta-Thalassemia: A Pharmacological Drug-Based Treatment. Drugs and Drug Candidates. 2024; 3(1):126-147. https://doi.org/10.3390/ddc3010008
Chicago/Turabian StyleBiswas, Shrabonti, Shanita Zaman Smrity, Md. Shimul Bhuia, Fatema Akter Sonia, Mst. Asma Aktar, Raihan Chowdhury, Tawhida Islam, Muhammad Torequl Islam, Gabriel Gonçalves Alencar, Cicera Laura Roque Paulo, and et al. 2024. "Beta-Thalassemia: A Pharmacological Drug-Based Treatment" Drugs and Drug Candidates 3, no. 1: 126-147. https://doi.org/10.3390/ddc3010008
APA StyleBiswas, S., Smrity, S. Z., Bhuia, M. S., Sonia, F. A., Aktar, M. A., Chowdhury, R., Islam, T., Islam, M. T., Gonçalves Alencar, G., Paulo, C. L. R., Diniz Gurgel, A. P. A., & Coutinho, H. D. M. (2024). Beta-Thalassemia: A Pharmacological Drug-Based Treatment. Drugs and Drug Candidates, 3(1), 126-147. https://doi.org/10.3390/ddc3010008