
31st Annual GP2A Medicinal Chemistry Conference

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
- -
- Best Presentation by a Postdoctoral Researcher: Dr. Danica Walsh. Helmholtz Institute of Pharmaceutical Research Saarland (HIPS). Prize sponsored by the European Journal of Medicinal Chemistry and MDPI Drugs and Drug Candidates.In addition, as the winner of the prize, Dr. Walsh had the opportunity to present her work at the EFMC Young Medicinal Chemists’ Symposium in Rome, Italy on 5–6 September 2024. EFMC-YMCS is supported by the European Federation for Medicinal Chemistry and Chemical Biology.
- -
- Best Presentation by a PhD Student: Nathan Broudic. University of Rouen. Prize sponsored by the ARC Foundation.
- -
- Best Poster Presentations: Florian Schwalen (CERMN, University of Caen) and Thomas Zimmermann (University of Würzburg). Prizes sponsored by the French association of medicinal chemistry teachers (Association Française des Enseignants de Chimie Thérapeutique—AFECT).
2. Keynote Lectures
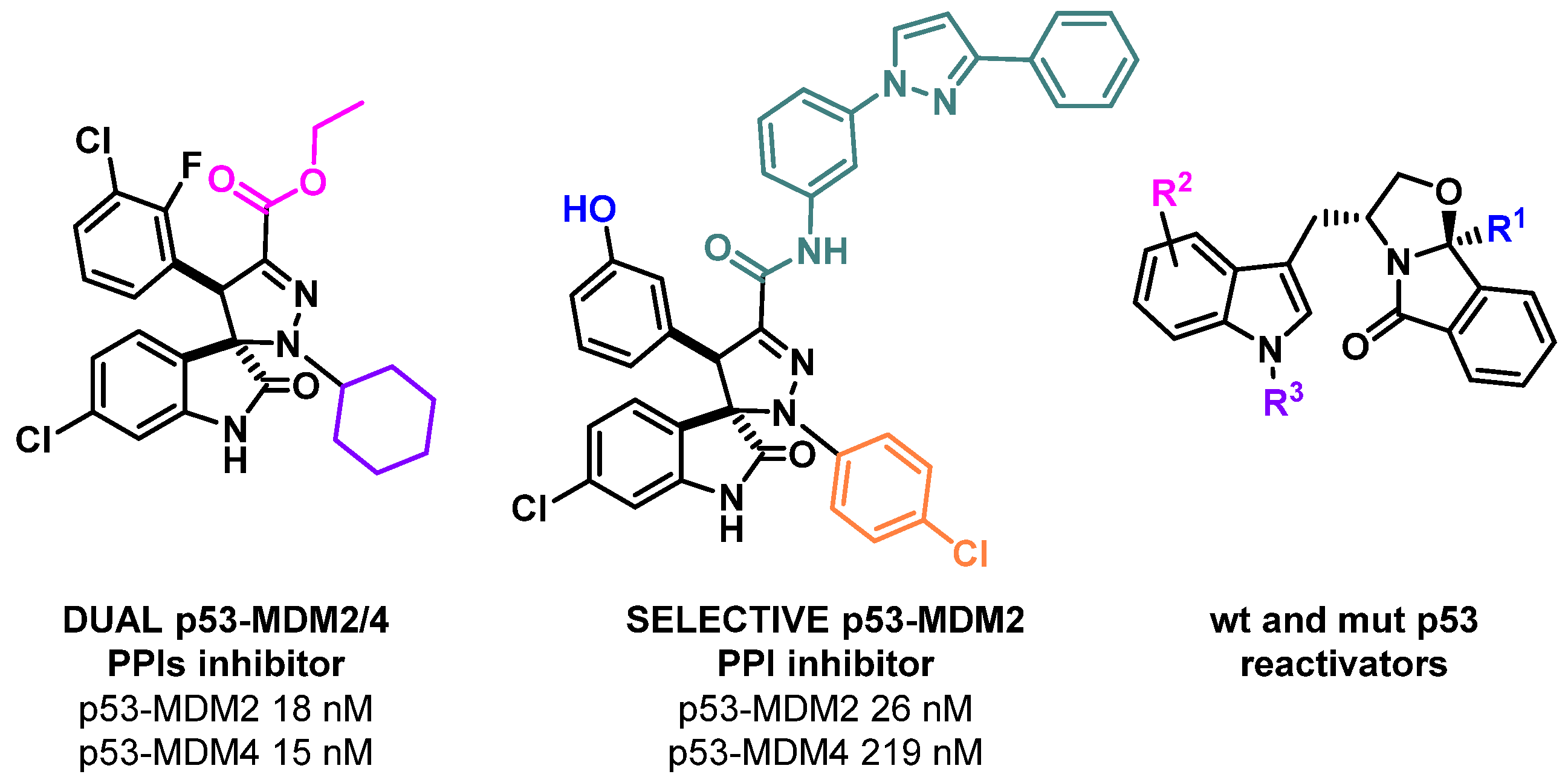
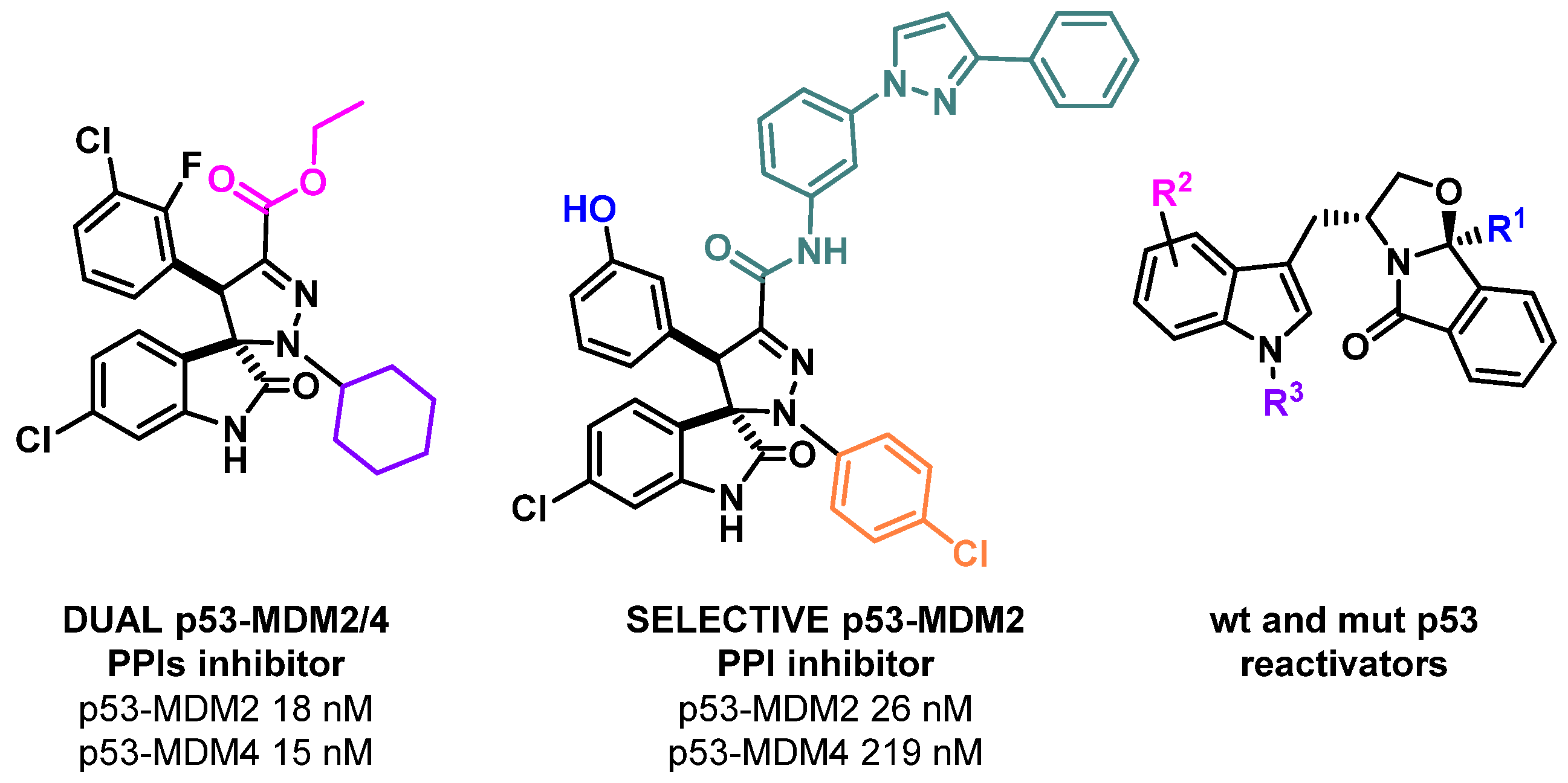
2.1. Development of a New Generation of p53 Activators to Tackle Cancer
- Maria M. M. Santos
- Research Institute for Medicines, Faculty of Pharmacy, Universidade de Lisboa, 1649-003 Lisboa, Portugal; mariasantos@ff.ulisboa.pt
2.2. Mimicking Nature’s Design—From Antimicrobial Siderophore-Drug Conjugates and Artificial Glucosinolates
- Philipp Klahn 1,2
- Department of Chemistry and Molecular Biology, University of Gothenburg, Göteborg, Sweden; philipp.klahn@gu.se
- Laboratories of Natural Product and Conjugation Chemistry (naconLabs)—A Technology Transfer Center of iTUBSmbH, Braunschweig, Germany.
2.3. Drug Discovery Approaches Targeting the Parasite or the Host Cell in the Treatment of Leishmaniasis
- Philippe M. Loiseau 1,*, Sébastien Pomel 1, Christian Cavé 1 and Gillian Barratt 2
- Chimiothérapie antiparasitaire (PARACHEM), UMR 8076 CNRS BioCIS; philippe.loiseau@universite-paris-saclay.fr
- IGPS, UMR 8612 CNRS, Faculté de Pharmacie, Université Paris-Saclay, France
2.4. Contilisant, a Small Molecule Designed for Alzheimer’s Disease Therapy
- José Marco-Contelles
- Laboratory of Medicinal Chemistry (Institute of General Organic Chemistry, CSIC), C/Juan de la Cierva, 3, 28006-Madrid, Spain; iqoc21@iqog.csic.es
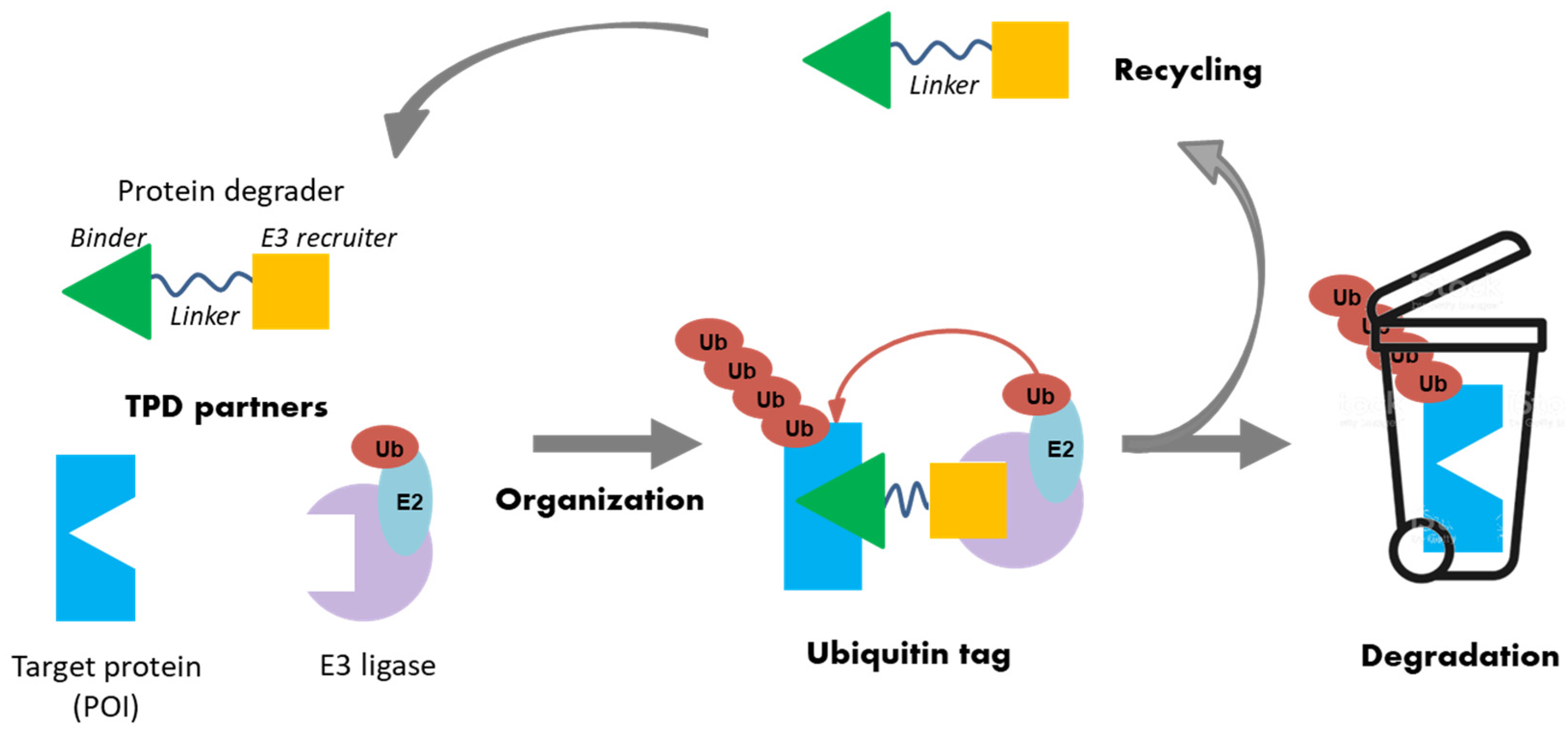
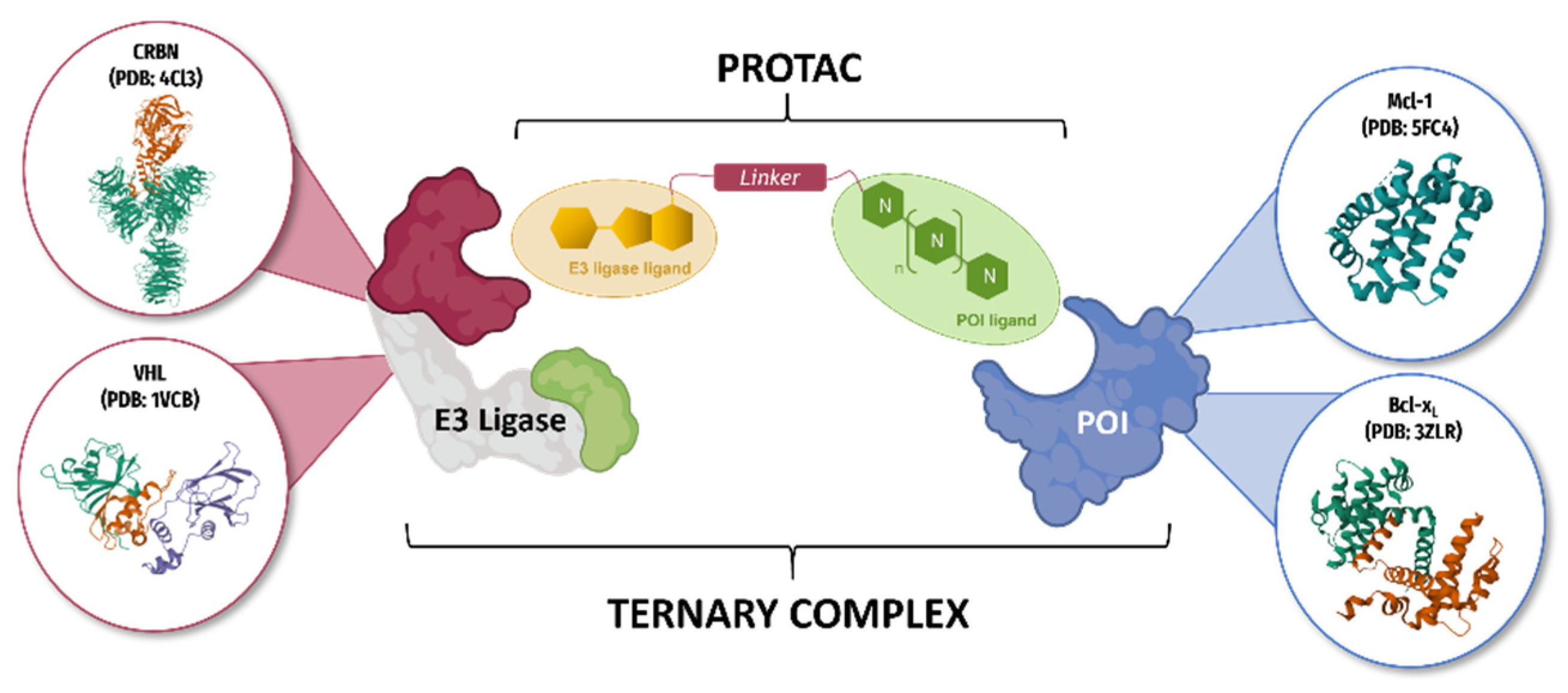
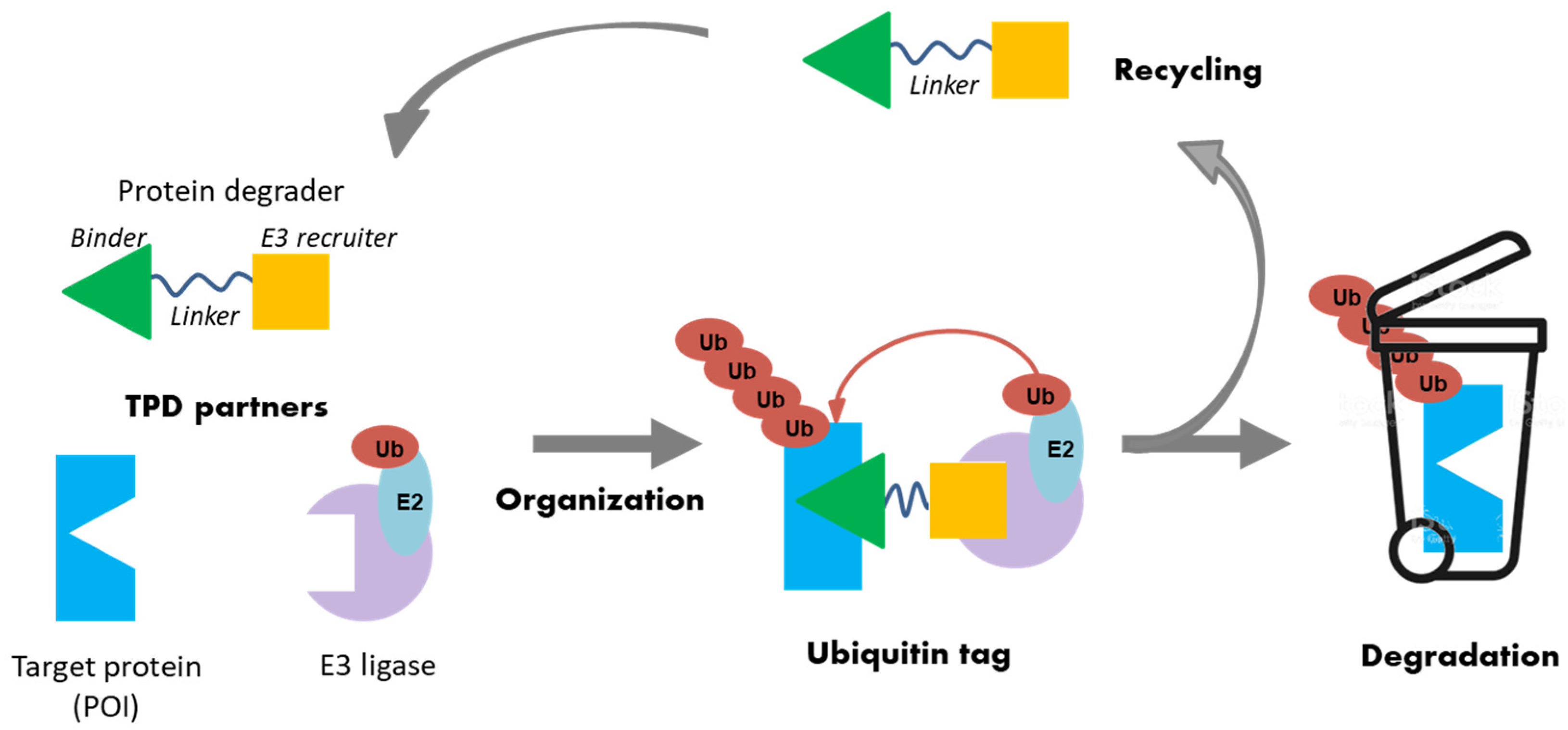
2.5. Emerging Approaches to Targeted Protein Degradation for an Innovative Drug Discovery Strategy
- Anne Sophie Voisin-Chiret
- Centre d’Etudes et de Recherche sur le Médicament de Normandie, UFR SANTE, University of Caen Normandie, 14032 Caen, France; anne-sophie.voisin@unicaen.fr
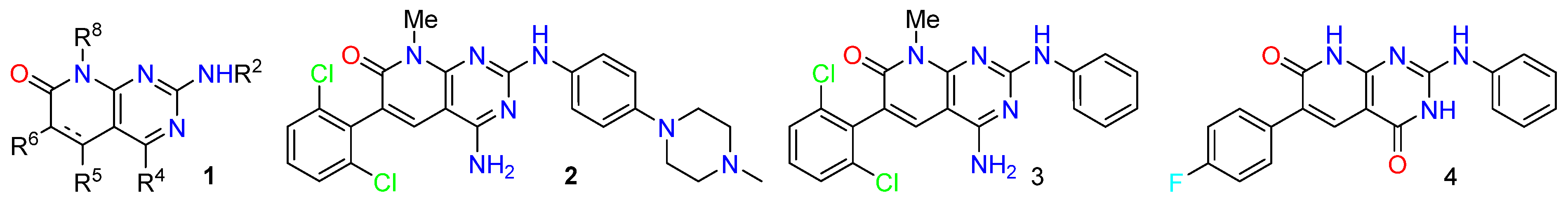
2.6. Pyrido[2,3-d]pyrimidin-7(8H)-ones: A Convenient Scaffold for the Synthesis of Bioactive Compounds
- José I. Borrell
- IQS, Universitat Ramon Llull, E-08017 Barcelona, Spain; jose.borrell@iqs.url.edu
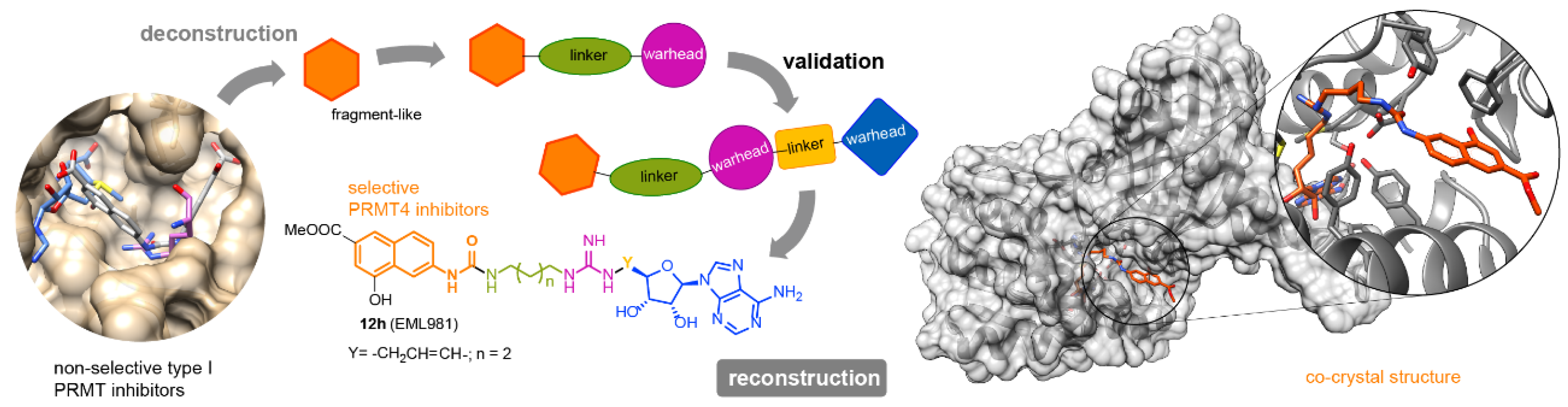
2.7. The Long and Winding Road: Discovering and Validating Protein Arginine Methyltransferase Inhibitors
- Gianluca Sbardella
- Department of Pharmacy, Epigenetic Med Chem Lab, University of Salerno, I-84084, Italy; gsbardella@unisa.it
2.8. Addressing Current Challenges in Antiviral Therapies against Emerging RNA Viruses
- Karine Alvarez
- Laboratoire d’Architecture et Fonction des Macromolécules Biologiques, UMR 7257 CNRS, Campus de Luminy, 13288 Marseille CEDEX 09, France; karine.alvarez@univ-amu.fr
3. Oral Communications
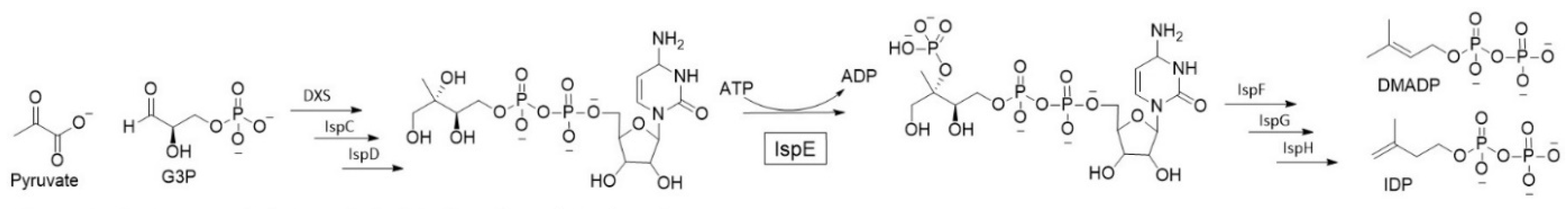
3.1. Design and Synthesis of Novel Anti-Infective Inhibitors of the Target lspE
- Danica Walsh, Rawia Hamid, Mostafa Hamed, Diana Estrada and Anna K.H. Hirsch
- Helmholtz Institute for Pharmaceutical Research Saarland, Saarbrücken, Germany 66123; danicajade.walsh@helmholtz-hips.de
3.2. Design, Synthesis and Biological Evaluation of Self-Immolative Pleiotropic Prodrugs as Potential Treatment against Alzheimer’s Disease
- Valentin Travers—Lesage 1, François-Xavier Toublet 1, Marc Since 1, Audrey Davis 1, Xavier Brazzolotto 2, Florian Nachon 2, Patrick Dallemagne 1 and Christophe Rochais 1
- Normandie Univ, UNICAEN, Centre d’Études et de Recherche sur le Médicament de Normandie—UR4258, Caen, 14000, France; valentin.travers--lesage@unicaen.fr
- Institut de Recherche Biomédicale des Armées, Département des Plateformes et Recherches Technologiques, Unité Développements Analytiques et Bioanalyse, 91223 Brétigny sur Orge, France
3.3. Pharmaceutical Applications of Thermoresponsive Liquid Crystals with Particular Emphasis on Skin Drug Delivery
- Mariia Nesterkina 1, Iryna Kravchenko 1, Anna K.H. Hirsch 1,2 and Claus-Michael Lehr 1,2
- Helmholtz Institute for Pharmaceutical Research Saarland (HIPS)—Helmholtz Centre for Infection Research (HZI), 66123 Saarbrücken, Germany; mariia.nesterkina@helmholtz-hips.de
- Department of Pharmacy, Saarland University, 66123 Saarbrücken, Germany
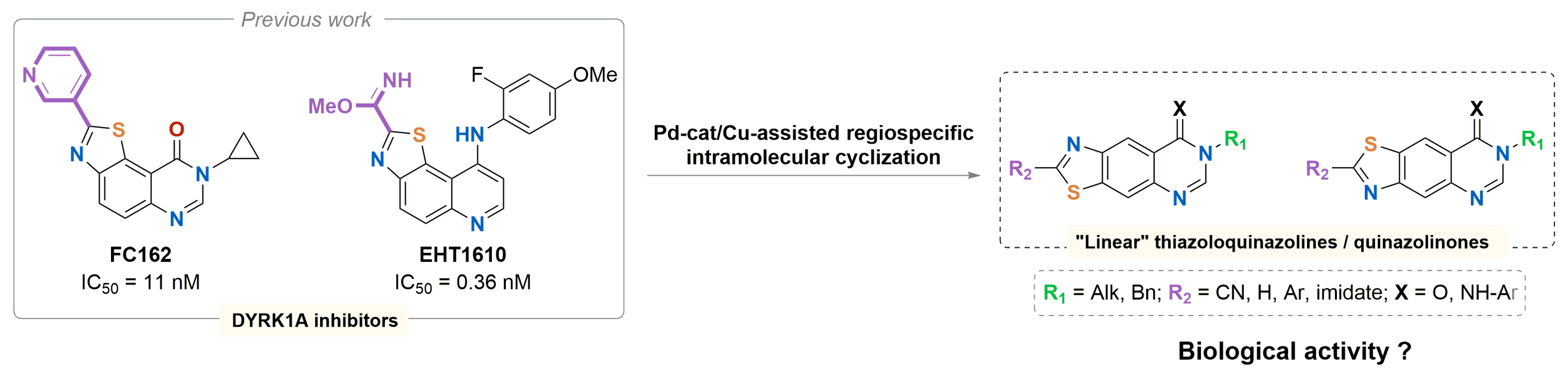
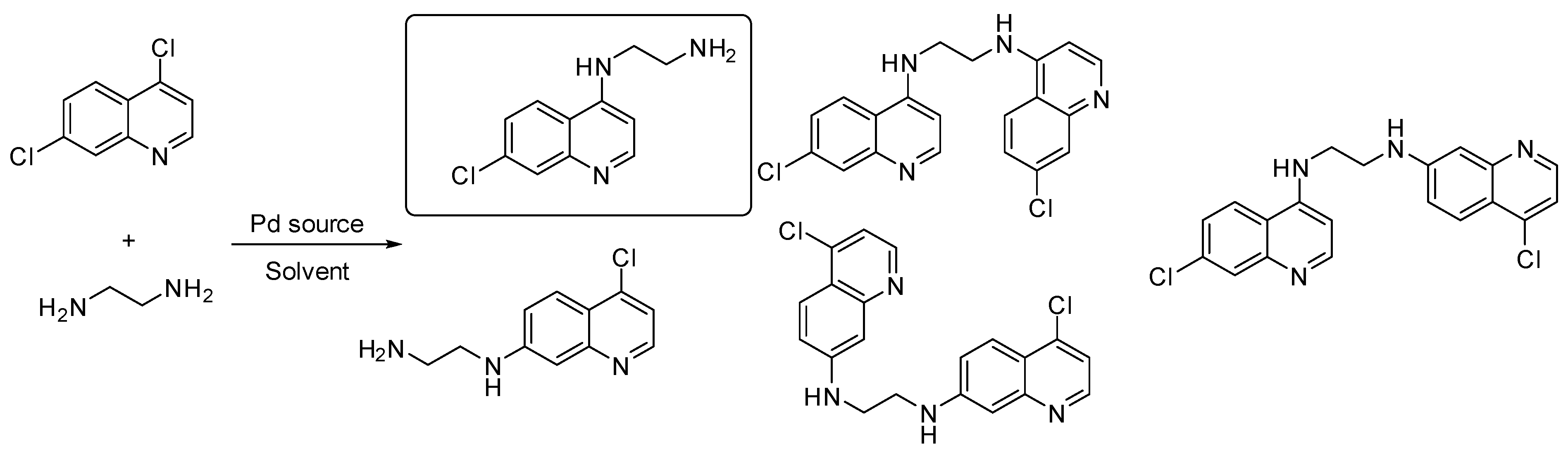
3.4. Synthesis and Biological Evaluation of Linear Thiazolo[4,5-g] and [5,4-g] Quinazolines, Analogues of V-Shaped DYRK1A Inhibitors EHT1610 and FC162
- Nathan Broudic 1, Alexandra Pacheco-Benichou 1, Thomas Robert 2,3,4, Stéphane Bach 2,3,4, Hélène Solhi 5, Rémi Le Guével 5, Corinne Fruit 1 and Thierry Besson 1
- Univ Rouen Normandie, INSA Rouen Normandie, CNRS, COBRA UMR 6014, 76000 Rouen, France; nathan.broudic@univ-rouen.fr
- Sorbonne Université, CNRS, UMR 8227, Station Biologique de Roscoff, Roscoff, France
- Sorbonne Université, CNRS, FR 2424, Plateforme de Criblage KISSf, Station Biologique de Roscoff, Roscoff, France
- Centre of Excellence for Pharmaceutical Sciences, North-West University, Potchefstroom, South Africa
- Plateforme ImPACcell, UAR BIOSIT, Université de Rennes, Campus de Villejean, 2 Avenue du Pr. Leon Bernard CS34317, 35043 Rennes cedex, France
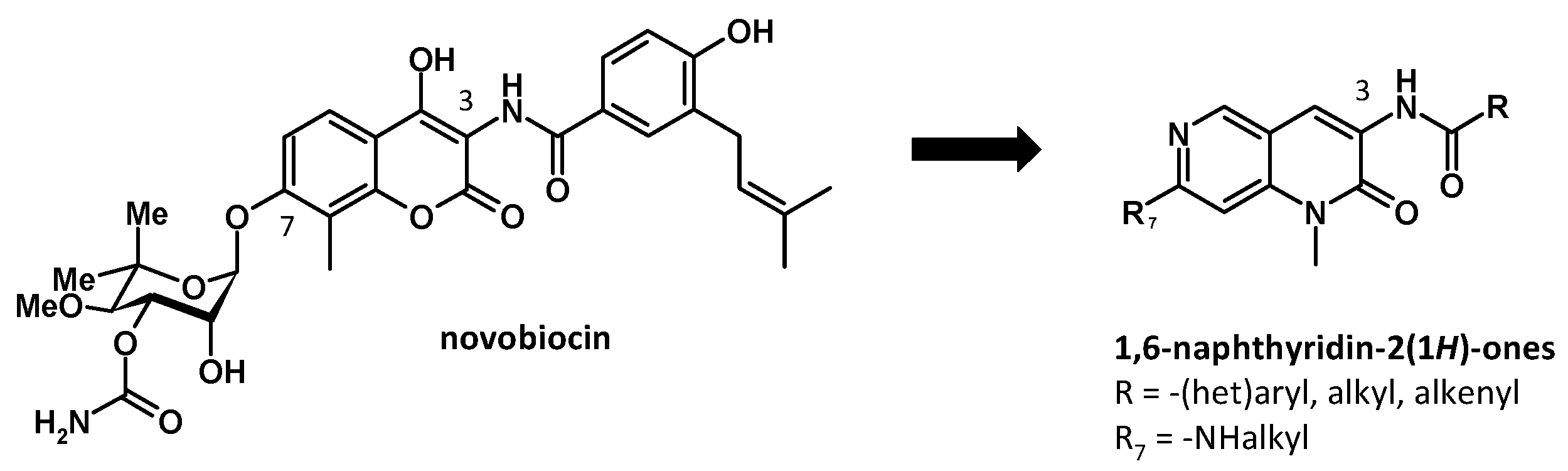
3.5. Synthesis and Biological Evaluation of 1,6-Naphtyridin-2(1H)-one Derivatives as Potential Hsp90 C-Terminal Inhibitors
- David Montoir 1, Eléonore Lepvrier 2, Alain Tonnerre 1, Cyrille Garnier 2, Sophie Barillé-Nion 3, Marc-Antoine Bazin 1
- Nantes Université, Cibles et médicaments des infections et de l’immunité, IICiMed, UR 1155, F-44000 Nantes, France; marc-antoine.bazin@univ-nantes.fr
- IRSET, Team DREAM, UMR_S 1085, University of Rennes 1, F-35000 Rennes, France
- Nantes Université, CRCI2NA, Team SATE, UMR_S 1307, F-44000 Nantes, France
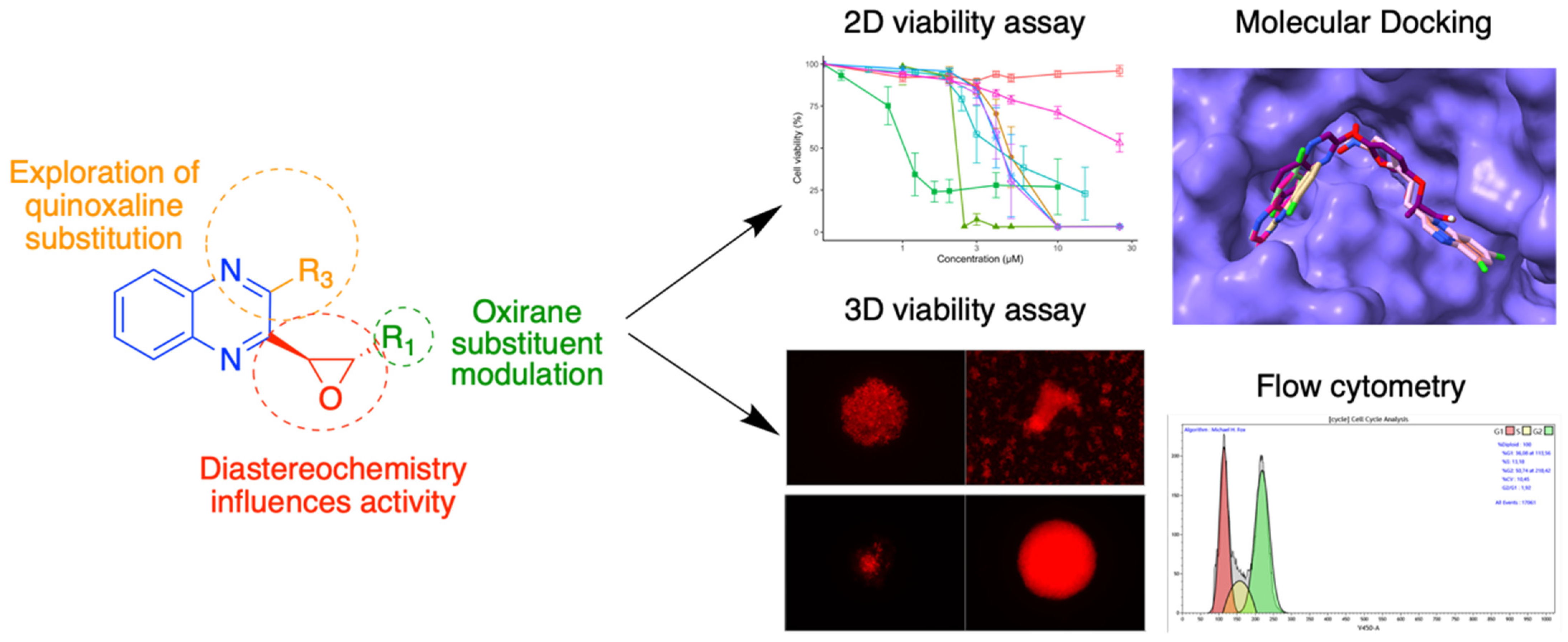
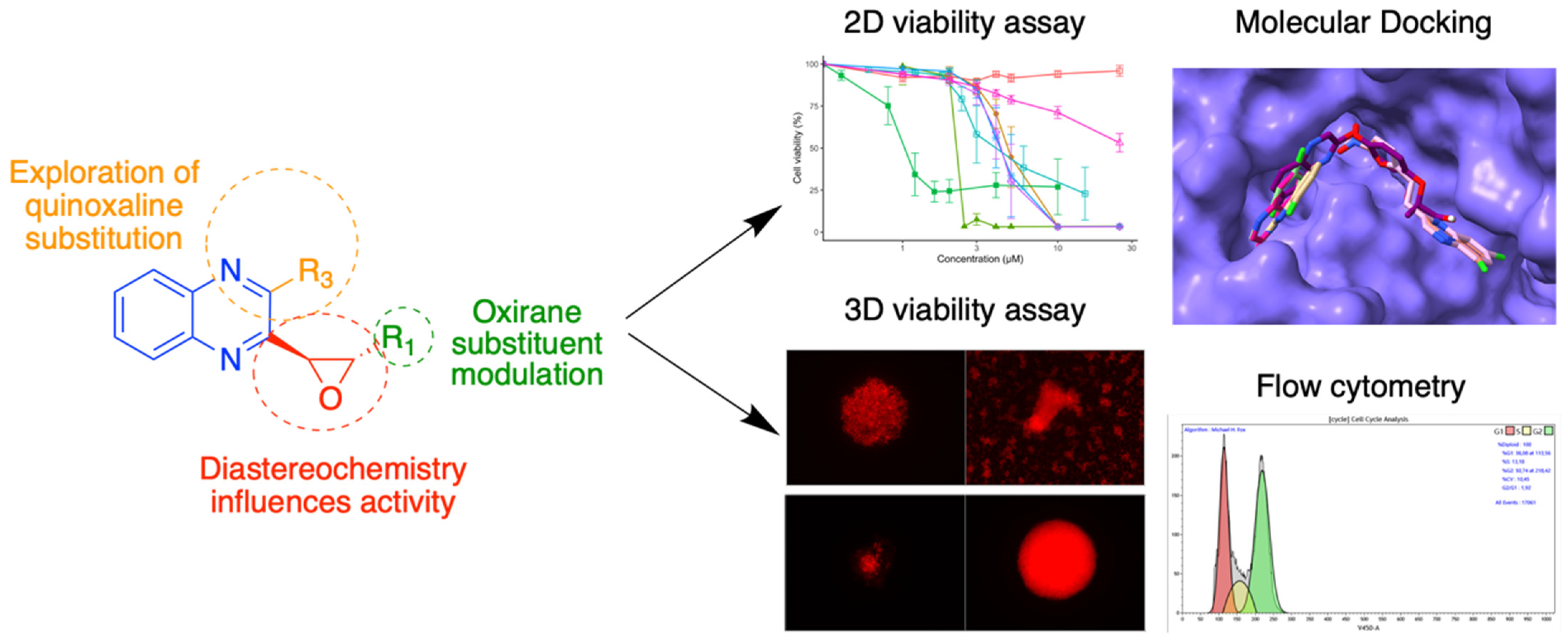
3.6. Quinoxaline Derivatives against Nervous System Tumors
- Vincent Montero 1, Marc Montana 1, Omar Khoumeri 1, Mélanie Matteudi 2, Christine Roux 2, Marie-Pierre Montero 2, Marie-Anne Estève 3, Manon Carré 2 and Patrice Vanelle 1
- Institut de Chimie Radicalaire (ICR), AMU, CNRS—Faculté de Pharmacie, Marseille, France; vincent.montero@etu.univ-amu.fr
- Centre de Recherche en Cancérologie de Marseille (CRCM), AMU, Inserm, CNRS—Faculté de Pharmacie, Marseille, France
- Institut de Neurophysiopathologie (INP), AMU, CNRS UMR 7051—Faculté de Médecine, Marseille, France
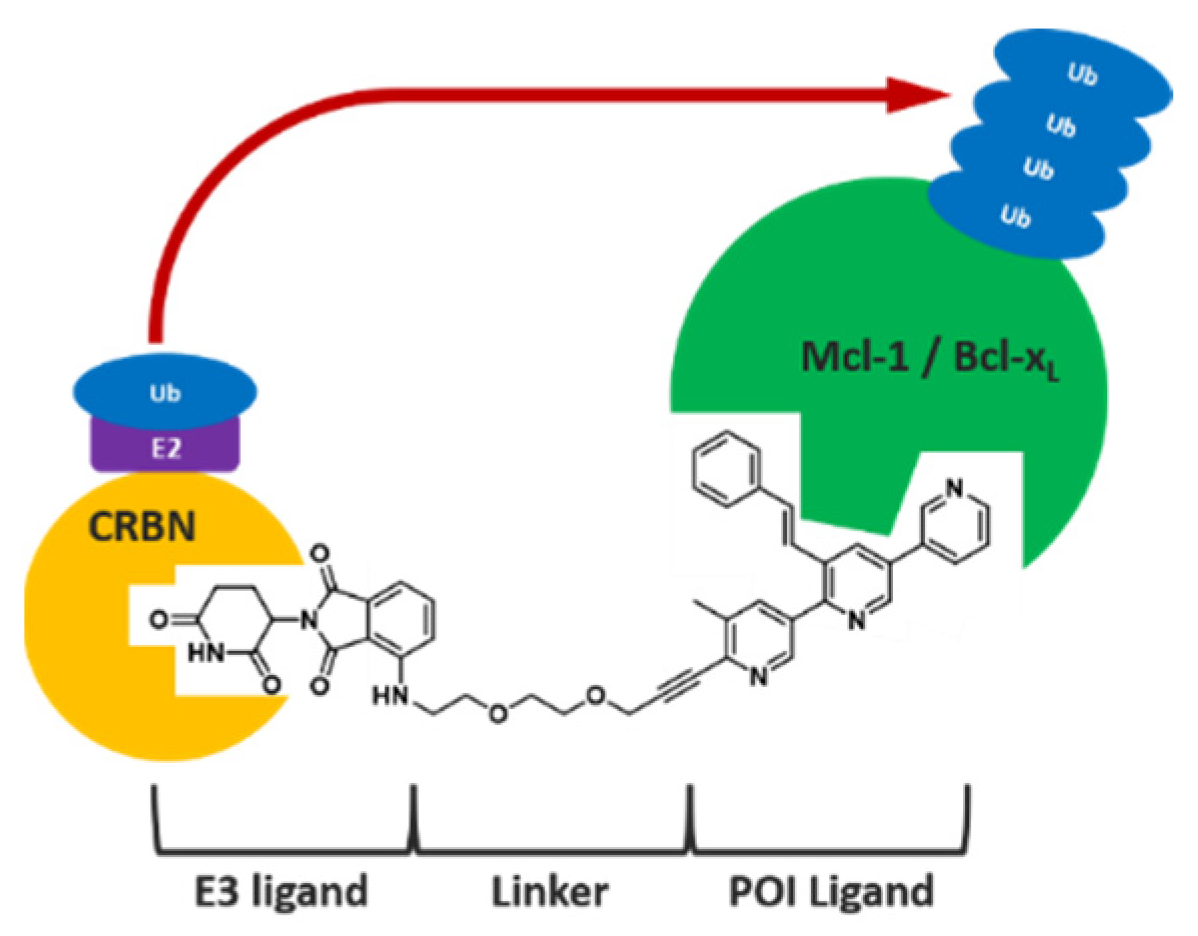
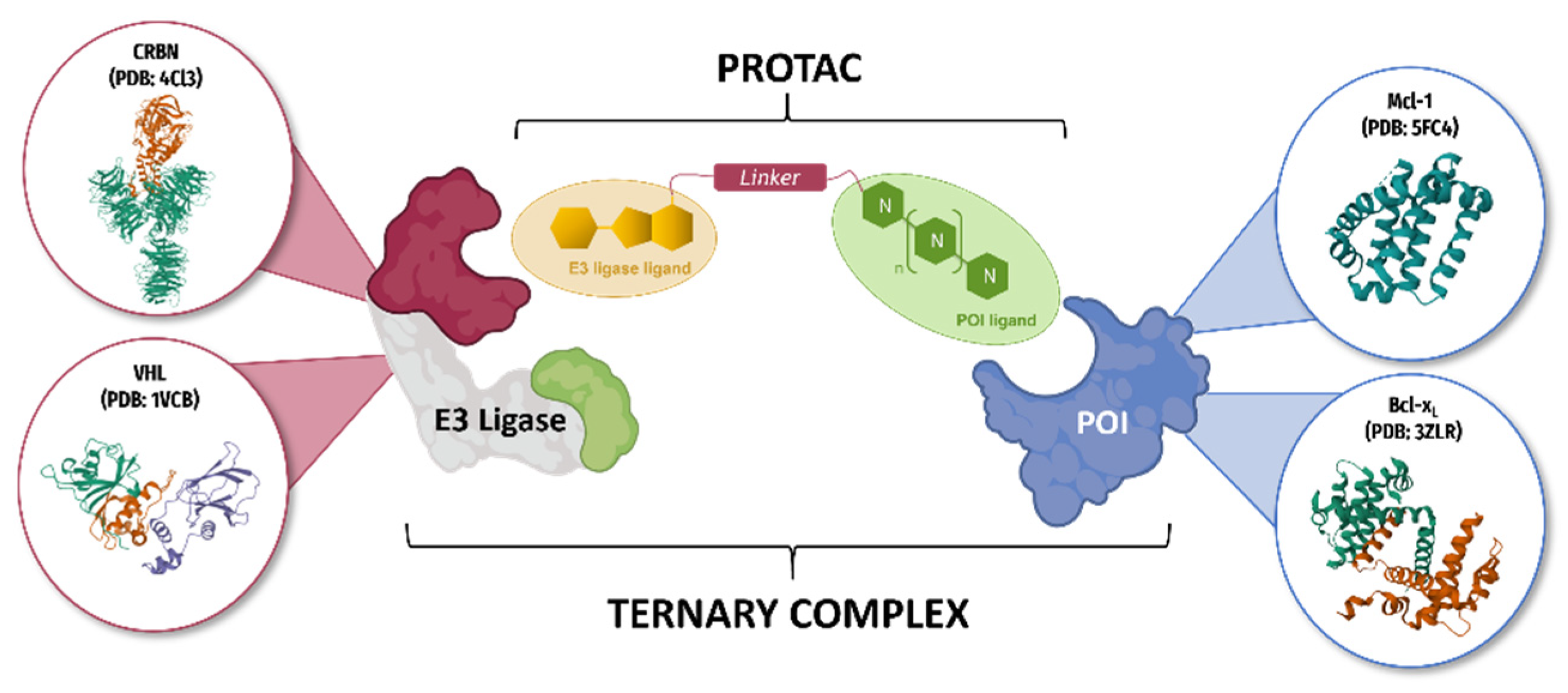
3.7. Proof of Concept and Optimisation of PROTAC Molecules Based on Pyridoclax Recycling against Mcl-1 and Bcl-xL in Ovarian Cancer
- Marie Cornu 1, Jocelyn Pezeril 2, Laurent Poulain 2, Charline Kieffer 1 and Anne Sophie Voisin-Chiret 1
- Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), University of Caen, France; marie.cornu@unicaen.fr
- Inserm U1086 ANTICIPE, University of Caen, France
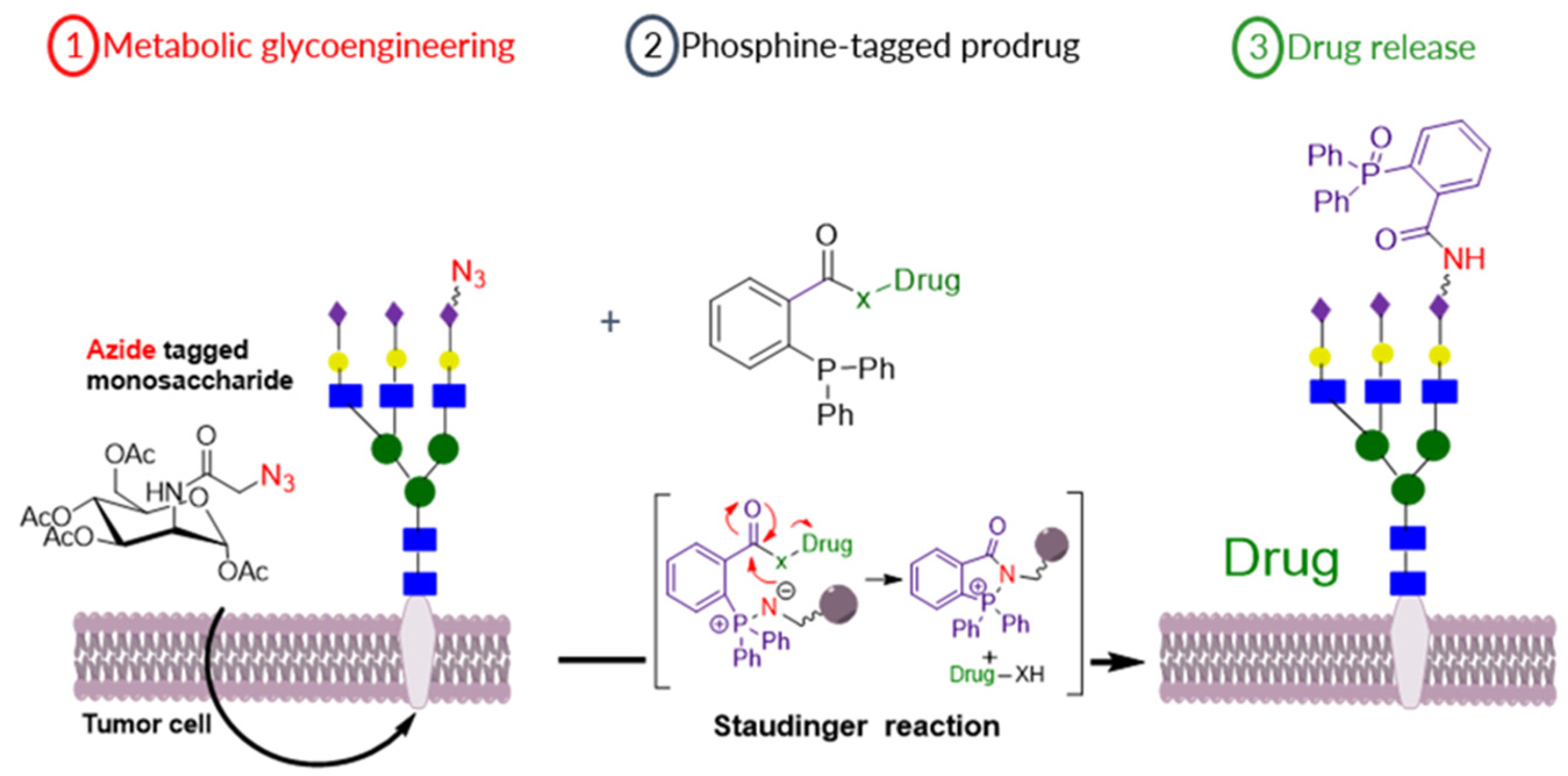
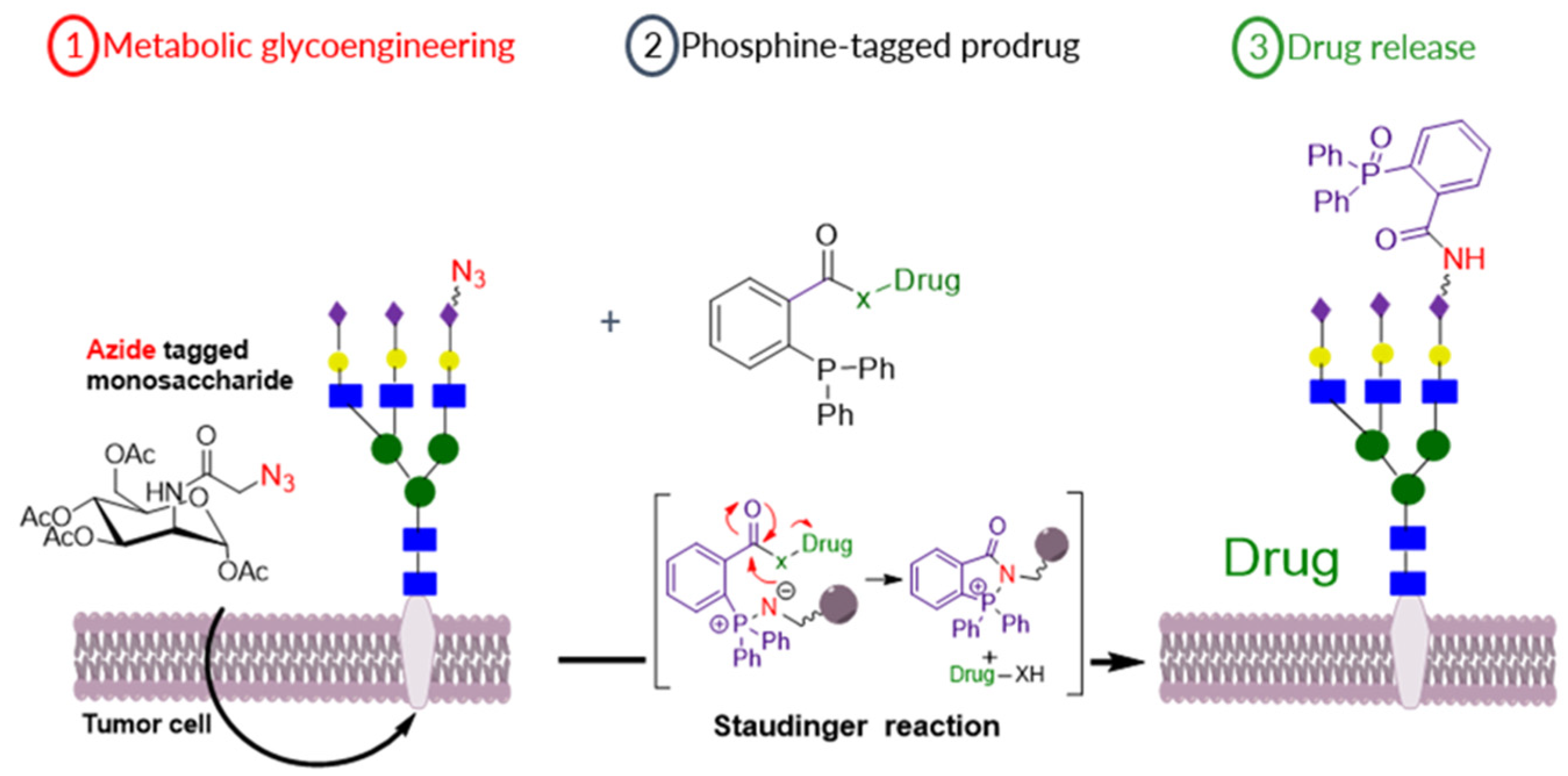
3.8. Bio-orthogonal Activation of Prodrugs, for the Potential Treatment of Breast Cancer, Using the Staudinger Reaction
- Madonna M. A. Mitry 1,2, Samuel Y Boateng 3, Francesca Greco 1 and Helen M.I. Osborn 1
- Reading School of Pharmacy, University of Reading, Whiteknights, Reading, RG6 6AD, UK; m.m.adeebmitry@pgr.reading.ac.uk
- Dept. of Pharmaceutical Chemistry, Faculty of Pharmacy, Ain Shams University, Cairo 11566, Egypt.
- School of Biological Sciences, University of Reading, Whiteknights, Reading, RG6 6UB, UK
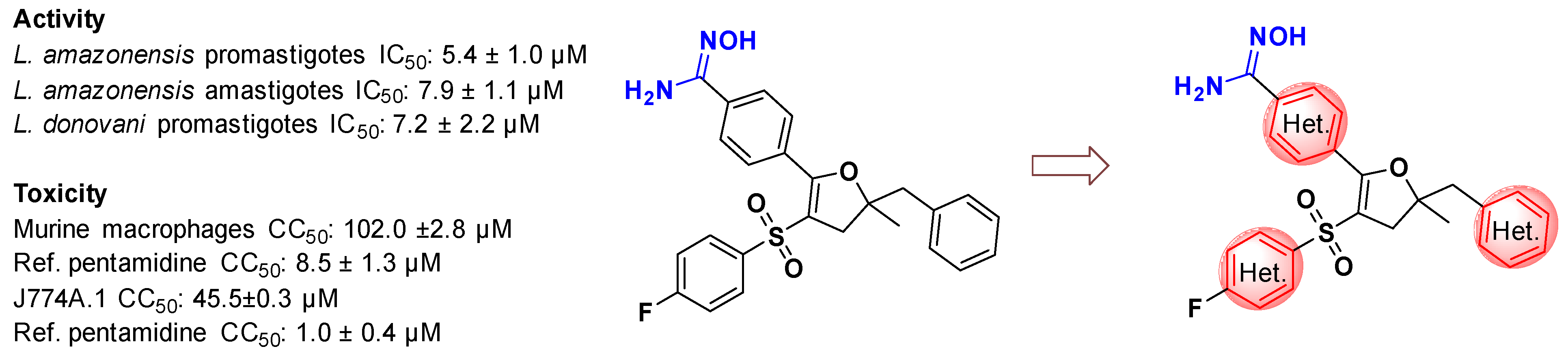
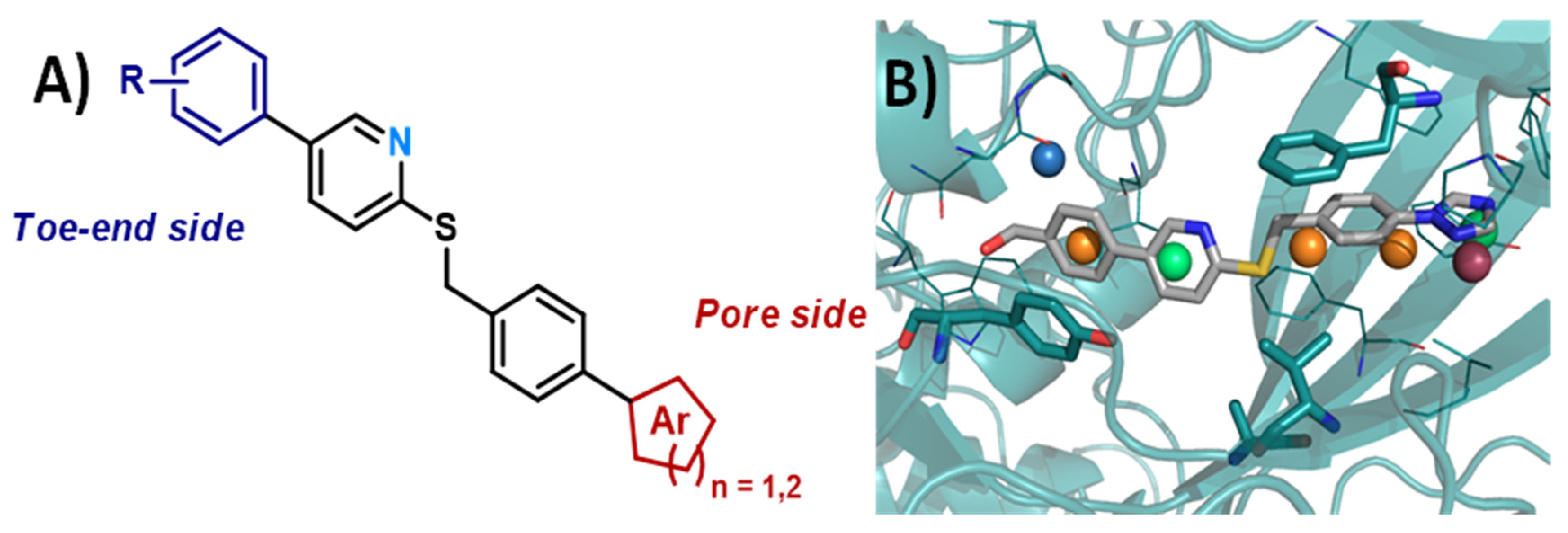
3.9. Antileishmanial Amidoximes, Exploring the Pyridine Effect Using a Ligand-Based Approach
- Oscar Leonardo Avendano Leon 1, Christophe Curti 1, Fabiana Maia Santos Urbancg Moncorvo 2, Youssef Kabri 1, Sébastien Redon 1, Eduardo Caio Torres-Santos 2 and Patrice Vanelle 1
- Equipe Pharmaco-Chimie Radicalaire, CNRS, ICR UMR 7273, Aix Marseille Univ, 13385 Marseille, France; oscar-leonardo.avendano-leon@etu.univ-amu.fr
- Laboratório de Bioquímica de Tripanossomatídeos, Fundação Oswaldo Cruz, Rio de Janeiro 21040-900, Brazil
3.10. About the Creation of the European Federation of National Academic Compound Collections (Eu-FNACC)
- Jean-Luc Galzi 1,2
- GPCRs, pain and inflammation, UMR CNRS 7242 Biotechnologie et signalisation cellulaire, Université de Strasbourg, Illkirch, France; galzi@unistra.fr
- ChemBioFrance-Chimiothèque Nationale UAR3035, 8 Rue de L’école Normale, CEDEX 05, 34296 Montpellier, France.
- Background:
- Vision:
4. Poster Presentations
4.1. Antioxidant Evaluation of Bio-Mimetic Chitosan Nanofibrous Mats as Wound Dressings
- Iacob Andreea-Teodoraa, Ionescu Oana-Mariaa, Lupașcu Florentinaa, Drăgan Mariaa, Apotrosoaei Mariaa, Vasincu Ioanaa and Profire Lenuțaa
- University of Medicine and Pharmacy “Grigore T. Popa”, Faculty of Pharmacy, Iasi 700115, Romania; panzariu.andreea.teodora@gmail.com
4.2. A New Type of Nitrogen Containing Cyclotriveratrylene: Its Synthesis for Application in Hyperpolarized Xenon-129 MRI
- Arsène Château 1, William Richard 1, Thomas Cailly 1,2,3,4, Emmanuelle Dubost 1,2 and Frederic Fabis 1
- Normandie Univ, UNICAEN, Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), 14000 Caen, France; arsene.chateau@unicaen.fr
- Physiopathology and Imaging of Neurological Disorders (PhIND), 14000 Caen, France
- Normandie Univ, UNICAEN, IMOGERE, 14000 Caen, France
- Department of Nuclear Medicine, CHU Côte de Nacre, 14000 Caen, France
4.3. Open Source Mycetoma (MycetOS): Discovery and Development of Novel Classes of Antifungal Agents against the Neglected Mycotic Disease
- Dmitrij Melechov, Matthew Todd, Open Source Mycetoma Consortium
- Department of Pharmaceutical and Biological Chemistry, UCL School of Pharmacy, London WC1N 1AX, UK; dmitrij.melechov.17@ucl.ac.uk
4.4. 3D Pharmacophoric Model Approach Based-New Antituberculosis Discovery
- Maximilien Fil 1, Elnur Garayev 2, Béatrice Baghdikian 2, Sok-Siya Bun-Llopet 2, Célia Breaud 2, Jean-Michel Bolla 1, Gérard Boyer 1 and Sandrine Alibert 2
- Aix-Marseille Université, UMR_MD1, INSERM U1261,MCT, Marseille, France
- IMBE UMR_7263, DFME, Marseille, France; sandrine.alibert@univ-amu.fr
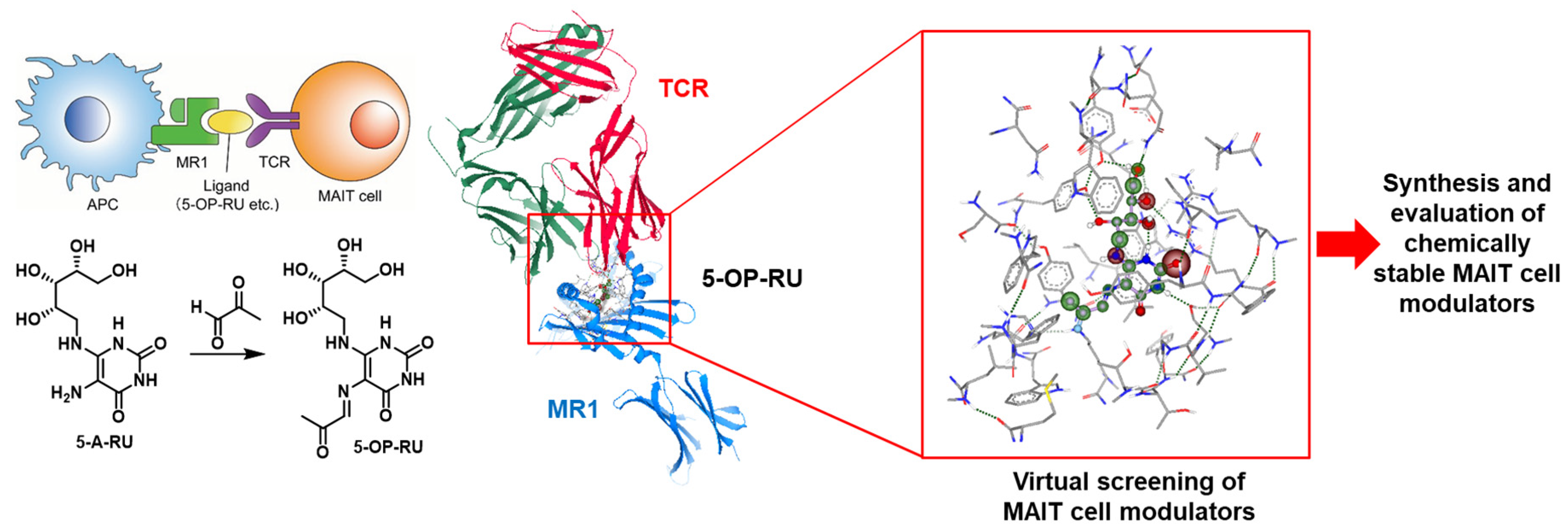
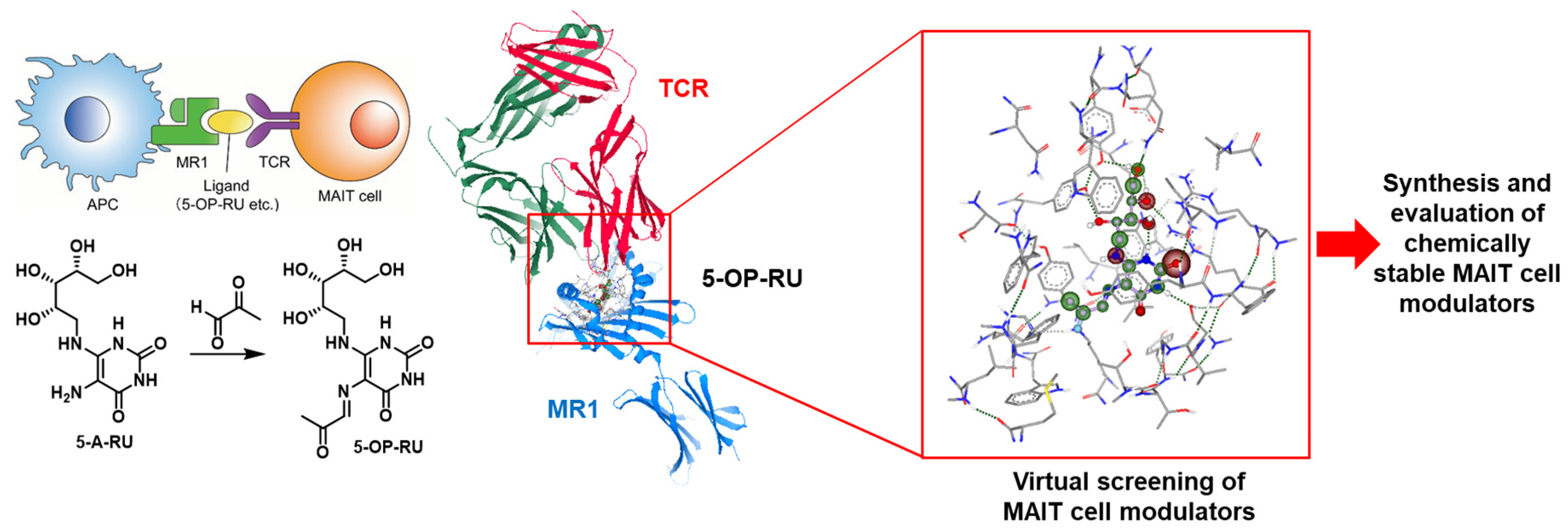
4.5. Development of Artificial OP-5-RU-Derived MAIT Cell Modulators for AICs against Microbial Infection
- Charity S. G. Ganskow, Philipp Klahn
- Department of Chemistry and Molecular Biology, University of Gothenburg, Sweden; charity.ganskow@gu.se
4.6. Arylsubstituted-isothiazol-3(2H)-one Pharmacophore as Part of Molecular Hybridization Agents against PDE4
- Boryana Borisova 1,2, Marie Laronze-Cochard 1, Veronika Nemska 2, Dancho Danalev 2 and Stéphane Gérard 1
- University of Reims Champagne-Ardenne, Faculty of Pharmacy, ICMR UMR CNRS 7312, 51 rue Cognacq Jay, 51096 Reims, France; stephane.gerard@univ-reims.fr
- University of Chemical Technology and Metallurgy, Department of Biotechnology, 8 Kliment Ohridski blvd., 1756 Sofia, Bulgaria
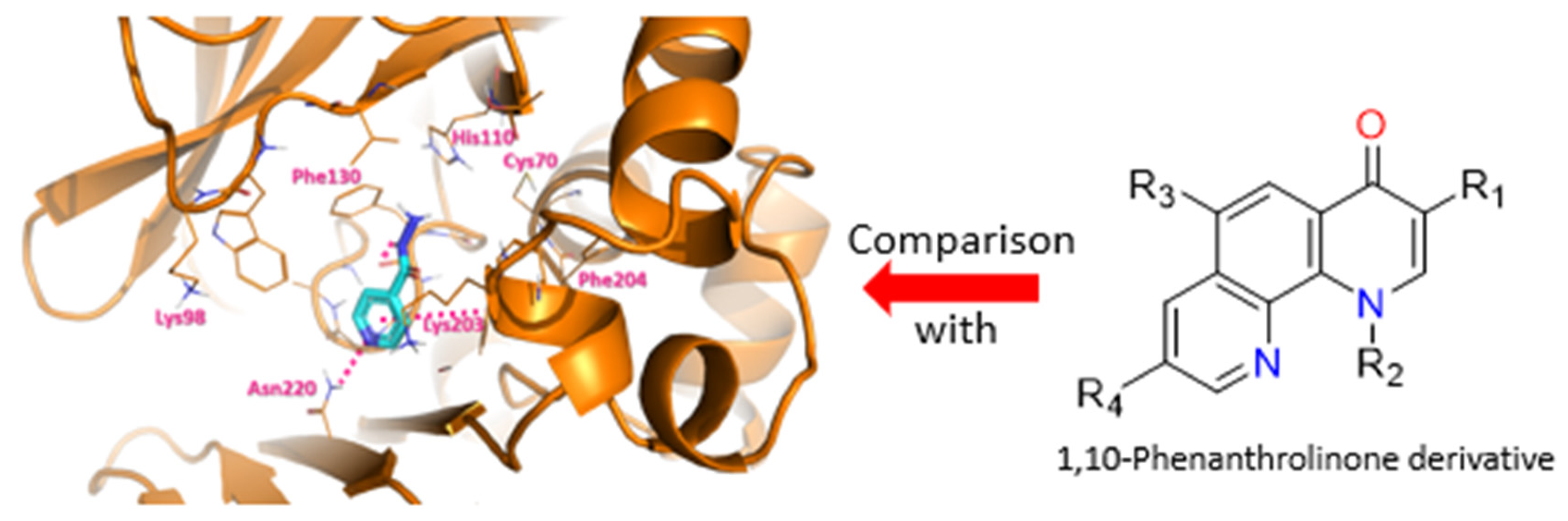
4.7. Targeting Persistence Cause to Enhance Tuberculosis Treatment
- Olivier Hebert, Alban Lepailleur, Cédric Lecoutey, Patrick Dallemagne and Christophe Rochais
- Normandie Univ., UNICAEN, Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Caen, France; olivier.hebert@unicaen.fr
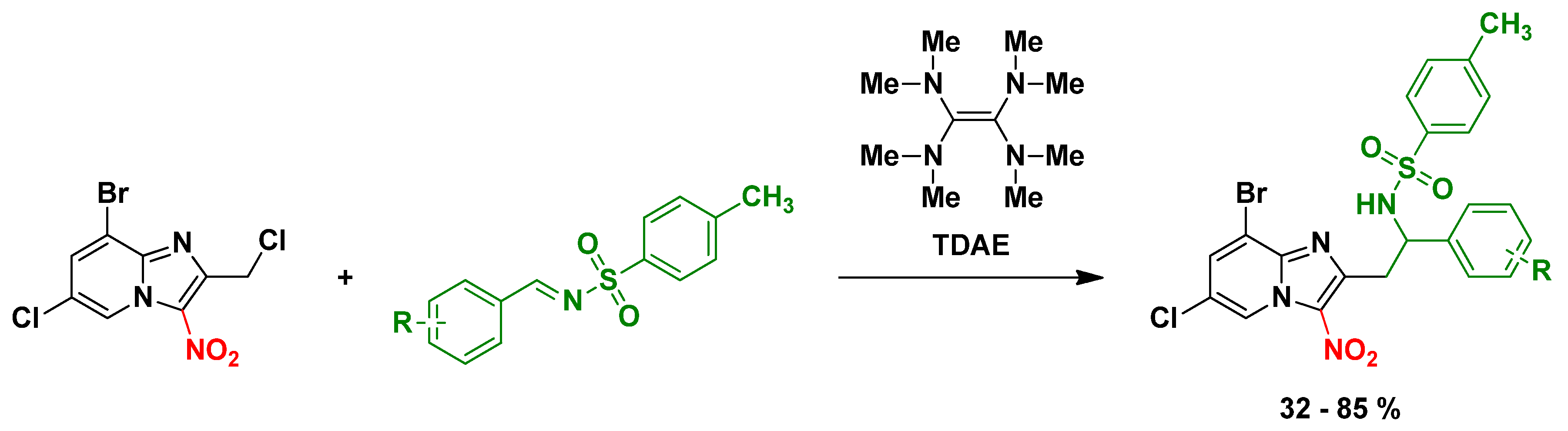
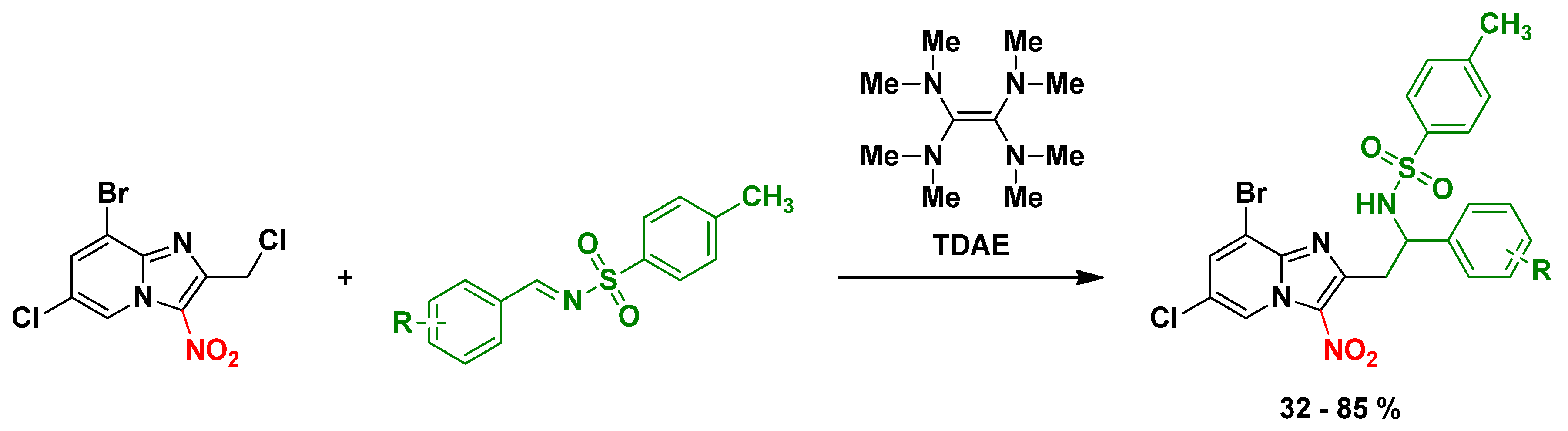
4.8. Development of an Original Synthetic Method Using TDAE to Modulate the Position 2 of Anti-Infective 3-Nitroimidazo[1,2-a]pyridine Derivatives
- Inès Jacquet 1, Romain Paoli-Lombardo 1, Hugo Pomares 1, Caroline Castera-Ducros 1, Pierre Verhaeghe 2, Pascal Rathelot 1, Nicolas Primas 1 and Patrice Vanelle 1
- Aix Marseille Univ, CNRS, ICR UMR 7273, PCR, Marseille, 13385; nicolas.primas@univ-amu.fr
- Université Grenoble Alpes, UMR CNRS 5063, COMET, Grenoble, 38400
4.9. Dual Mcl-1 and Bcl-xL PROTAC Degraders for the Treatment of Chemoresistant Ovarian Cancer
- Thomas Lemaitre, Charline Kieffer, Marc Since and Anne Sophie Voisin-Chiret
- CERMN, UNICAEN, 14000 Caen, France; thomas.lemaitre@unicaen.fr
4.10. Pharmacophore Validation of CTN1122, Imidazo[1,2-a]pyrazine Lead Compound Endowed with Antileishmanial Activity and Targeting Leishmania Casein Kinase 1
- Lhana Tisseur 1, Marc-Antoine Bazin 1, Sandrine Cojean 2, Fabrice Pagniez 1, Cédric Logé 1, Guillaume Bernadat 2, Christian Cavé 2, Isabelle Ourliac-Garnier 1, Carine Picot 1, Olivier Leclercq 3, Blandine Baratte 4, Najma Rachidi 3, Stéphane Bach 4, Philippe Loiseau 2, Patrice Le Pape 1 and Pascal Marchand 1
- Nantes Université, Cibles et médicaments des infections et de l’immunité, IICiMed, UR 1155, Nantes, France; lhana.tisseur@etu.univ-nantes.fr
- Université Paris-Saclay, Chimiothérapie Antiparasitaire, BIOmolécules: Conception, Isolement et Synthèse—BioCIS UMR 8076 CNRS, Châtenay-Malabry, France
- Institut Pasteur and INSERM U1201, Unité de Parasitologie Moléculaire et Signalisation, Paris, France
- Sorbonne Universités, UPMC Paris 06, CNRS USR3151 “Protein Phosphorylation and Human Disease” group, Station Biologique, Roscoff, France
4.11. Iodination of Aminopyridines to Access 5-Fluoro 4- or 7-Azaindoles
- Marine Ortillon, François Hallé and Thierry Lomberget
- Université de Lyon, Université Lyon 1, CNRS UMR 5246 Institut de Chimie et Biochimie Moléculaires et Supramoléculaires (ICBMS),Faculté de Pharmacie-ISPB, 8, Avenue Rockefeller, F-69373, Lyon, Cedex 08, France; marine.ortillon@univ-lyon1.fr
4.12. Development of a Fluorescent Ligand for the Intracellular Binding Site of NTSR1
- Patrick Shinkwin 1, Max Huber 1, Hannah Vogt 1, Matthias Schiedel 2, Dorothée Weikert 1 and Peter Gmeiner 1
- Department of Chemistry and Pharmacy, Friedrich-Alexander University Erlangen, 91058 Erlangen, Germany; patrick.shinkwin@fau.de
- Institute of Medicinal and Pharmaceutical Chemistry, Technical University Braunschweig, 38106 Braunschweig, Germany
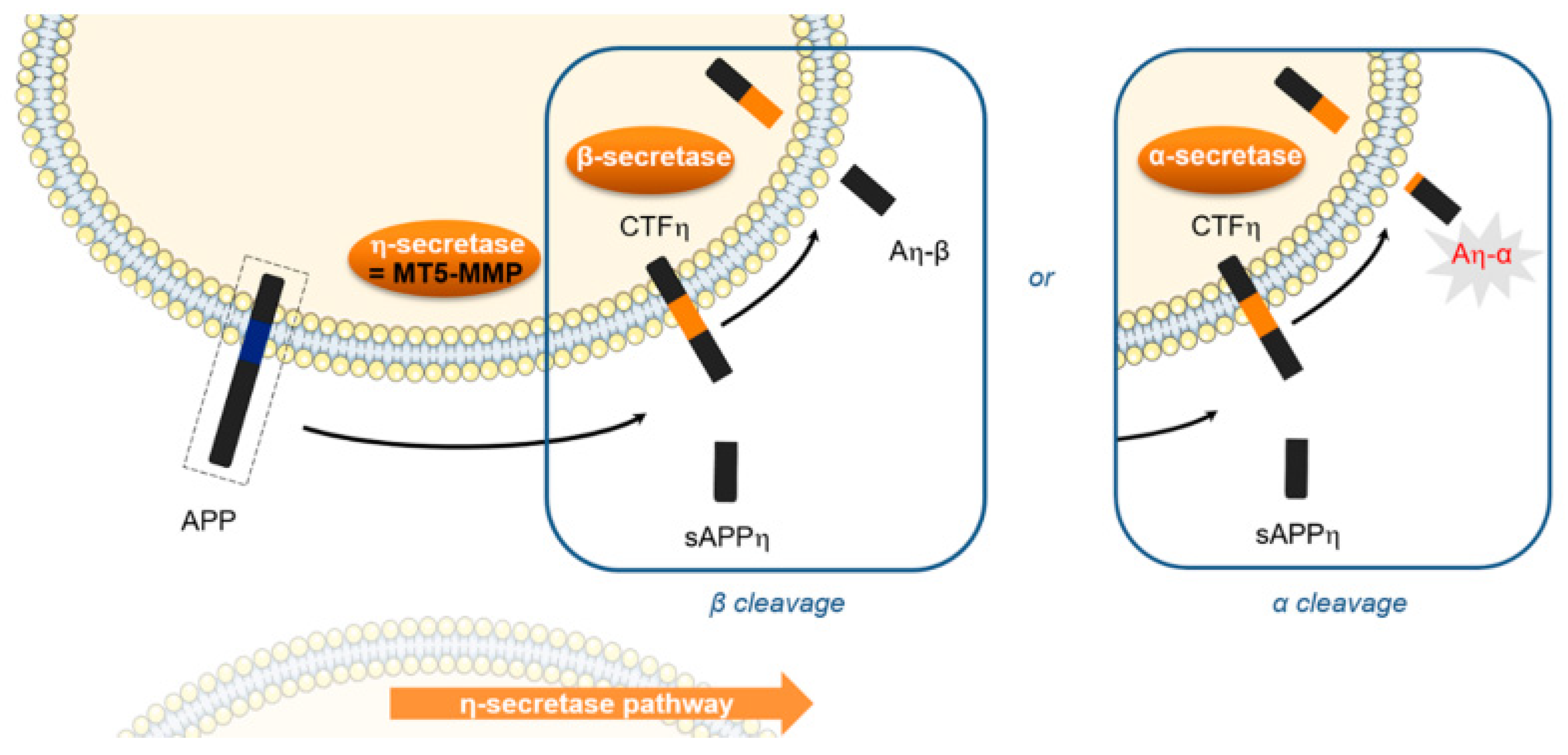
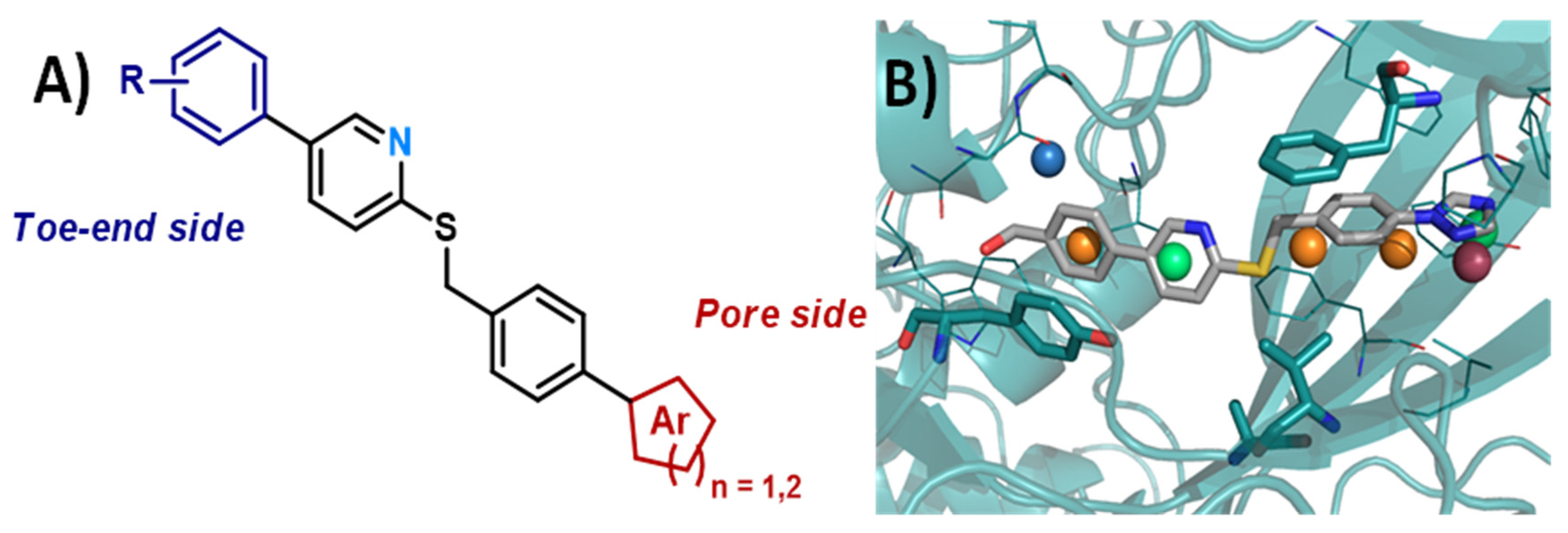
4.13. Synthesis and Biological Evaluation of MT5-MMP (Membrane-Type 5 Matrix MetalloProteinase) Inhibitors for a New Potential Treatment of Alzheimer’s Disease
- Chloé Rémondin, Audrey Davis, Alban Lepailleur, Christophe Rochais and Patrick Dallemagne
- Normandie Univ., Université de Caen Normandie, Centre d’Etudes et de Recherche sur le Médicament de Normandie, Caen, France; chloe.remondin@unicaen.fr
4.14. Synthesis and SAR Study of New 5- and 7-Substituted 3-Nitroimidazo[1,2-a]pyridine Derivatives as Potent Antileishmanial Molecules
- Romain Paoli-Lombardo 1, Nicolas Primas 1, Sébastien Hutter 2, Alix Sournia-Saquet 3, Caroline Castera-Ducros 1, Inès Jacquet 1, Pierre Verhaeghe 4, Nadine Azas 2, Pascal Rathelot 1 and Patrice Vanelle 1
- Aix Marseille Univ, CNRS, ICR UMR 7273, PCR, Marseille, 13385; nicolas.primas@univ-amu.fr
- Aix Marseille Univ, IHU Méditerranée Infection, UMR VITROME, Marseille, 13005
- Université Paul Sabatier, CNRS UPR 8241, LCC, Toulouse, 31077
- Université Grenoble Alpes, UMR CNRS 5063, COMET, Grenoble, 38400
4.15. Enterovirus Inhibitors: Development of New Broad-Spectrum Capsid Binders
- Hugo Roux 1, Antonio Coluccia 2, Franck Touret 3, Laurent Bourguignon 4, Omar Khoumeri 1, Florence Gattacceca 5, Sarah Giacometti 6, Raphaelle Fanciullino 7, Romano Silvestri 2, Antoine Nougairede 3, Patrice Vanelle 1 and Manon Roche 1
- Aix-Marseille Université, CNRS, ICR UMR 7273, PCR, Faculté de Pharmacie, 13005 Marseille, France; hugo.roux@centrale-marseille.fr
- Sapienza University, Department of Drug Chemistry and Technologies, Rome, Italy
- Aix-Marseille Univ., UVE-IRD_190-Inserm_1207 EFS-IRBA of Medical Research, Marseille, France
- UMR CNRS 5558, Laboratoire de Biométrie et Biologie Évolutive, Villeurbanne, France
- Aix-Marseille Univ., COMPO Centre Inria Sophia Antipolis CRCM Inserm U1068, Institut Paoli-Calmettes
- Aix-Marseille Université, Plate-forme SMARTc, CRCM--INSERM-1068, Marseille, France
4.16. Developing New Cytotoxic Molecules toward Chemoresistant Ovarian Cancer Thanks to the PROTAC Technology
- Florian Schwalen, Charline Kieffer and Anne-Sophie Voisin-Chiret
- Centre d’Etudes et de Recherche sur le Médicament de Normandie, Normandie University, Caen, France, 14000; florian.schwalen@unicaen.fr
4.17. Design and Synthesis of New Polyamine Quinoline Antibiotic Enhancers to Fight Resistant Gram-Negative P. aeruginosa Bacteria
- Thomas Troïa 1, Jacques Siad 1, Carole Di Giorgio 2 and Jean Michel Brunel 1
- Aix Marseille Univ, INSERM, SSA, MCT, 13385 Marseille, France; jean-michel.brunel@inserm.fr
- Aix Marseille Univ, CNRS, IRD, IMBE UMR 7263, Laboratoire de Mutagénèse Environnementale, 13385 Marseille, France
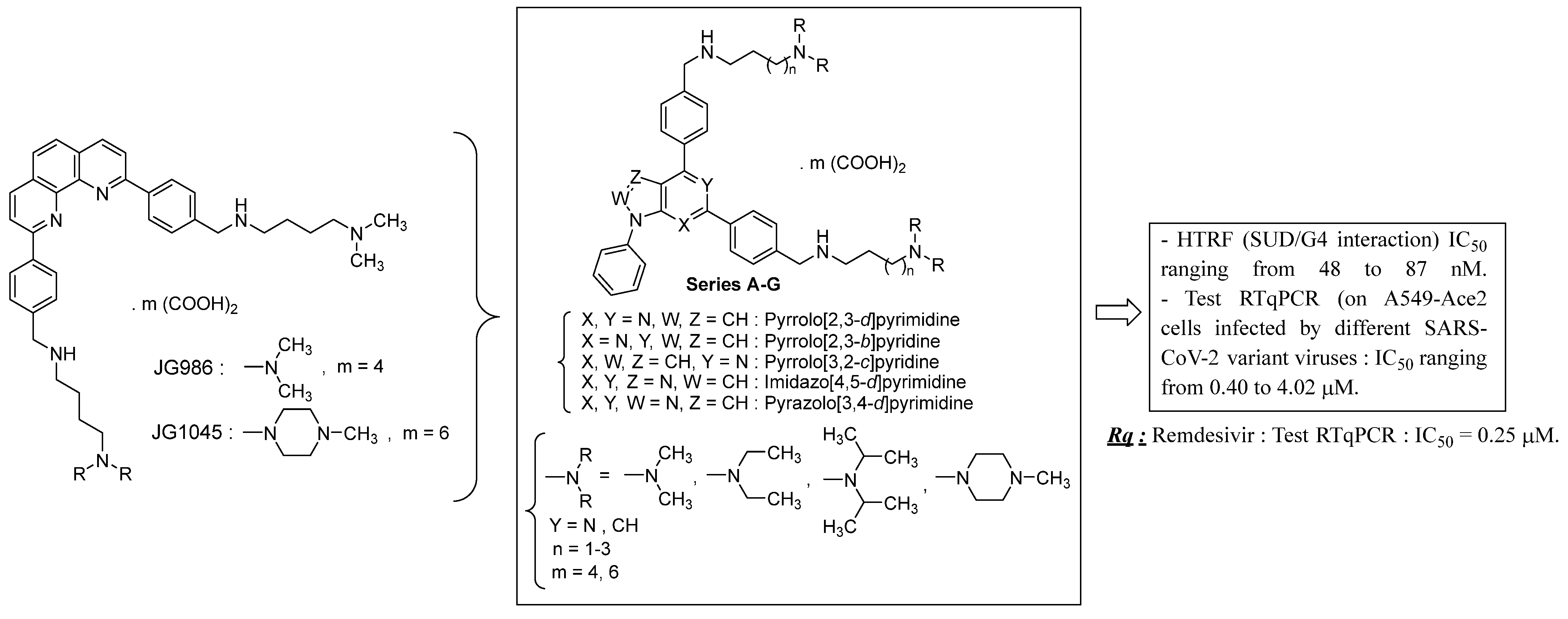
4.18. Biochemical and Functional Characterization of SARS-CoV-2 Unique Domain (SUD) in Nsp3—RNA G4 Complexes and Therapeutic Properties of G4-Ligands Inhibiting Their Formation
- Jean Guillon 1, Solène Savrimoutou 1, Amina Gaouar 1, Sandra Albenque-Rubio 1, Stéphane Moreau 1, Olivier Helynck 2, Jeanne Chiaravalli 2, Rofia Boudria-Souilah 3, Noël Pinaud 4, Luisa Ronga 5, Jean-Louis Mergny 6, Hélène Munier-Lehmann 2 and Marc Lavigne 3
- Univ. Bordeaux, laboratoire ARNA, INSERM U1212, CNRS UMR5320, UFR de Pharmacie, Bordeaux, France; jean.guillon@u-bordeaux.fr
- Institut Pasteur, Université de Paris, CNRS UMR3523, PF-CCB, Paris, France
- Institut Pasteur, Université de Paris, Dpt de Virologie, Paris, France
- Univ. Bordeaux, ISM—CNRS UMR5255, Talence Cedex, France
- Université de Pau et des Pays de l’Adour, E2S UPPA, CNRS, IPREM, Pau, France
- Laboratoire d’optique et Biosciences, Ecole Polytechnique, Inserm U1182, CNRS UMR7645, Institut Polytechnique de Paris, Palaiseau, France
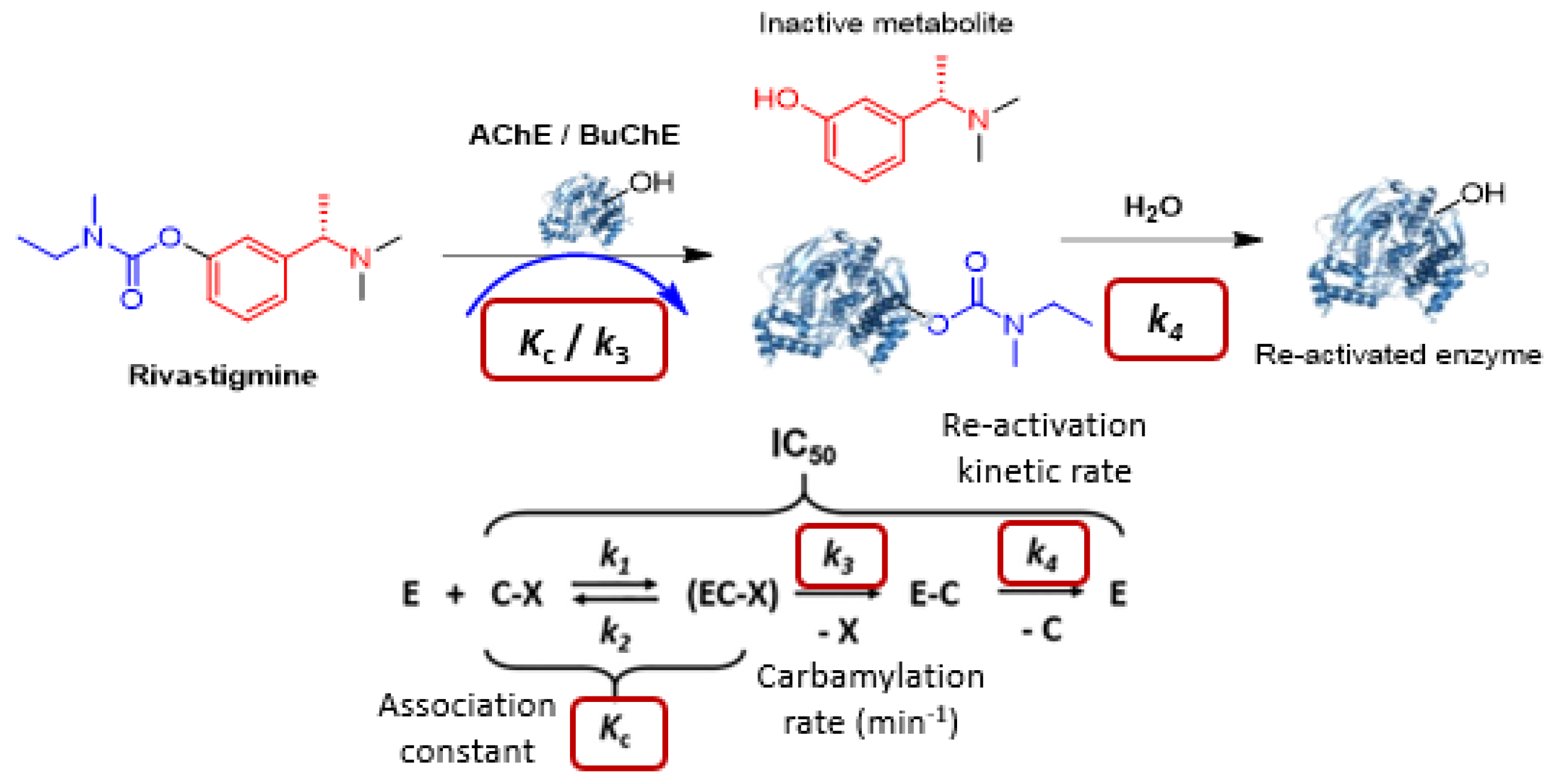
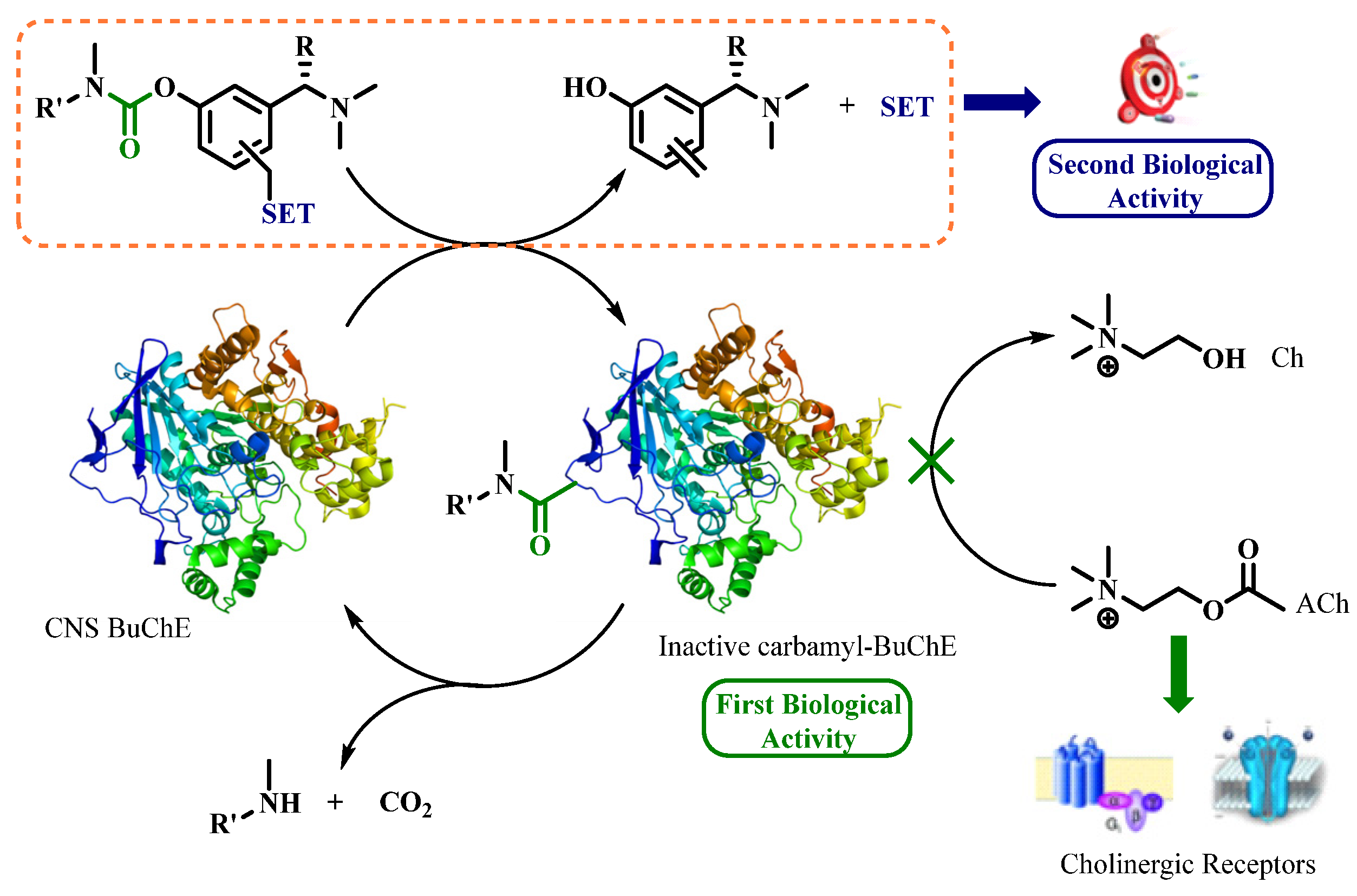
4.19. Cholinesterases Pseudo-Irreversible Covalent Inhibitors to Treat Alzheimer’s Disease: Mechanism of Enzyme Regeneration Process
- Alice Wang 1, Marc Since 1, Valentin Travers--Lesage 1, Audrey Davis 1, Florian Nachon 2, Xavier Brazzolotto 2, Patrick Dallemagne 1 and Christophe Rochais 1
- Normandie Univ, Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), 14000 Caen, France; alice.wang@unicaen.fr
- Institut de Recherche Biomédicale des Armées, Département des Plateformes et Recherches Technologiques, Unité Développements Analytiques et Bioanalyse, 91223 Brétigny sur Orge, France
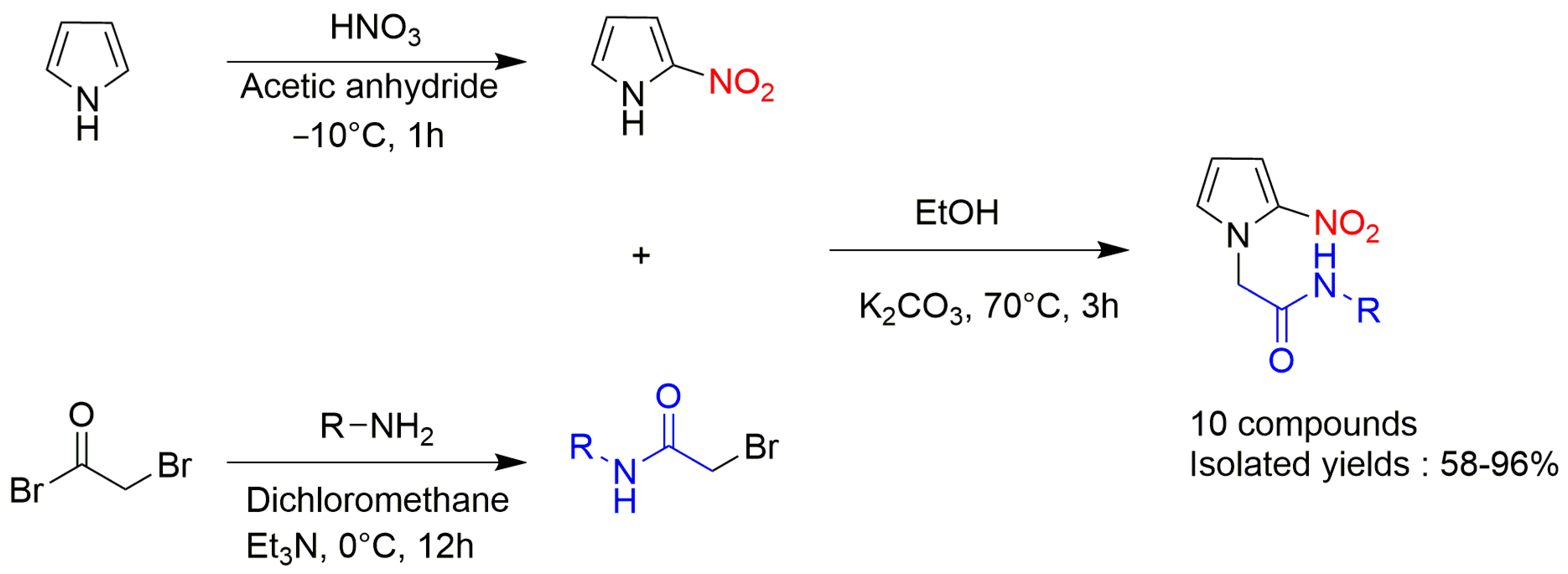
4.20. Synthesis and Anti-Trypanosoma Cruzi Biological Evaluation of Novel 2-Nitropyrrole Derivatives
- Fanny Mathias 1, Youssef Kabri 1, Damien Brun 1, Nicolas Primas 1, Carole Di Giorgio 2 and Patrice Vanelle 1
- Aix-Marseille Université, Institut de Chimie Radicalaire ICR, UMR CNRS 7273, Laboratoire de Pharmaco-Chimie Radicalaire, Faculté de Pharmacie, 27 Boulevard Jean Moulin—CS 30064, 13385 Marseille Cedex 05, France; fanny.mathias@univ-amu.fr
- Laboratoire de Mutagénèse Environnementale, CNRS, IRD, Aix Marseille University, IMBE UMR 7263, Avignon University, 13385 Marseille, France
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Espadinha, M.; Lopes, E.A.; Marques, V.; Amaral, J.D.; Dos Santos, D.J.V.A.; Mori, M.; Daniele, S.; Piccarducci, R.; Zappelli, E.; Martini, C.; et al. Discovery of MDM2-P53 and MDM4-P53 Protein-Protein Interactions Small Molecule Dual Inhibitors. Eur. J. Med. Chem. 2022, 241, 114637. [Google Scholar] [CrossRef]
- Barcherini, V.; Almeida, J.; Lopes, E.A.; Wang, M.; Magalhães E Silva, D.; Mori, M.; Wang, S.; Saraiva, L.; Santos, M.M.M. Potency and Selectivity Optimization of Tryptophanol-Derived Oxazoloisoindolinones: Novel P53 Activators in Human Colorectal Cancer. ChemMedChem 2021, 16, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Zscherp, R.; Coetzee, J.; Vornweg, J.; Grunenberg, J.; Herrmann, J.; Müller, R.; Klahn, P. Biomimetic Enterobactin Analogue Mediates Iron-Uptake and Cargo Transport into E. coli and P. aeruginosa. Chem. Sci. 2021, 12, 10179–10190. [Google Scholar] [CrossRef] [PubMed]
- Rohrbacher, C.; Zscherp, R.; Weck, S.C.; Klahn, P.; Ducho, C. Synthesis of an Antimicrobial Enterobactin-Muraymycin Conjugate for Improved Activity Against Gram-Negative Bacteria. Chemistry 2023, 29, e202202408. [Google Scholar] [CrossRef] [PubMed]
- Jimidar, C.C.; Wiese, L.; Stirz, M.; Beutling, U.; Schallmey, A.; Brönstrup, M.; Klahn, P. pseudo-Glucosinolates (psGSLs)—Exploiting the chemistry of glucosinolates (GSLs). In Proceedings of the the Irseer Naturstofftage, Kloster Irsee, Germany, 16–18 February 2022. [Google Scholar]
- Balaraman, K.; Vieira, N.C.; Moussa, F.; Vacus, J.; Cojean, S.; Pomel, S.; Bories, C.; Figadère, B.; Kesavan, V.; Loiseau, P.M. In Vitro and in Vivo Antileishmanial Properties of a 2-n-Propylquinoline Hydroxypropyl β-Cyclodextrin Formulation and Pharmacokinetics via Intravenous Route. Biomed. Pharmacother. 2015, 76, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Daligaux, P.; Lazar, N.; Ha-Duong, T.; Cavé, C.; van Tilbeurgh, H.; Loiseau, P.M.; Pomel, S. Biochemical Analysis of Leishmanial and Human GDP-Mannose Pyrophosphorylases and Selection of Inhibitors as New Leads. Sci. Rep. 2017, 7, 751. [Google Scholar] [CrossRef]
- Loiseau, P.M.; Balaraman, K.; Barratt, G.; Pomel, S.; Durand, R.; Frézard, F.; Figadère, B. The Potential of 2-Substituted Quinolines as Antileishmanial Drug Candidates. Molecules 2022, 27, 2313. [Google Scholar] [CrossRef]
- Pomel, S.; Cojean, S.; Pons, V.; Cintrat, J.-C.; Nguyen, L.; Vacus, J.; Pruvost, A.; Barbier, J.; Gillet, D.; Loiseau, P.M. An Adamantamine Derivative as a Drug Candidate for the Treatment of Visceral Leishmaniasis. J. Antimicrob. Chemother. 2021, 76, 2640–2650. [Google Scholar] [CrossRef]
- Bautista-Aguilera, Ó.M.; Hagenow, S.; Palomino-Antolin, A.; Farré-Alins, V.; Ismaili, L.; Joffrin, P.-L.; Jimeno, M.L.; Soukup, O.; Janočková, J.; Kalinowsky, L.; et al. Multitarget-Directed Ligands Combining Cholinesterase and Monoamine Oxidase Inhibition with Histamine H3 R Antagonism for Neurodegenerative Diseases. Angew. Chem. Int. Ed. Engl. 2017, 56, 12765–12769. [Google Scholar] [CrossRef]
- Bautista-Aguilera, Ó.M.; Budni, J.; Mina, F.; Medeiros, E.B.; Deuther-Conrad, W.; Entrena, J.M.; Moraleda, I.; Iriepa, I.; López-Muñoz, F.; Marco-Contelles, J. Contilisant, a Tetratarget Small Molecule for Alzheimer’s Disease Therapy Combining Cholinesterase, Monoamine Oxidase Inhibition, and H3R Antagonism with S1R Agonism Profile. J. Med. Chem. 2018, 61, 6937–6943. [Google Scholar] [CrossRef]
- López, M.F.; Marco, C.J.L.; Stark, H.; Hagenow, S.; Ramsay, R.R. New Compounds with Antioxidant Capacity That Combine the Inhibition of Monoaminoxidases and Cholinesterase Enzymes and the Interaction with the Histamine 3 Receptor, Its Obtaining Procedure and Pharmaceutical Compositions Containing Them, ES2701954 (A1). 2019. Available online: https://worldwide.espacenet.com/publicationDetails/biblio?FT=D&date=20190226&DB=&locale=fr_EP&CC=ES&NR=2701954A1&KC=A1&ND=4 (accessed on 12 January 2024).
- Lai, A.; Crews, C. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Guedeney, N.; Cornu, M.; Schwalen, F.; Kieffer, C.; Voisin-Chiret, A.S. PROTAC Technology: A New Drug Design for Chemical Biology with Many Challenges in Drug Discovery. Drug Discov. Today 2023, 28, 103395. [Google Scholar] [CrossRef]
- Jubete, G.; Puig de la Bellacasa, R.; Estrada-Tejedor, R.; Teixidó, J.; Borrell, J.I. Pyrido[2,3-d]Pyrimidin-7(8H)-Ones: Synthesis and Biomedical Applications. Molecules 2019, 24, 4161. [Google Scholar] [CrossRef] [PubMed]
- Puig de la Bellacasa, R.; Roué, G.; Balsas, P.; Pérez-Galán, P.; Teixidó, J.; Colomer, D.; Borrell, J.I. 4-Amino-2-Arylamino-6-(2,6-Dichlorophenyl)-Pyrido[2,3-d]Pyrimidin-7-(8H)-Ones as BCR Kinase Inhibitors for B Lymphoid Malignancies. Eur. J. Med. Chem. 2014, 86, 664–675. [Google Scholar] [CrossRef]
- Molina, V.M.Á.; García, R.S.; Borrell, B.J.I.; Teixido, C.J.; Estrada, T.R.; Puig, D.L.B.C.R. 4-Amino-6-(2,6-dichlorophenyl)-8-methyl-2-(phenylamino)-pyrido[2,3-d]pyrimidin-7(8h)-one for Treatment of Solid Cancers. EP3120851 (A1). 2017. Available online: https://worldwide.espacenet.com/publicationDetails/biblio?FT=D&date=20170125&DB=EPODOC&locale=fr_EP&CC=EP&NR=3120851A1&KC=A1&ND=4 (accessed on 12 January 2024).
- Camarasa, M.; Puig de la Bellacasa, R.; González, À.L.; Ondoño, R.; Estrada, R.; Franco, S.; Badia, R.; Esté, J.; Martínez, M.Á.; Teixidó, J.; et al. Design, Synthesis and Biological Evaluation of Pyrido[2,3-d]Pyrimidin-7-(8H)-Ones as HCV Inhibitors. Eur. J. Med. Chem. 2016, 115, 463–483. [Google Scholar] [CrossRef]
- Iannelli, G.; Milite, C.; Marechal, N.; Cura, V.; Bonnefond, L.; Troffer-Charlier, N.; Feoli, A.; Rescigno, D.; Wang, Y.; Cipriano, A.; et al. Turning Nonselective Inhibitors of Type I Protein Arginine Methyltransferases into Potent and Selective Inhibitors of Protein Arginine Methyltransferase 4 through a Deconstruction-Reconstruction and Fragment-Growing Approach. J. Med. Chem. 2022, 65, 11574–11606. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef]
- George, A. Antimicrobial Resistance, Trade, Food Safety and Security. One Health 2018, 5, 6–8. [Google Scholar] [CrossRef]
- Walesch, S.; Birkelbach, J.; Jézéquel, G.; Haeckl, F.P.J.; Hegemann, J.D.; Hesterkamp, T.; Hirsch, A.K.H.; Hammann, P.; Müller, R. Fighting Antibiotic Resistance-Strategies and (Pre)Clinical Developments to Find New Antibacterials. EMBO Rep. 2023, 24, e56033. [Google Scholar] [CrossRef]
- Frank, A.; Groll, M. The Methylerythritol Phosphate Pathway to Isoprenoids. Chem. Rev. 2017, 117, 5675–5703. [Google Scholar] [CrossRef]
- Eady, N.; Sheehan, R.; Rantell, K.; Sinai, A.; Bernal, J.; Bohnen, I.; Bonell, S.; Courtenay, K.; Dodd, K.; Gazizova, D.; et al. Impact of Cholinesterase Inhibitors or Memantine on Survival in Adults with Down Syndrome and Dementia: Clinical Cohort Study. Br. J. Psychiatry 2018, 212, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Frautschy, S.A.; Cole, G.M. Why Pleiotropic Interventions Are Needed for Alzheimer’s Disease. Mol. Neurobiol. 2010, 41, 392–409. [Google Scholar] [CrossRef]
- Kandiah, N.; Pai, M.-C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The Advantages of Dual Inhibition of Acetylcholinesterase and Butyrylcholinesterase and Its Role in Subcortical Vascular Dementia and Parkinson’s Disease Dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Travers-Lesage, V.; Mignani, S.M.; Dallemagne, P.; Rochais, C. Advances in Prodrug Design for Alzheimer’s Disease: The State of the Art. Expert Opin. Drug Discov. 2022, 17, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Toublet, F.-X.; Lalut, J.; Hatat, B.; Lecoutey, C.; Davis, A.; Since, M.; Corvaisier, S.; Freret, T.; Sopková-de Oliveira Santos, J.; Claeysen, S.; et al. Pleiotropic Prodrugs: Design of a Dual Butyrylcholinesterase Inhibitor and 5-HT6 Receptor Antagonist with Therapeutic Interest in Alzheimer’s Disease. Eur. J. Med. Chem. 2021, 210, 113059. [Google Scholar] [CrossRef] [PubMed]
- Foucourt, A.; Hédou, D.; Dubouilh-Benard, C.; Girard, A.; Taverne, T.; Casagrande, A.-S.; Désiré, L.; Leblond, B.; Besson, T. Design and Synthesis of Thiazolo[5,4-f]Quinazolines as DYRK1A Inhibitors, Part II. Molecules 2014, 19, 15411–15439. [Google Scholar] [CrossRef] [PubMed]
- Couly, F.; Harari, M.; Dubouilh-Benard, C.; Bailly, L.; Petit, E.; Diharce, J.; Bonnet, P.; Meijer, L.; Fruit, C.; Besson, T. Development of Kinase Inhibitors via Metal-Catalyzed C−H Arylation of 8-Alkyl-Thiazolo[5,4-f]-Quinazolin-9-Ones Designed by Fragment-Growing Studies. Molecules 2018, 23, 2181. [Google Scholar] [CrossRef]
- Chaikuad, A.; Diharce, J.; Schröder, M.; Foucourt, A.; Leblond, B.; Casagrande, A.-S.; Désiré, L.; Bonnet, P.; Knapp, S.; Besson, T. An Unusual Binding Model of the Methyl 9-Anilinothiazolo[5,4-f] Quinazoline-2-Carbimidates (EHT 1610 and EHT 5372) Confers High Selectivity for Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases. J. Med. Chem. 2016, 59, 10315–10321. [Google Scholar] [CrossRef]
- Broudic, N.; Pacheco-Benichou, A.; Fruit, C.; Besson, T. Synthesis of 2-Cyanobenzothiazoles via Pd-Catalyzed/Cu-Assisted C-H Functionalization/Intramolecular C-S Bond Formation from N-Arylcyanothioformamides. Molecules 2022, 27, 8426. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Carnero, A.; Paz-Ares, L. Inhibition of HSP90 Molecular Chaperones: Moving into the Clinic. Lancet Oncol. 2013, 14, e358–e369. [Google Scholar] [CrossRef]
- Zhao, H.; Garg, G.; Zhao, J.; Moroni, E.; Girgis, A.; Franco, L.S.; Singh, S.; Colombo, G.; Blagg, B.S.J. Design, Synthesis and Biological Evaluation of Biphenylamide Derivatives as Hsp90 C-Terminal Inhibitors. Eur. J. Med. Chem. 2015, 89, 442–466. [Google Scholar] [CrossRef] [PubMed]
- Kusuma, B.R.; Khandelwal, A.; Gu, W.; Brown, D.; Liu, W.; Vielhauer, G.; Holzbeierlein, J.; Blagg, B.S.J. Synthesis and Biological Evaluation of Coumarin Replacements of Novobiocin as Hsp90 Inhibitors. Bioorg Med. Chem. 2014, 22, 1441–1449. [Google Scholar] [CrossRef]
- Zhao, H.; Moroni, E.; Colombo, G.; Blagg, B.S.J. Identification of a New Scaffold for Hsp90 C-Terminal Inhibition. ACS Med. Chem. Lett. 2014, 5, 84–88. [Google Scholar] [CrossRef]
- Montoir, D.; Barillé-Nion, S.; Tonnerre, A.; Juin, P.; Duflos, M.; Bazin, M.-A. Novel 1,6-Naphthyridin-2(1H)-Ones as Potential Anticancer Agents Targeting Hsp90. Eur. J. Med. Chem. 2016, 119, 17–33. [Google Scholar] [CrossRef]
- Montana, M.; Montero, V.; Khoumeri, O.; Vanelle, P. Quinoxaline Derivatives as Antiviral Agents: A Systematic Review. Molecules 2020, 25, 2784. [Google Scholar] [CrossRef] [PubMed]
- Montana, M.; Montero, V.; Khoumeri, O.; Vanelle, P. Quinoxaline Moiety: A Potential Scaffold against Mycobacterium Tuberculosis. Molecules 2021, 26, 4742. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.A.; Pessoa, A.M.; Cordeiro, M.N.D.S.; Fernandes, R.; Prudêncio, C.; Noronha, J.P.; Vieira, M. Quinoxaline, Its Derivatives and Applications: A State of the Art Review. Eur. J. Med. Chem. 2015, 97, 664–672. [Google Scholar] [CrossRef]
- Montana, M.; Mathias, F.; Terme, T.; Vanelle, P. Antitumoral Activity of Quinoxaline Derivatives: A Systematic Review. Eur. J. Med. Chem. 2019, 163, 136–147. [Google Scholar] [CrossRef]
- Montana, M.; Correard, F.; Khoumeri, O.; Esteve, M.-A.; Terme, T.; Vanelle, P. Synthesis of New Quinoxalines Containing an Oxirane Ring by the TDAE Strategy and in Vitro Evaluation in Neuroblastoma Cell Lines. Molecules 2014, 19, 14987–14998. [Google Scholar] [CrossRef]
- Montero, V.; Montana, M.; Khoumeri, O.; Correard, F.; Estève, M.-A.; Vanelle, P. Synthesis, In Vitro Antiproliferative Activity, and In Silico Evaluation of Novel Oxiranyl-Quinoxaline Derivatives. Pharmaceuticals 2022, 15, 781. [Google Scholar] [CrossRef] [PubMed]
- Brotin, E.; Meryet-Figuière, M.; Simonin, K.; Duval, R.E.; Villedieu, M.; Leroy-Dudal, J.; Saison-Behmoaras, E.; Gauduchon, P.; Denoyelle, C.; Poulain, L. Bcl-XL and MCL-1 Constitute Pertinent Targets in Ovarian Carcinoma and Their Concomitant Inhibition Is Sufficient to Induce Apoptosis. Int. J. Cancer 2010, 126, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Simonin, K.; N’Diaye, M.; Lheureux, S.; Loussouarn, C.; Dutoit, S.; Briand, M.; Giffard, F.; Brotin, E.; Blanc-Fournier, C.; Poulain, L. Platinum Compounds Sensitize Ovarian Carcinoma Cells to ABT-737 by Modulation of the Mcl-1/Noxa Axis. Apoptosis 2013, 18, 492–508. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Gloaguen, C.; Voisin-Chiret, A.S.; Sopkova-de Oliveira Santos, J.; Fogha, J.; Gautier, F.; De Giorgi, M.; Burzicki, G.; Perato, S.; Pétigny-Lechartier, C.; Simonin-Le Jeune, K.; et al. First Evidence That Oligopyridines, α-Helix Foldamers, Inhibit Mcl-1 and Sensitize Ovarian Carcinoma Cells to Bcl-xL-Targeting Strategies. J. Med. Chem. 2015, 58, 1644–1668. [Google Scholar] [CrossRef] [PubMed]
- Voisin-Chiret, A.-S.; Kieffer, C.; Guedeney, N.; Denis, C.; De, P.M.; Poulain, L.; Weiswald, L.; Paysant, H. Bcl-2 Family Proteins Modulating Compounds for Cancer Treatment. WO2021180966 (A1). 2021. Available online: https://worldwide.espacenet.com/publicationDetails/biblio?FT=D&date=20210916&DB=EPODOC&locale=fr_EP&CC=WO&NR=2021180966A1&KC=A1&ND=4 (accessed on 12 January 2024).
- Mahato, R.; Tai, W.; Cheng, K. Prodrugs for Improving Tumor Targetability and Efficiency. Adv. Drug Deliv. Rev. 2011, 63, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Saxon, E.; Bertozzi, C.R. Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef]
- Saxon, E.; Luchansky, S.J.; Hang, H.C.; Yu, C.; Lee, S.C.; Bertozzi, C.R. Investigating Cellular Metabolism of Synthetic Azidosugars with the Staudinger Ligation. J. Am. Chem. Soc. 2002, 124, 14893–14902. [Google Scholar] [CrossRef]
- Tuon, F.F.; Dantas, L.R.; de Souza, R.M.; Ribeiro, V.S.T.; Amato, V.S. Liposomal Drug Delivery Systems for the Treatment of Leishmaniasis. Parasitol. Res. 2022, 121, 3073–3082. [Google Scholar] [CrossRef]
- Mathison, B.A.; Bradley, B.T. Review of the Clinical Presentation, Pathology, Diagnosis, and Treatment of Leishmaniasis. Lab. Med. 2023, 54, 363–371. [Google Scholar] [CrossRef]
- Bouhlel, A.; Curti, C.; Dumètre, A.; Laget, M.; Crozet, M.D.; Azas, N.; Vanelle, P. Synthesis and Evaluation of Original Amidoximes as Antileishmanial Agents. Bioorg. Med. Chem. 2010, 18, 7310–7320. [Google Scholar] [CrossRef]
- Islam, M.B.; Islam, M.I.; Nath, N.; Emran, T.B.; Rahman, M.R.; Sharma, R.; Matin, M.M. Recent Advances in Pyridine Scaffold: Focus on Chemistry, Synthesis, and Antibacterial Activities. Biomed. Res. Int. 2023, 2023, 9967591. [Google Scholar] [CrossRef] [PubMed]
- Daemi, H.; Mashayekhi, M.; Pezeshki Modaress, M. Facile Fabrication of Sulfated Alginate Electrospun Nanofibers. Carbohydr. Polym. 2018, 198, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, G.; Bonde, G.V.; Mittal, P.; Khan, G.; Pandey, V.K.; Bakade, B.V.; Mishra, B. Biomimetic PCL-Gelatin Based Nanofibers Loaded with Ciprofloxacin Hydrochloride and Quercetin: A Potential Antibacterial and Anti-Oxidant Dressing Material for Accelerated Healing of a Full Thickness Wound. Int. J. Pharm. 2019, 567, 118480. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, O.M.; Iacob, A.-T.; Mignon, A.; Van Vlierberghe, S.; Baican, M.; Danu, M.; Ibănescu, C.; Simionescu, N.; Profire, L. Design, Preparation and in Vitro Characterization of Biomimetic and Bioactive Chitosan/Polyethylene Oxide Based Nanofibers as Wound Dressings. Int. J. Biol. Macromol. 2021, 193, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.; Stewart, N.J.; Chan, H.-F.; Rao, M.; Norquay, G.; Wild, J.M. In Vivo Methods and Applications of Xenon-129 Magnetic Resonance. Prog. Nucl. Magn. Reson. Spectrosc. 2021, 122, 42–62. [Google Scholar] [CrossRef] [PubMed]
- Shepelytskyi, Y.; Grynko, V.; Rao, M.R.; Li, T.; Agostino, M.; Wild, J.M.; Albert, M.S. Hyperpolarized 129 Xe Imaging of the Brain: Achievements and Future Challenges. Magn. Reson. Med. 2022, 88, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Huber, G.; Beguin, L.; Desvaux, H.; Brotin, T.; Fogarty, H.A.; Dutasta, J.-P.; Berthault, P. Cryptophane-Xenon Complexes in Organic Solvents Observed through NMR Spectroscopy. J. Phys. Chem. A 2008, 112, 11363–11372. [Google Scholar] [CrossRef]
- Vigier, C.; Fossé, P.; Fabis, F.; Cailly, T.; Dubost, E. Controlled Access to C1-Symmetrical Cyclotriveratrylenes (CTVs) by Using a Sequential Barluenga Boronic Coupling (BBC) Approach. Adv. Synth. Catal. 2021, 363, 3756–3761. [Google Scholar] [CrossRef]
- Patriciu, O.-I.; Fînaru, A.-L.; Massip, S.; Leger, J.-M.; Jarry, C.; Guillaumet, G. Smiles Rearrangement as a Tool for the Preparation of Dihydrodipyridopyrazines. Org. Lett. 2009, 11, 5502–5505. [Google Scholar] [CrossRef]
- Vigier, C.; Fayolle, D.; El Siblani, H.; Sopkova-de Oliveira Santos, J.; Fabis, F.; Cailly, T.; Dubost, E. Synthesis and Physicochemical Properties of Cryptophazane-A Soluble and Functionalizable C1 -Symmetrical Cryptophane. Angew. Chem. Int. Ed. Engl. 2022, 61, e202208580. [Google Scholar] [CrossRef]
- Hospenthal, D.R. Agents of Mycetoma. In Mandell, Douglas and Bennett’s Principles and Practice of Infectious Diseases; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 2929–2933. ISBN 9780323482554. [Google Scholar]
- Todd, M.H. Six Laws of Open Source Drug Discovery. ChemMedChem 2019, 14, 1804–1809. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Melse, Y.; Konings, M.; Phat Duong, H.; Eadie, K.; Laleu, B.; Perry, B.; Todd, M.H.; Ioset, J.-R.; van de Sande, W.W.J. Addressing the Most Neglected Diseases through an Open Research Model: The Discovery of Fenarimols as Novel Drug Candidates for Eumycetoma. PLoS Negl. Trop. Dis. 2018, 12, e0006437. [Google Scholar] [CrossRef] [PubMed]
- Bagcchi, S. WHO’s Global Tuberculosis Report 2022. Lancet Microbe 2023, 4, e20. [Google Scholar] [CrossRef] [PubMed]
- WHO Consolidated Guidelines on Tuberculosis: Module 4: Treatment-Drug-Susceptible Tuberculosis Treatment; WHO Guidelines Approved by the Guidelines Review Committee; World Health Organization: Geneva, Switzerland, 2022.
- Fernandes, G.F.S.; Thompson, A.M.; Castagnolo, D.; Denny, W.A.; Dos Santos, J.L. Tuberculosis Drug Discovery: Challenges and New Horizons. J. Med. Chem. 2022, 65, 7489–7531. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Stevens, C.M.; Zhang, L.; Zgurskaya, H.I.; Niederweis, M. Transporters Involved in the Biogenesis and Functionalization of the Mycobacterial Cell Envelope. Chem. Rev. 2021, 121, 5124–5157. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Besra, G.S. Mycobacterial Cell Wall Biosynthesis: A Multifaceted Antibiotic Target. Parasitology 2018, 145, 116–133. [Google Scholar] [CrossRef] [PubMed]
- Toubal, A.; Nel, I.; Lotersztajn, S.; Lehuen, A. Mucosal-Associated Invariant T Cells and Disease. Nat. Rev. Immunol. 2019, 19, 643–657. [Google Scholar] [CrossRef]
- Yan, J.; Allen, S.; McDonald, E.; Das, I.; Mak, J.Y.W.; Liu, L.; Fairlie, D.P.; Meehan, B.S.; Chen, Z.; Corbett, A.J.; et al. MAIT Cells Promote Tumor Initiation, Growth, and Metastases via Tumor MR1. Cancer Discov. 2020, 10, 124–141. [Google Scholar] [CrossRef]
- Gütschow, M.; Pietsch, M.; Themann, A.; Fahrig, J.; Schulze, B. 2,4,5-Triphenylisothiazol-3 (2H)-One 1,1-Dioxides as Inhibitors of Human Leukocyte Elastase. J. Enzyme Inhib. Med. Chem. 2005, 20, 341–347. [Google Scholar] [CrossRef]
- Ngo, H.X.; Shrestha, S.K.; Green, K.D.; Garneau-Tsodikova, S. Development of Ebsulfur Analogues as Potent Antibacterials against Methicillin-Resistant Staphylococcus Aureus. Bioorg. Med. Chem. 2016, 24, 6298–6306. [Google Scholar] [CrossRef]
- Cornelio, B.; Laronze-Cochard, M.; Miambo, R.; De Grandis, M.; Riccioni, R.; Borisova, B.; Dontchev, D.; Machado, C.; Ceruso, M.; Fontana, A.; et al. 5-Arylisothiazol-3(2H)-One-1,(1)-(Di)Oxides: A New Class of Selective Tumor-Associated Carbonic Anhydrases (hCA IX and XII) Inhibitors. Eur. J. Med. Chem. 2019, 175, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic Targeting of 3′,5′-Cyclic Nucleotide Phosphodiesterases: Inhibition and Beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef] [PubMed]
- Barberot, C.; Moniot, A.; Allart-Simon, I.; Malleret, L.; Yegorova, T.; Laronze-Cochard, M.; Bentaher, A.; Médebielle, M.; Bouillon, J.-P.; Hénon, E.; et al. Synthesis and Biological Evaluation of Pyridazinone Derivatives as Potential Anti-Inflammatory Agents. Eur. J. Med. Chem. 2018, 146, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Tuberculosis. Available online: https://www.who.int/health-topics/tuberculosis (accessed on 12 January 2024).
- Extensively Drug-Resistant Tuberculosis (XDR TB) Fact Sheet, CDC. Available online: https://www.cdc.gov/tb/publications/factsheets/drtb/xdrtb.htm (accessed on 12 January 2024).
- Gillespie, S.H. The Role of Moxifloxacin in Tuberculosis Therapy. Eur. Respir. Rev. 2016, 25, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, S.; Cimino, M.; Ouattara, M.; Lecoutey, C.; Buchieri, M.V.; Alonso-Rodriguez, N.; Briffotaux, J.; Mornico, D.; Gicquel, B.; Rochais, C.; et al. Phenanthrolinic Analogs of Quinolones Show Antibacterial Activity against M. Tuberculosis. Eur. J. Med. Chem. 2020, 207, 112821. [Google Scholar] [CrossRef]
- Burkholder, C.; Dolbier, W.R.; Médebielle, M.; Ndedi, A. Tetrakis(Dimethylamino)Ethylene (TDAE) as a Useful Reductant of Some Chlorodifluoromethylated Ketones. A New Approach for the Synthesis of α,α-Difluoroketone Derivatives. Tet. Lett. 1998, 39, 8853–8856. [Google Scholar] [CrossRef]
- Giuglio-Tonolo, G.; Terme, T.; Médebielle, M.; Vanelle, P. Original Reaction of P-Nitrobenzyl Chloride with Aldehydes Using Tetrakis(Dimethylamino)Ethylene (TDAE). Tet. Lett. 2003, 44, 6433–6435. [Google Scholar] [CrossRef]
- Marhadour, S.; Marchand, P.; Pagniez, F.; Bazin, M.-A.; Picot, C.; Lozach, O.; Ruchaud, S.; Antoine, M.; Meijer, L.; Rachidi, N.; et al. Synthesis and Biological Evaluation of 2,3-Diarylimidazo[1,2-a]Pyridines as Antileishmanial Agents. Eur. J. Med. Chem. 2012, 58, 543–556. [Google Scholar] [CrossRef]
- Fersing, C.; Boudot, C.; Pedron, J.; Hutter, S.; Primas, N.; Castera-Ducros, C.; Bourgeade-Delmas, S.; Sournia-Saquet, A.; Moreau, A.; Cohen, A.; et al. 8-Aryl-6-Chloro-3-Nitro-2-(Phenylsulfonylmethyl)Imidazo[1,2-a]Pyridines as Potent Antitrypanosomatid Molecules Bioactivated by Type 1 Nitroreductases. Eur. J. Med. Chem. 2018, 157, 115–126. [Google Scholar] [CrossRef]
- Paoli-Lombardo, R.; Primas, N.; Bourgeade-Delmas, S.; Hutter, S.; Sournia-Saquet, A.; Boudot, C.; Brenot, E.; Castera-Ducros, C.; Corvaisier, S.; Since, M.; et al. Improving Aqueous Solubility and In Vitro Pharmacokinetic Properties of the 3-Nitroimidazo[1,2-a]Pyridine Antileishmanial Pharmacophore. Pharmaceuticals 2022, 15, 998. [Google Scholar] [CrossRef] [PubMed]
- Bolomsky, A.; Vogler, M.; Köse, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 Inhibitors, Fast-Lane Development of a New Class of Anti-Cancer Agents. J. Hematol. Oncol. 2020, 13, 173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nimmer, P.M.; Tahir, S.K.; Chen, J.; Fryer, R.M.; Hahn, K.R.; Iciek, L.A.; Morgan, S.J.; Nasarre, M.C.; Nelson, R.; et al. Bcl-2 Family Proteins Are Essential for Platelet Survival. Cell Death Differ. 2007, 14, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Marchand, P.; Bazin, M.-A.; Pagniez, F.; Rivière, G.; Bodero, L.; Marhadour, S.; Nourrisson, M.-R.; Picot, C.; Ruchaud, S.; Bach, S.; et al. Synthesis, Antileishmanial Activity and Cytotoxicity of 2,3-Diaryl- and 2,3,8-Trisubstituted Imidazo[1,2-a]Pyrazines. Eur. J. Med. Chem. 2015, 103, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Durieu, E.; Prina, E.; Leclercq, O.; Oumata, N.; Gaboriaud-Kolar, N.; Vougogiannopoulou, K.; Aulner, N.; Defontaine, A.; No, J.H.; Ruchaud, S.; et al. From Drug Screening to Target Deconvolution: A Target-Based Drug Discovery Pipeline Using Leishmania Casein Kinase 1 Isoform 2 To Identify Compounds with Antileishmanial Activity. Antimicrob. Agents Chemother. 2016, 60, 2822–2833. [Google Scholar] [CrossRef]
- Bazin, M.-A.; Cojean, S.; Pagniez, F.; Bernadat, G.; Cavé, C.; Ourliac-Garnier, I.; Nourrisson, M.-R.; Morgado, C.; Picot, C.; Leclercq, O.; et al. In Vitro Identification of Imidazo[1,2-a]Pyrazine-Based Antileishmanial Agents and Evaluation of L. Major Casein Kinase 1 Inhibition. Eur. J. Med. Chem. 2021, 210, 112956. [Google Scholar] [CrossRef]
- Motati, D.R.; Amaradhi, R.; Ganesh, T. Azaindole Therapeutic Agents. Bioorg Med. Chem. 2020, 28, 115830. [Google Scholar] [CrossRef]
- Motati, D.R.; Amaradhi, R.; Ganesh, T. Recent Developments in the Synthesis of Azaindoles from Pyridine and Pyrrole Building Blocks. Org. Chem. Front. 2021, 8, 466–513. [Google Scholar] [CrossRef]
- Radix, S.; Hallé, F.; Mahiout, Z.; Teissonnière, A.; Bouchez, G.; Auberger, L.; Barret, R.; Lomberget, T. A Journey through Hemetsberger–Knittel, Leimgruber–Batcho and Bartoli Reactions: Access to Several Hydroxy 5- and 6-Azaindoles. Helv. Chim. Acta 2022, 105, e202100211. [Google Scholar] [CrossRef]
- Alekseyev, R.; Amirova, S.; Terenin, V. Synthesis of 5-Chloro-7-Azaindoles by Fischer Reaction. Chem. Heterocycl. Compd. 2017, 53, 196–206. [Google Scholar] [CrossRef]
- Baranger, K.; Marchalant, Y.; Bonnet, A.E.; Crouzin, N.; Carrete, A.; Paumier, J.-M.; Py, N.A.; Bernard, A.; Bauer, C.; Charrat, E.; et al. MT5-MMP Is a New pro-Amyloidogenic Proteinase That Promotes Amyloid Pathology and Cognitive Decline in a Transgenic Mouse Model of Alzheimer’s Disease. Cell Mol. Life Sci. 2016, 73, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.; Rochais, C.; Baranger, K.; Rivera, S.; Dallemagne, P. Matrix Metalloproteinases as New Targets in Alzheimer’s Disease: Opportunities and Challenges. J. Med. Chem. 2020, 63, 10705–10725. [Google Scholar] [CrossRef] [PubMed]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef] [PubMed]
- Fersing, C.; Basmaciyan, L.; Boudot, C.; Pedron, J.; Hutter, S.; Cohen, A.; Castera-Ducros, C.; Primas, N.; Laget, M.; Casanova, M.; et al. Nongenotoxic 3-Nitroimidazo[1,2-a]Pyridines Are NTR1 Substrates That Display Potent in Vitro Antileishmanial Activity. ACS Med. Chem. Lett. 2019, 10, 34–39. [Google Scholar] [CrossRef]
- Brown, D.M.; Zhang, Y.; Scheuermann, R.H. Epidemiology and Sequence-Based Evolutionary Analysis of Circulating Non-Polio Enteroviruses. Microorganisms 2020, 8, 1856. [Google Scholar] [CrossRef]
- Wang, Y.; Zou, G.; Xia, A.; Wang, X.; Cai, J.; Gao, Q.; Yuan, S.; He, G.; Zhang, S.; Zeng, M.; et al. Enterovirus 71 Infection in Children with Hand, Foot, and Mouth Disease in Shanghai, China: Epidemiology, Clinical Feature and Diagnosis. Virol. J. 2015, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Midgley, C.M.; Watson, J.T.; Nix, W.A.; Curns, A.T.; Rogers, S.L.; Brown, B.A.; Conover, C.; Dominguez, S.R.; Feikin, D.R.; Gray, S.; et al. Severe Respiratory Illness Associated with a Nationwide Outbreak of Enterovirus D68 in the USA (2014): A Descriptive Epidemiological Investigation. Lancet Respir. Med. 2015, 3, 879–887. [Google Scholar] [CrossRef]
- Muehlenbachs, A.; Bhatnagar, J.; Zaki, S.R. Tissue Tropism, Pathology and Pathogenesis of Enterovirus Infection. J. Pathol. 2015, 235, 217–228. [Google Scholar] [CrossRef]
- Lacroix, C.; Querol-Audí, J.; Roche, M.; Franco, D.; Froeyen, M.; Guerra, P.; Terme, T.; Vanelle, P.; Verdaguer, N.; Neyts, J.; et al. A Novel Benzonitrile Analogue Inhibits Rhinovirus Replication. J. Antimicrob. Chemother. 2014, 69, 2723–2732. [Google Scholar] [CrossRef]
- Da Costa, L.; Scheers, E.; Coluccia, A.; Casulli, A.; Roche, M.; Di Giorgio, C.; Neyts, J.; Terme, T.; Cirilli, R.; La Regina, G.; et al. Structure-Based Drug Design of Potent Pyrazole Derivatives against Rhinovirus Replication. J. Med. Chem. 2018, 61, 8402–8416. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C.S. Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Walensky, L.D. BCL-2 in the Crosshairs: Tipping the Balance of Life and Death. Cell Death Differ. 2006, 13, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Debrincat, M.A.; Pleines, I.; Lebois, M.; Lane, R.M.; Holmes, M.L.; Corbin, J.; Vandenberg, C.J.; Alexander, W.S.; Ng, A.P.; Strasser, A.; et al. BCL-2 Is Dispensable for Thrombopoiesis and Platelet Survival. Cell Death Dis. 2015, 6, e1721. [Google Scholar] [CrossRef]
- Rasmussen, M.L.; Taneja, N.; Neininger, A.C.; Wang, L.; Robertson, G.L.; Riffle, S.N.; Shi, L.; Knollmann, B.C.; Burnette, D.T.; Gama, V. MCL-1 Inhibition by Selective BH3 Mimetics Disrupts Mitochondrial Dynamics Causing Loss of Viability and Functionality of Human Cardiomyocytes. iScience 2020, 23, 101015. [Google Scholar] [CrossRef] [PubMed]
- Troïa, T.; Siad, J.; Di Giorgio, C.; Brunel, J.M. Design and Synthesis of New Polyamine Quinoline Antibiotic Enhancers to Fight Resistant Gram-Negative P. Aeruginosa Bacteria. Eur. J. Med. Chem. Rep. 2022, 5, 100054. [Google Scholar] [CrossRef]
- Lavigne, M.; Helynck, O.; Rigolet, P.; Boudria-Souilah, R.; Nowakowski, M.; Baron, B.; Brülé, S.; Hoos, S.; Raynal, B.; Guittat, L.; et al. SARS-CoV-2 Nsp3 Unique Domain SUD Interacts with Guanine Quadruplexes and G4-Ligands Inhibit This Interaction. Nucleic Acids Res. 2021, 49, 7695–7712. [Google Scholar] [CrossRef]
- Chiaravalli, J.; Guillon, J.; Lavigne, M.; Munier-Lehmann, H.; Mergny, J.-L.; Meyer, B.; Savrimoutou, S. 6-6 or 5-6 Fused Bicyclic Compounds Comprising a Pyri(Mi)Dine Ring Useful in The|treatment of Infectious Diseases. WO2022129376 (A1). 2022. Available online: https://worldwide.espacenet.com/publicationDetails/biblio?FT=D&date=20220623&DB=EPODOC&locale=fr_EP&CC=WO&NR=2022129376A1&KC=A1&ND=4 (accessed on 12 January 2024).
- Toublet, F.-X.; Lecoutey, C.; Lalut, J.; Hatat, B.; Davis, A.; Since, M.; Corvaisier, S.; Freret, T.; Sopkova de Oliveira Santos, J.; Claeysen, S.; et al. Inhibiting Acetylcholinesterase to Activate Pleiotropic Prodrugs with Therapeutic Interest in Alzheimer’s Disease. Molecules 2019, 24, 2786. [Google Scholar] [CrossRef]
- Scheiner, M.; Hoffmann, M.; He, F.; Poeta, E.; Chatonnet, A.; Monti, B.; Maurice, T.; Decker, M. Selective Pseudo-Irreversible Butyrylcholinesterase Inhibitors Transferring Antioxidant Moieties to the Enzyme Show Pronounced Neuroprotective Efficacy In Vitro and In Vivo in an Alzheimer’s Disease Mouse Model. J. Med. Chem. 2021, 64, 9302–9320. [Google Scholar] [CrossRef]
- Košak, U.; Strašek, N.; Knez, D.; Jukič, M.; Žakelj, S.; Zahirović, A.; Pišlar, A.; Brazzolotto, X.; Nachon, F.; Kos, J.; et al. N-Alkylpiperidine Carbamates as Potential Anti-Alzheimer’s Agents. Eur. J. Med. Chem. 2020, 197, 112282. [Google Scholar] [CrossRef]
- WHO|World Health Organization. Available online: https://www.who.int/news/item/15-11-2021-who-and-bayer-renew-longstanding-collaboration-to-accelerate-control-and-elimination-of-neglected-tropical-diseases (accessed on 12 January 2024).







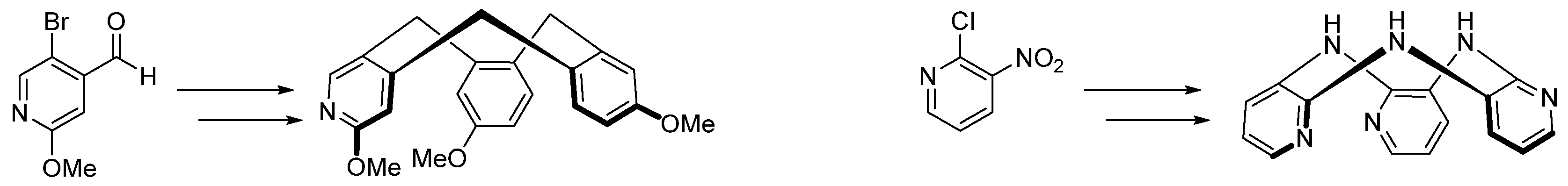







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Primas, N.; Castera-Ducros, C.; Paoli-Lombardo, R.; Curti, C.; Mathias, F.; Rathelot, P.; Marchand, P.; Vanelle, P. 31st Annual GP2A Medicinal Chemistry Conference. Drugs Drug Candidates 2024, 3, 209-243. https://doi.org/10.3390/ddc3010013
Primas N, Castera-Ducros C, Paoli-Lombardo R, Curti C, Mathias F, Rathelot P, Marchand P, Vanelle P. 31st Annual GP2A Medicinal Chemistry Conference. Drugs and Drug Candidates. 2024; 3(1):209-243. https://doi.org/10.3390/ddc3010013
Chicago/Turabian StylePrimas, Nicolas, Caroline Castera-Ducros, Romain Paoli-Lombardo, Christophe Curti, Fanny Mathias, Pascal Rathelot, Pascal Marchand, and Patrice Vanelle. 2024. "31st Annual GP2A Medicinal Chemistry Conference" Drugs and Drug Candidates 3, no. 1: 209-243. https://doi.org/10.3390/ddc3010013
APA StylePrimas, N., Castera-Ducros, C., Paoli-Lombardo, R., Curti, C., Mathias, F., Rathelot, P., Marchand, P., & Vanelle, P. (2024). 31st Annual GP2A Medicinal Chemistry Conference. Drugs and Drug Candidates, 3(1), 209-243. https://doi.org/10.3390/ddc3010013