

Effect of the N501Y Mutation on Ligands Complexed with SARS-CoV-2 RBD: Insights on Potential Drug Candidates for COVID-19

 ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Validation of Docking and ADMET Results
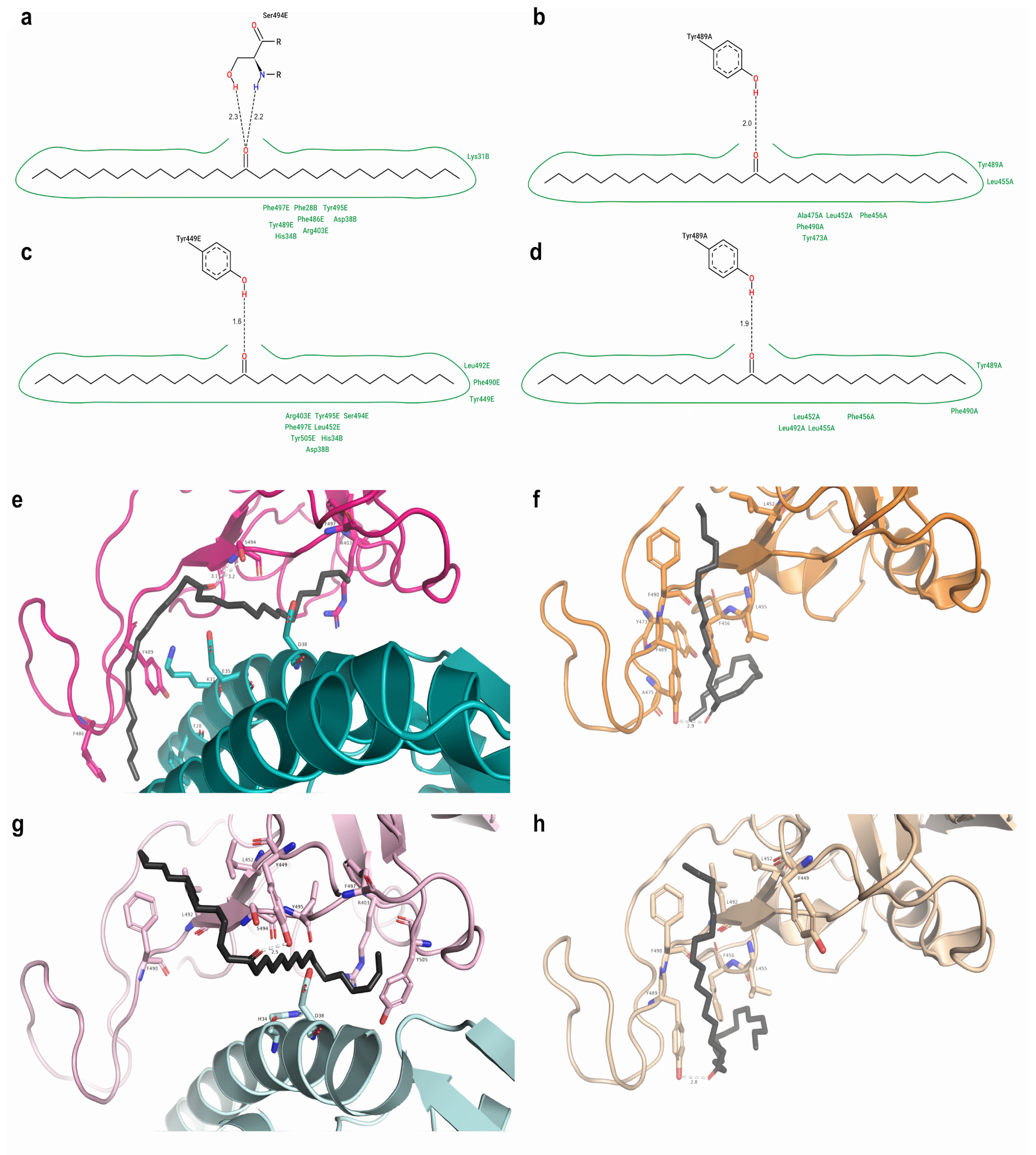
2.2. Protein–Ligand Docking
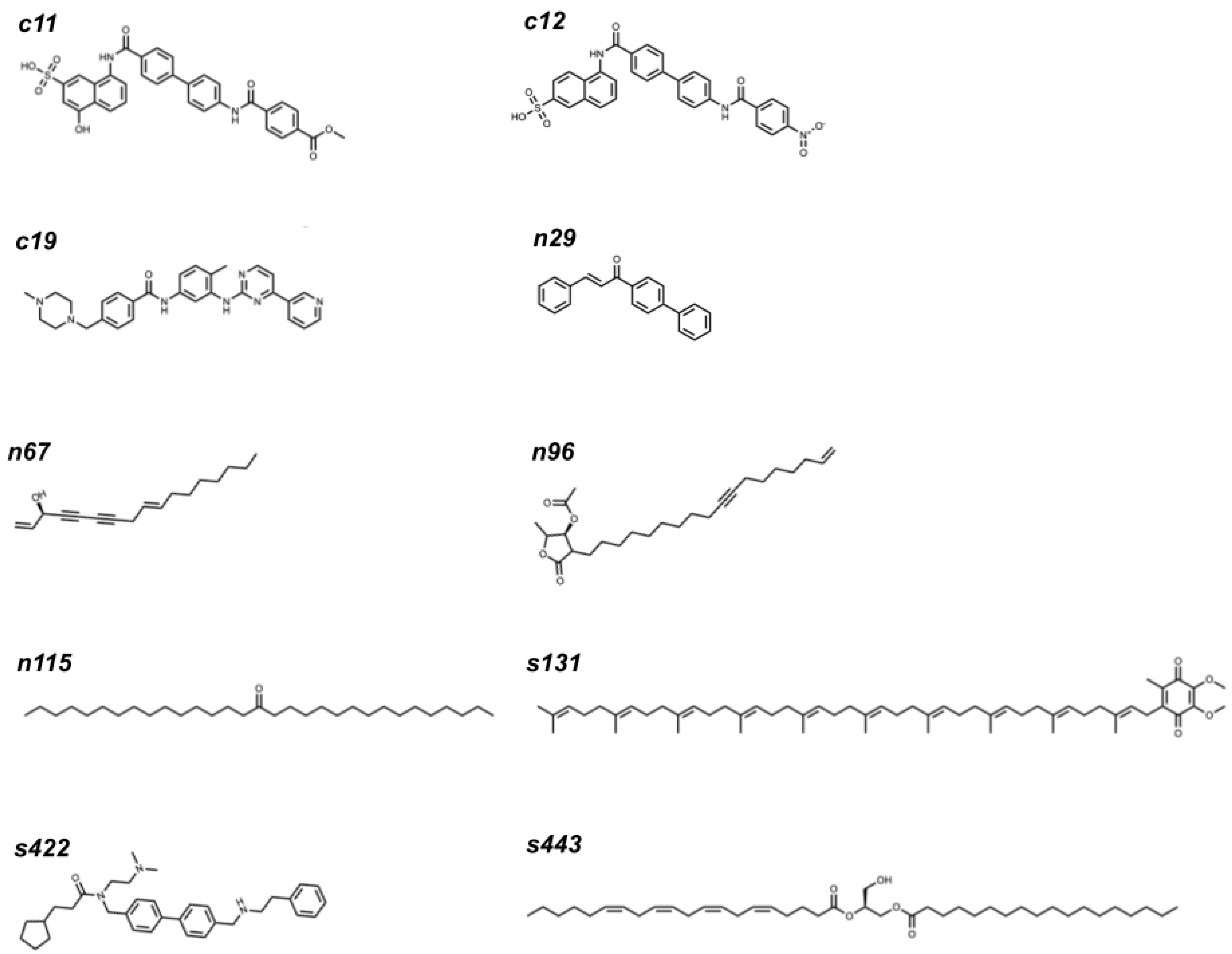
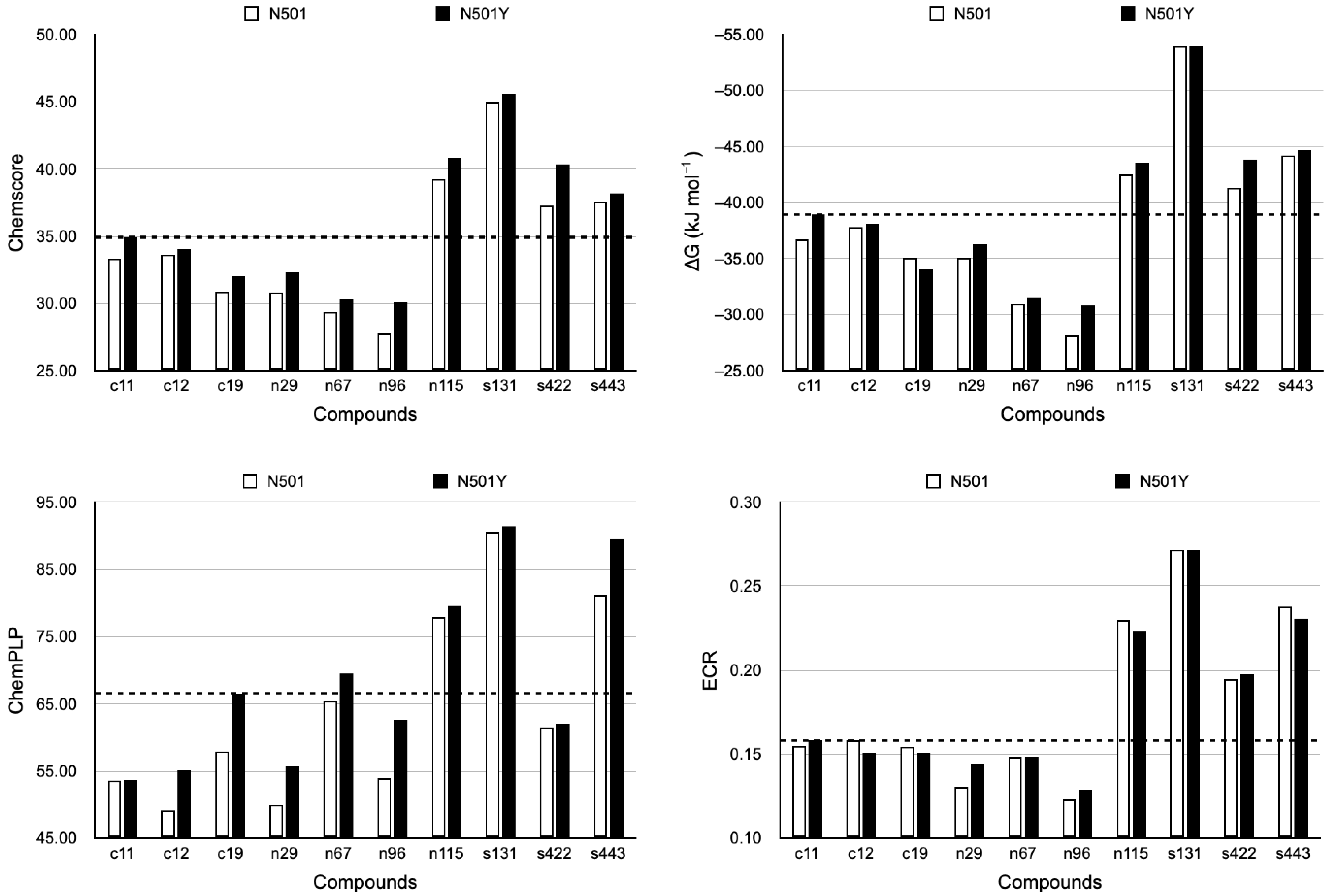
2.3. Top Three Hit Compounds from Docking Simulations
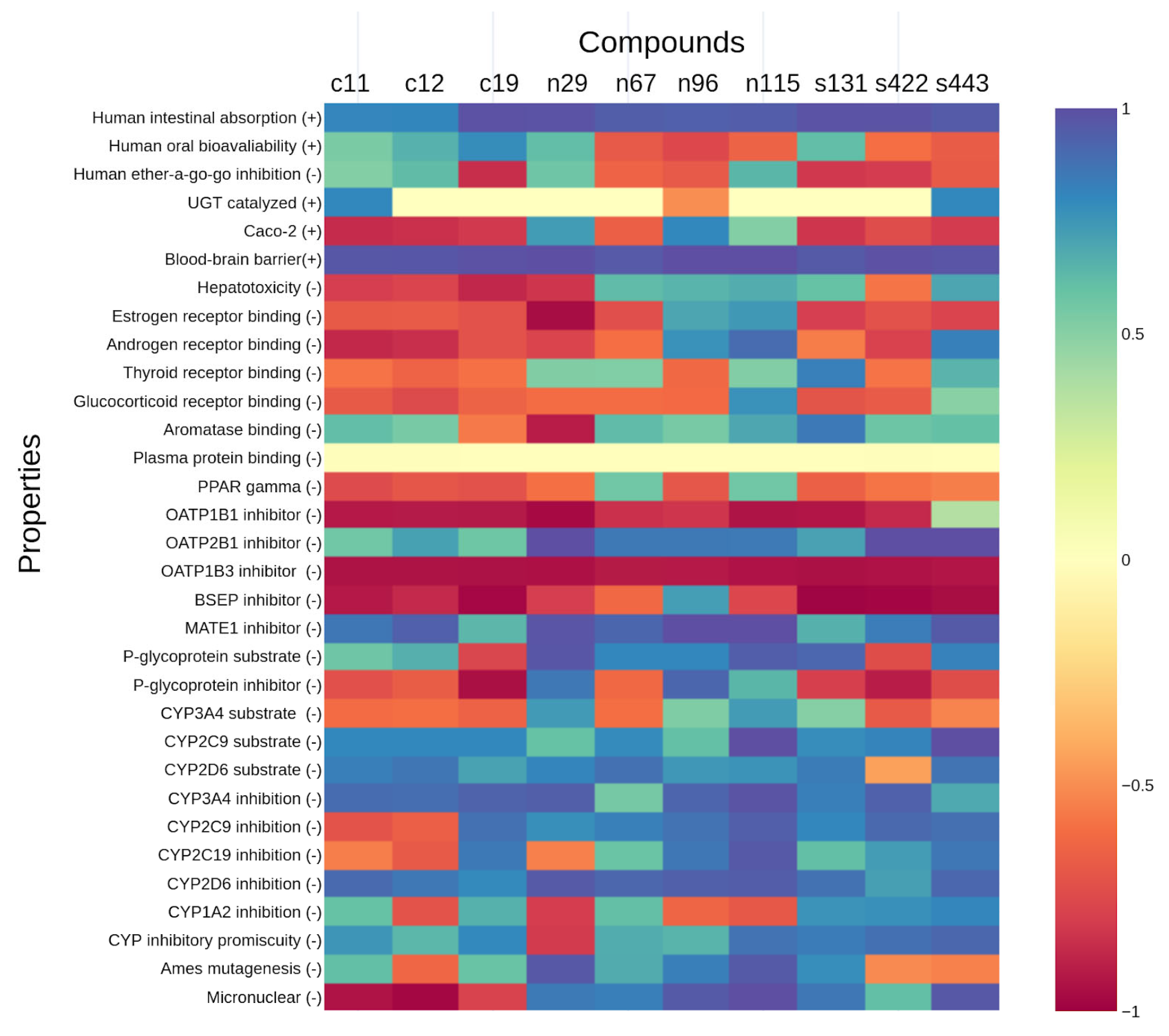
2.4. Analyses of ADMET Properties
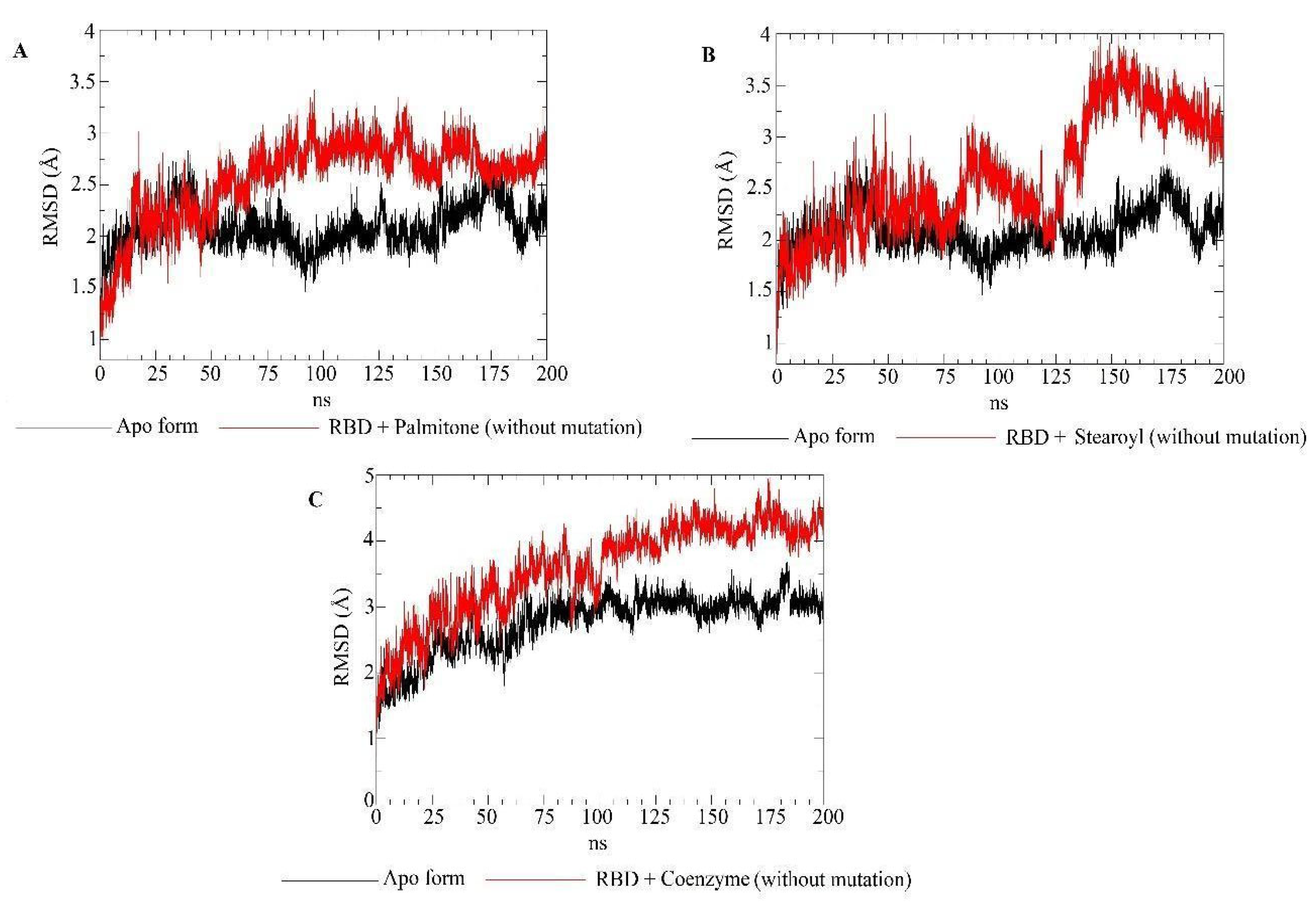
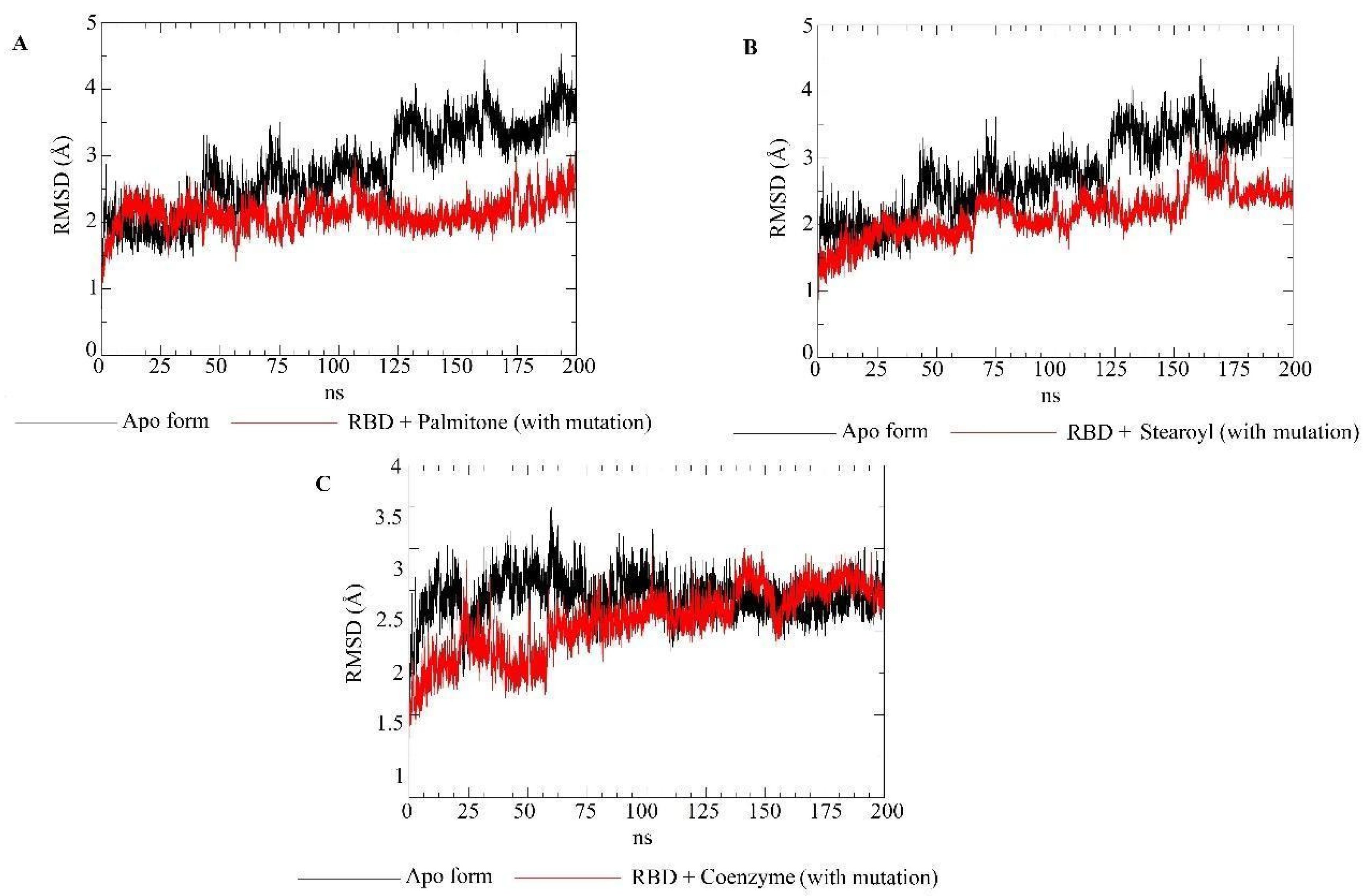
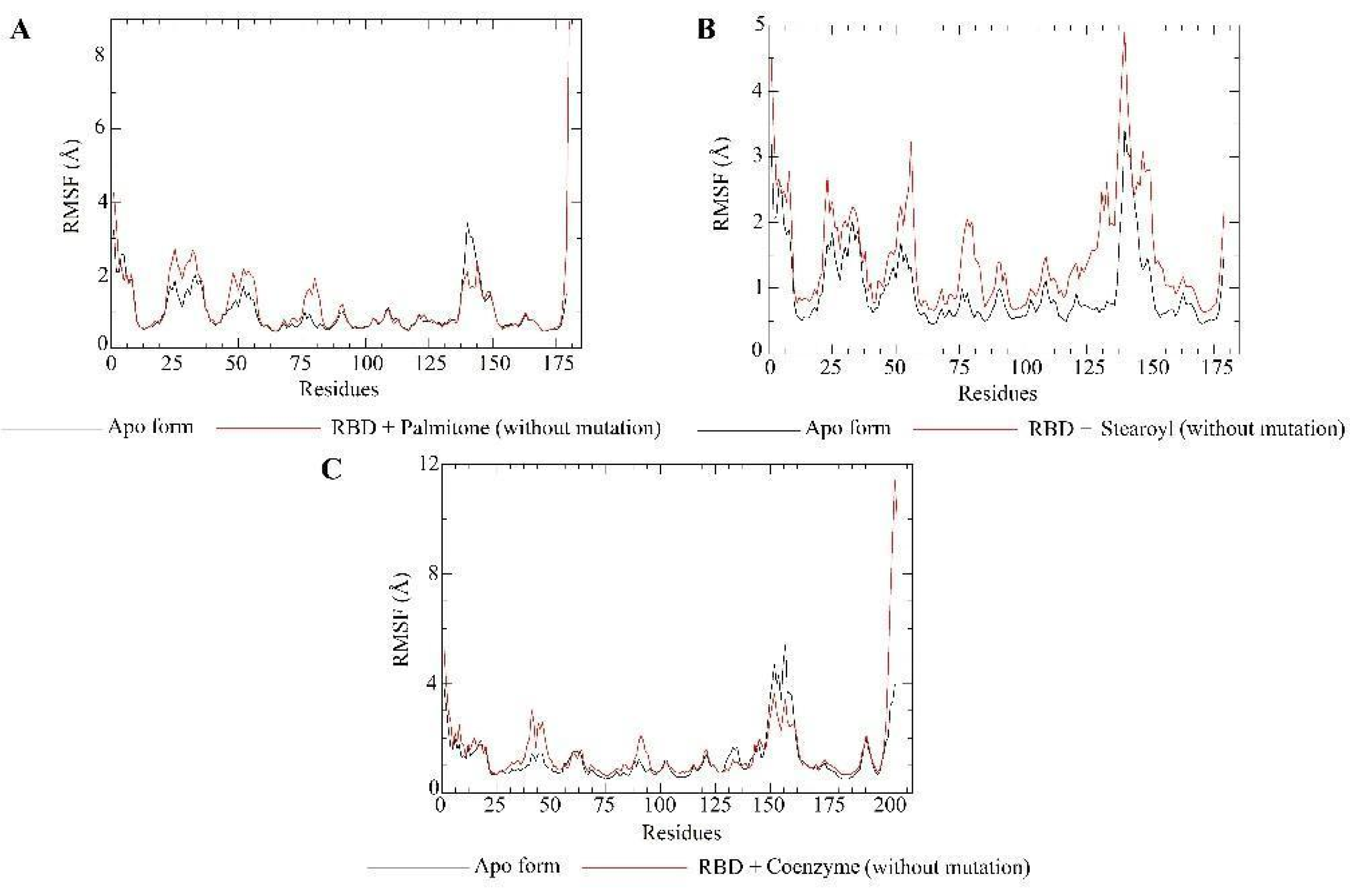
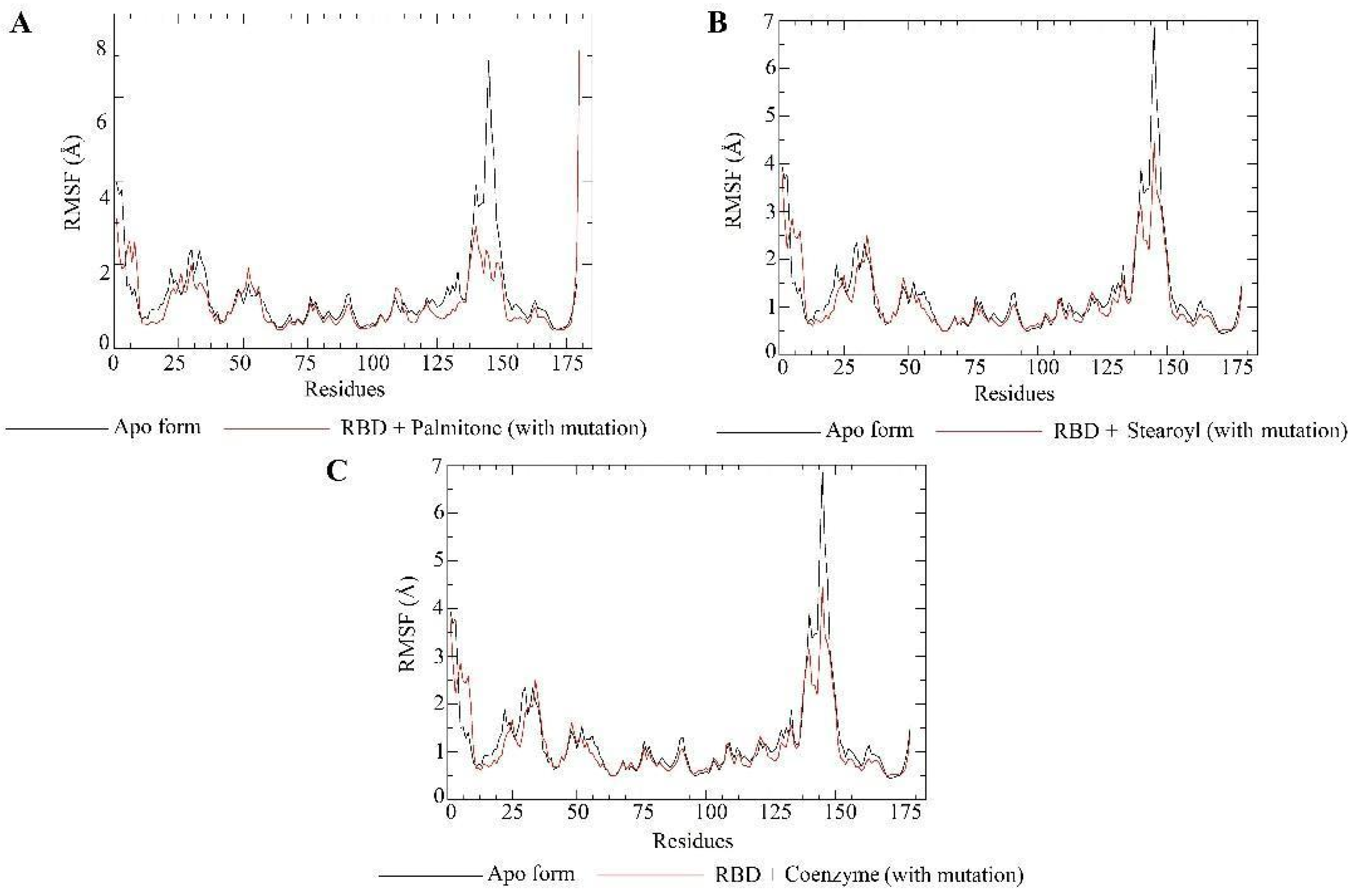
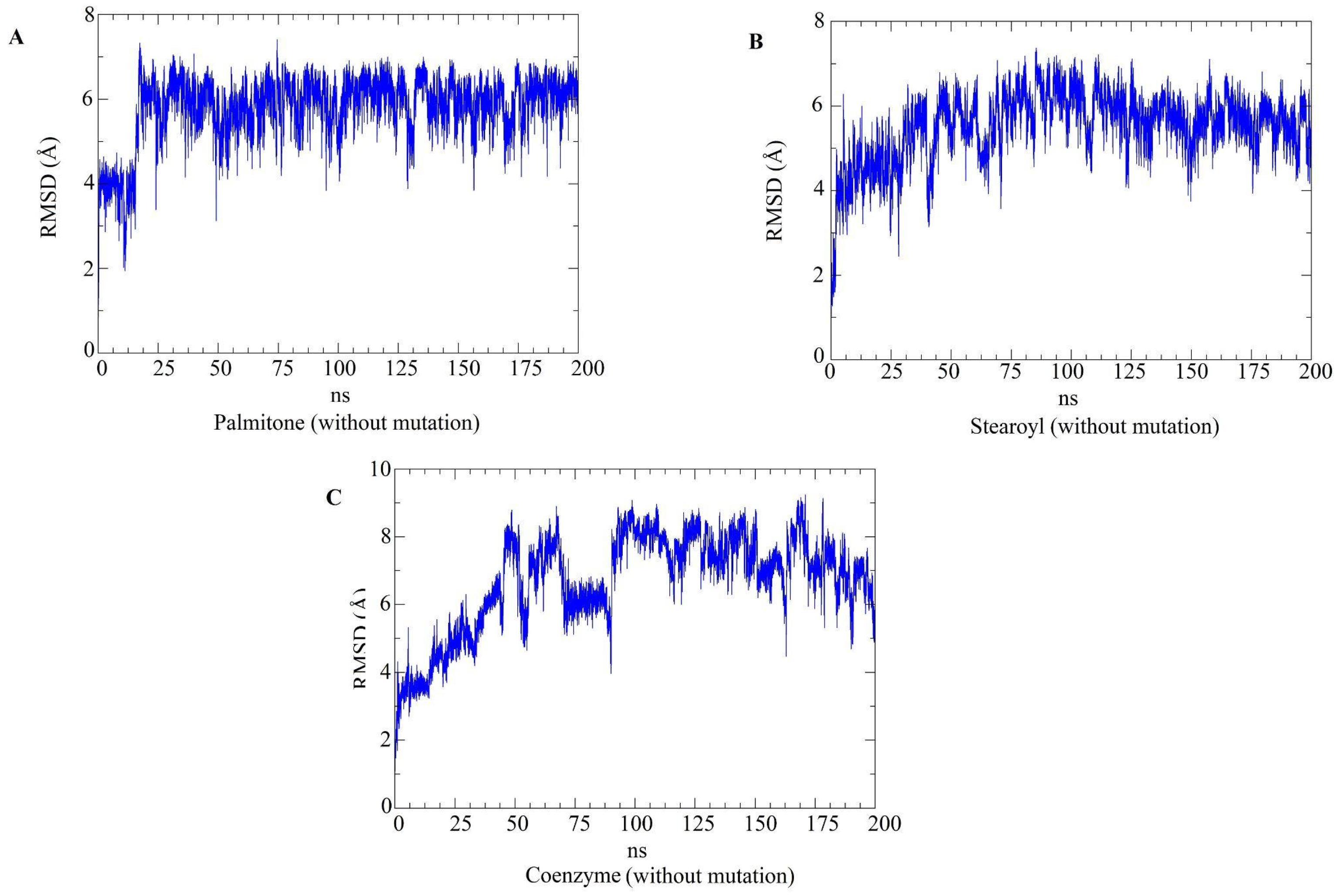
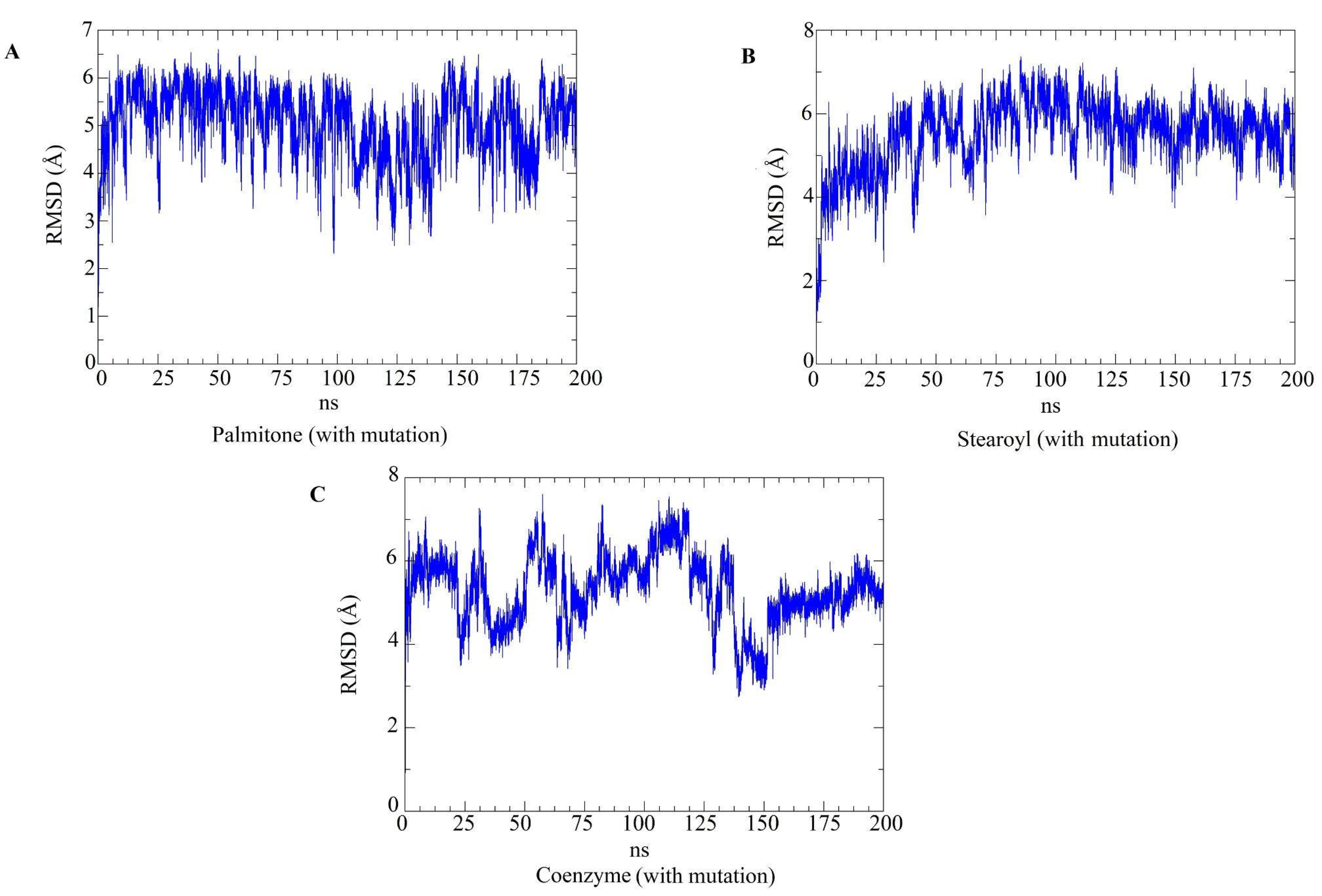
2.5. Molecular Dynamics Simulations
2.6. Free Energy Calculations
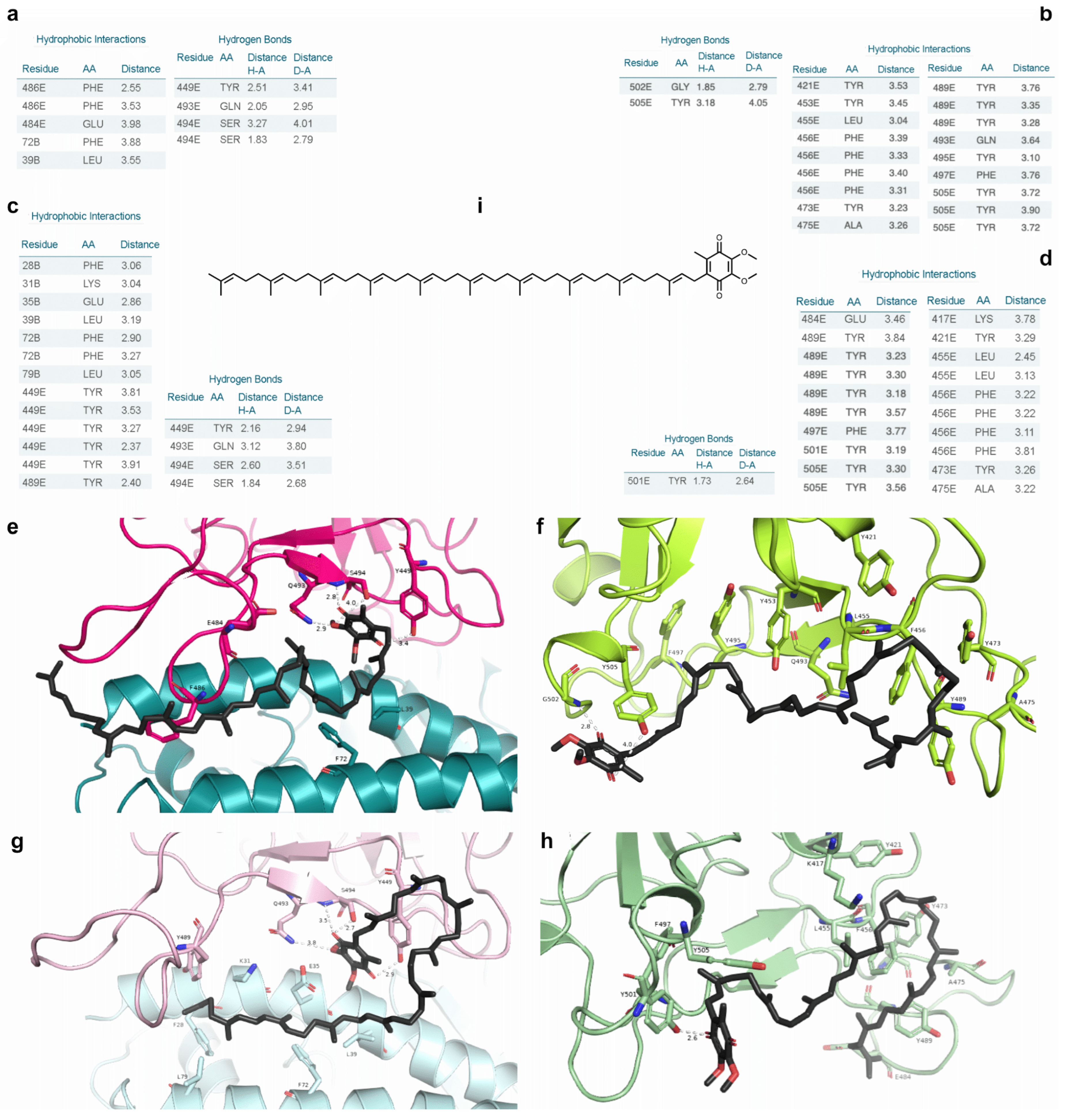
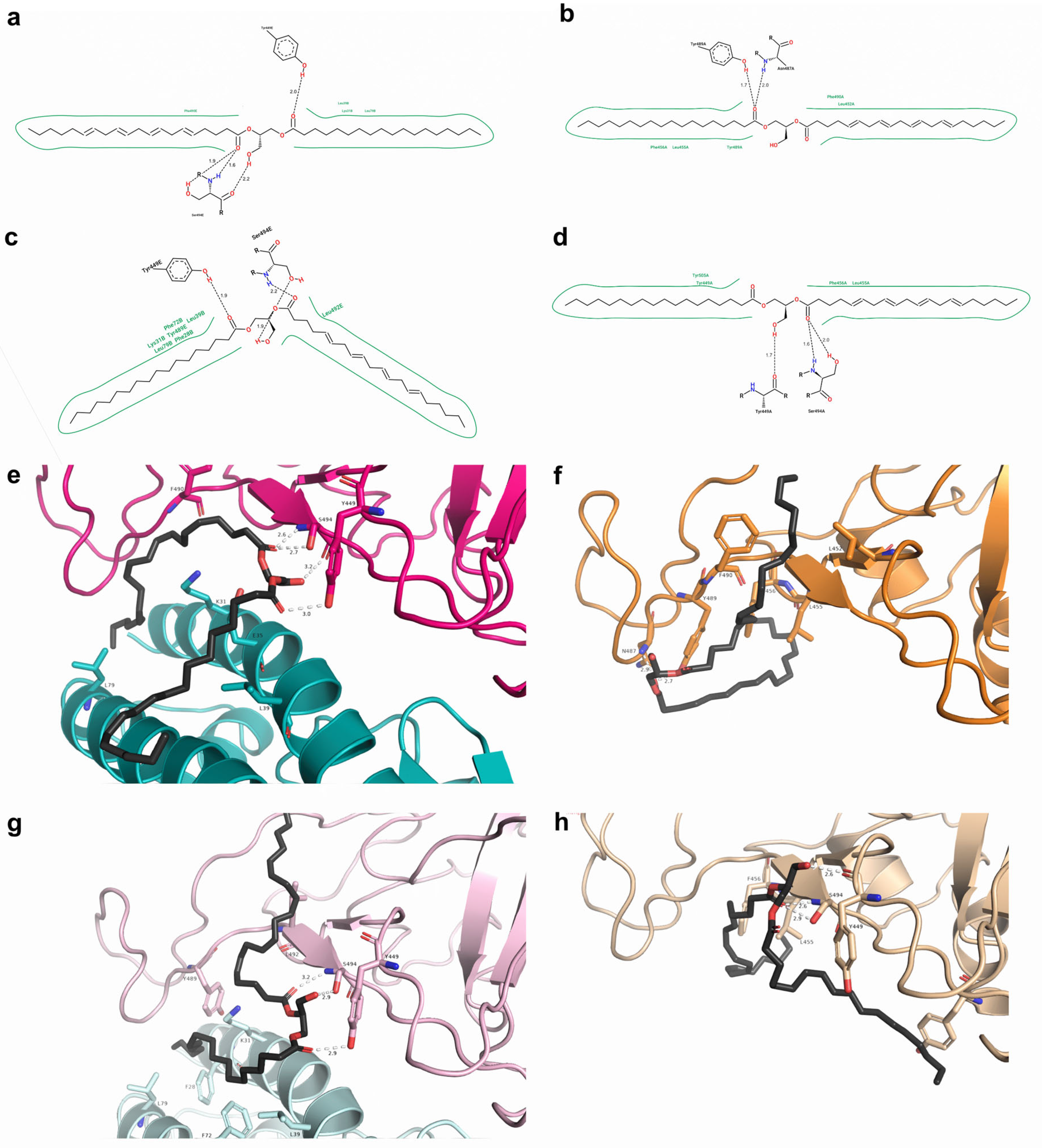
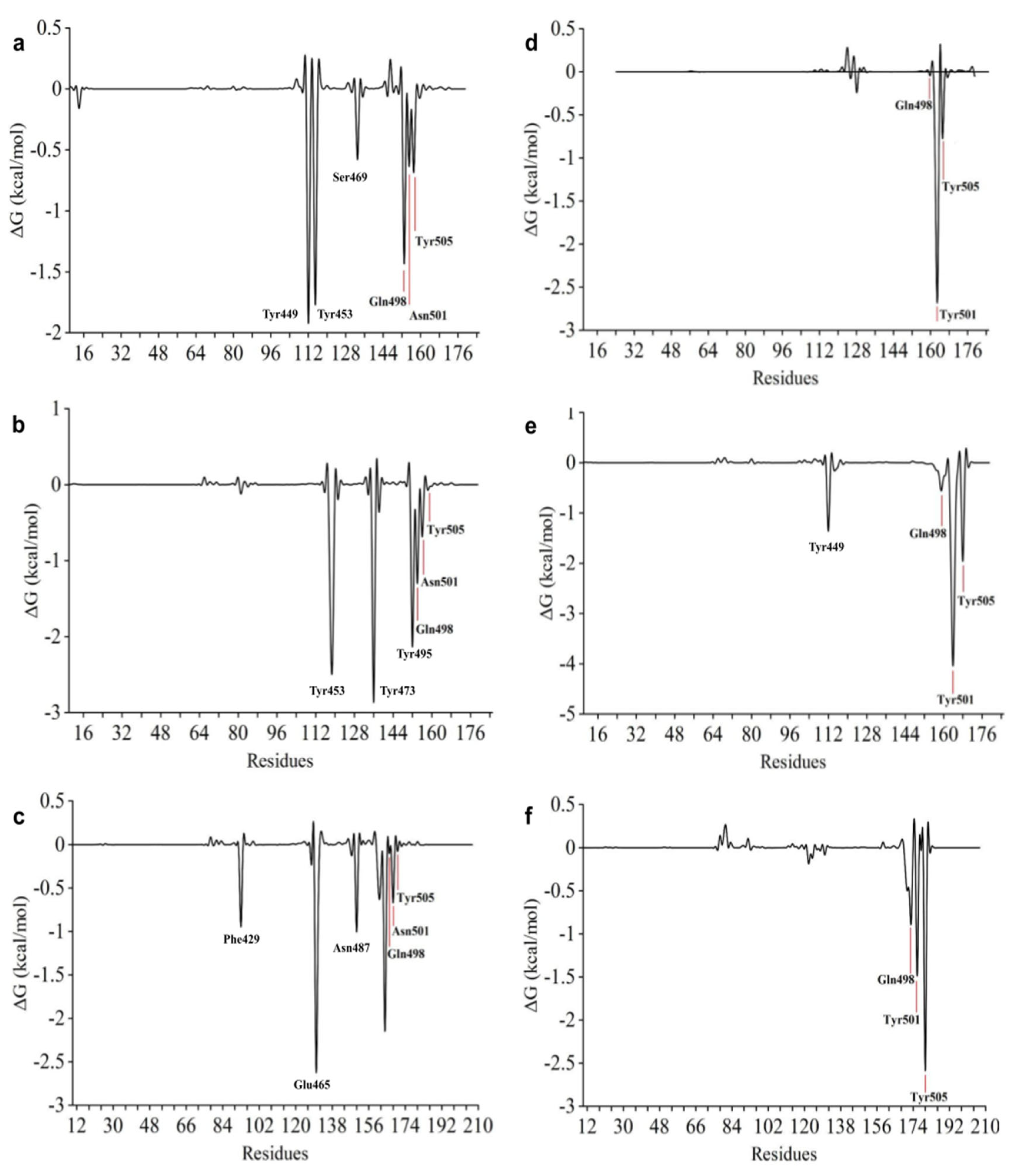
2.7. Free Energy Decomposition
2.8. Current Findings in the Literature on the Screened Molecules
3. Materials and Methods
3.1. Selection and Processing of Proteins and Ligands
3.2. Protein–Ligand Docking
3.3. ADMET Analyses
3.4. Validation of Docking and ADMET Results
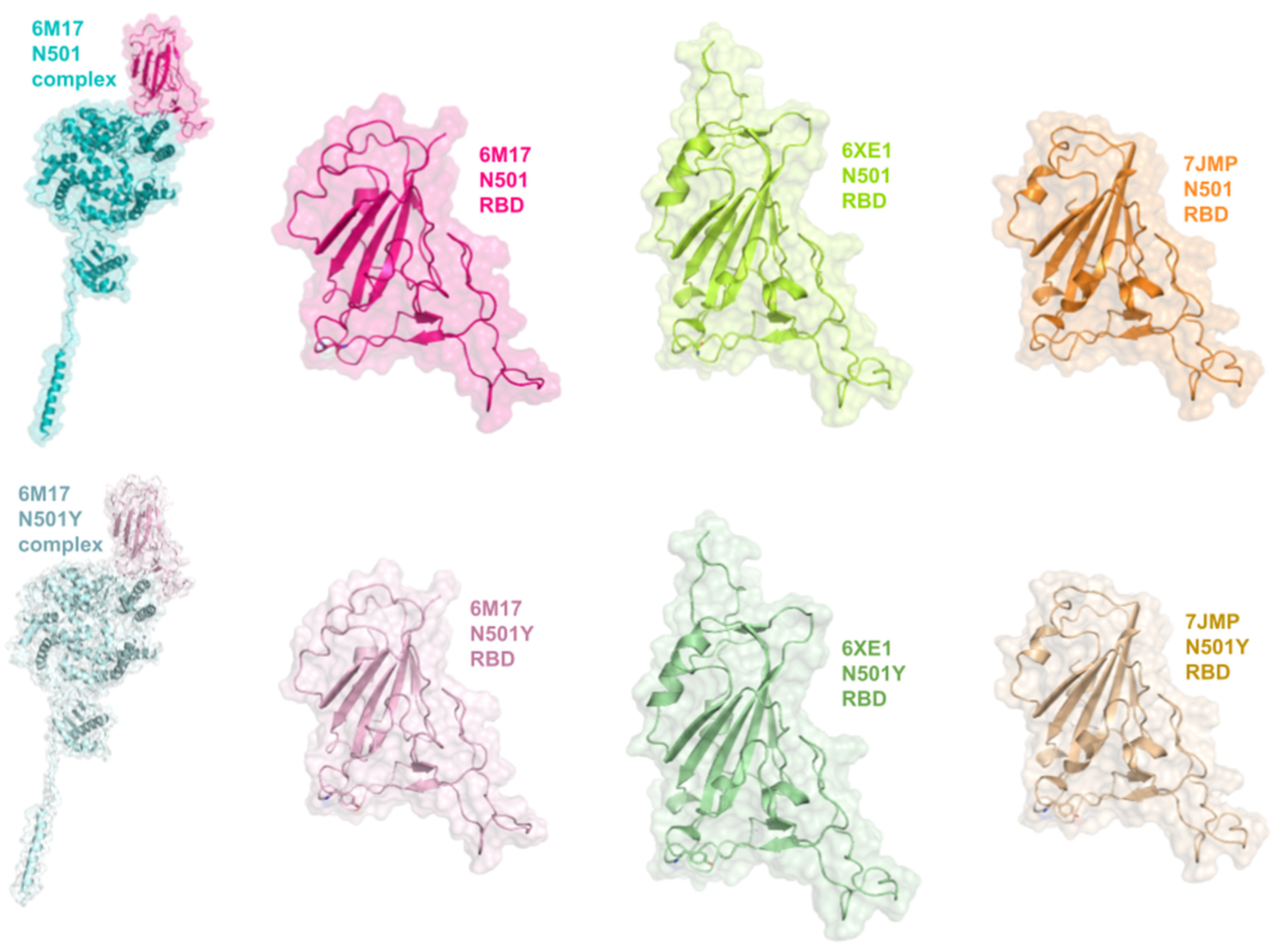
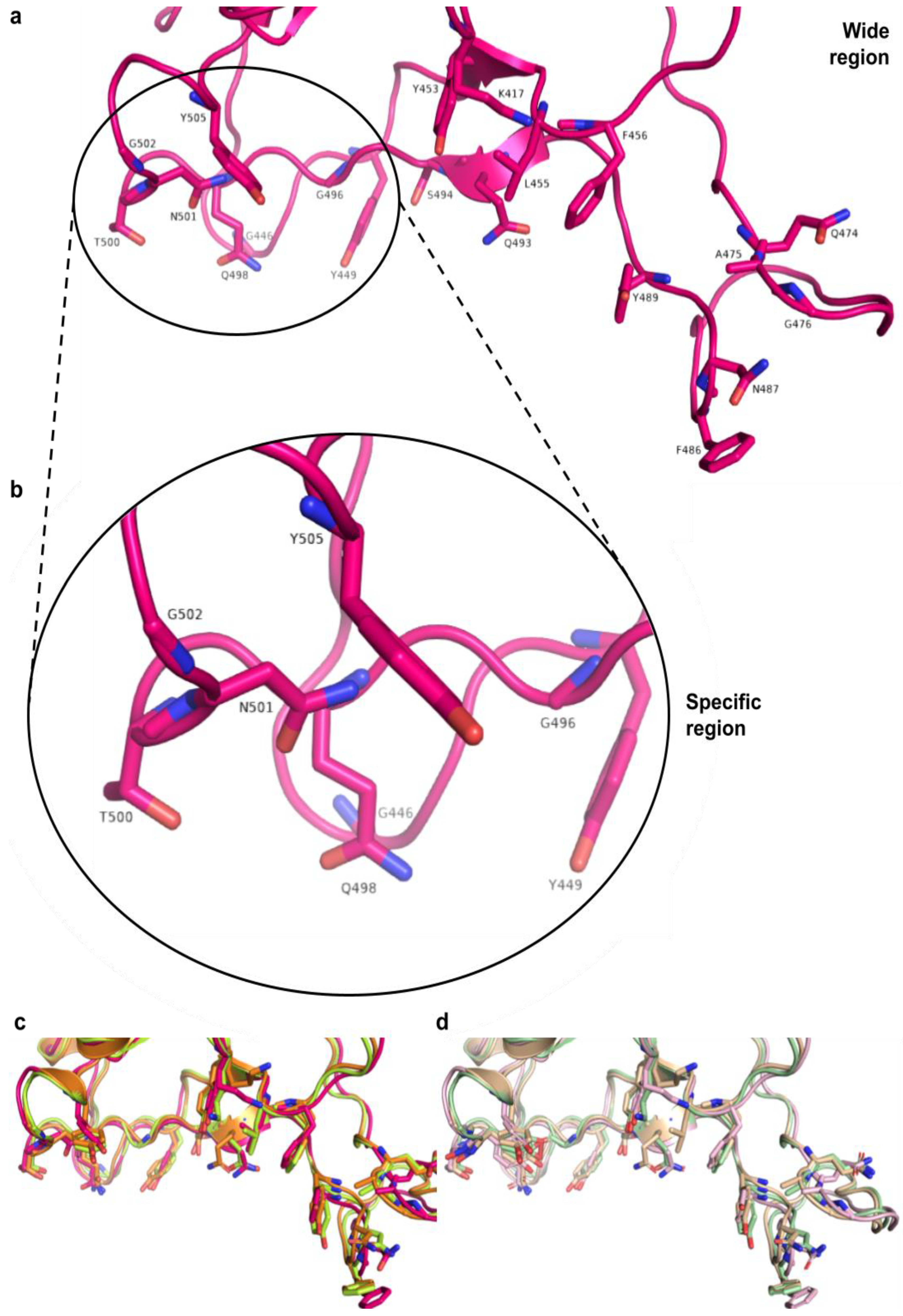
3.5. Interactions between RBD of Viral Spike and PD of ACE2 Receptor
3.6. Molecular Dynamics Simulations
3.7. Statistical Analysis of Docking Results
3.8. Structural Analysis of the Proteins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Coronavirus Disease (COVID-19): Situation Reports. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed on 23 February 2021).
- WHO. Summary of Probable SARS Cases with Onset of Illness. Available online: https://www.who.int/publications/m/item/summary-of-probable-sars-cases-with-onset-of-illness-from-1-november-2002-to-31-july-2003 (accessed on 23 February 2021).
- WHO. Middle East Respiratory Syndrome: MERS Situation Update. Available online: http://www.emro.who.int/health-topics/mers-cov/mers-outbreaks.html (accessed on 23 February 2021).
- WHO. Novel Coronavirus—China. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2020-DON233 (accessed on 23 February 2021).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Plowright, R.K.; Parrish, C.R.; McCallum, H.; Hudson, P.J.; Ko, A.I.; Graham, A.L.; Lloyd-Smith, J.O. Pathways to zo-onotic spillover. Nat. Rev. Microbiol. 2017, 15, 502–510. [Google Scholar] [CrossRef]
- Greger, M. The Human/Animal Interface: Emergence and Resurgence of Zoonotic Infectious Diseases. Crit. Rev. Microbiol. 2007, 33, 243–299. [Google Scholar] [CrossRef]
- Ye, Z.-W.; Yuan, S.; Yuen, K.-S.; Fung, S.-Y.; Chan, C.-P.; Jin, D.-Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Hanson, Q.M.; Wilson, K.M.; Shen, M.; Itkin, Z.; Eastman, R.T.; Shinn, P.; Hall, M.D. Targeting ACE2–RBD Interaction as a Platform for COVID-19 Therapeutics: Development and Drug-Repurposing Screen of an AlphaLISA Proximity Assay. ACS Pharmacol. Transl. Sci. 2020, 3, 1352–1360. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, F.; Shen, C.; Peng, W.; Li, D.; Zhao, C.; Li, Z.; Li, S.; Bi, Y.; Yang, Y.; et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 2020, 368, 1274–1278. [Google Scholar] [CrossRef]
- Grant, O.C.; Montgomery, D.; Ito, K.; Woods, R.J. Analysis of the SARS-CoV-2 spike protein glycan shield reveals implica-tions for immune recognition. Sci. Rep. 2020, 10, 14991. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- WHO. Therapeutics and COVID-19: Living Guideline. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2022.4 (accessed on 15 July 2022).
- Singh, B.; Ryan, H.; Kredo, T.; Chaplin, M.; Fletcher, T. Chloroquine or hydroxychloroquine for prevention and treatment of COVID-19. Cochrane Database Syst. Rev. 2021, 2, CD013587. [Google Scholar] [CrossRef]
- Ghazy, R.M.; Almaghraby, A.; Shaaban, R.; Kamal, A.; Beshir, H.; Moursi, A.; Ramadan, A.; Taha, S.H.N. A systematic review and meta-analysis on chloroquine and hydroxychloroquine as monotherapy or combined with azithromycin in COVID-19 treatment. Sci. Rep. 2020, 10, 22139. [Google Scholar] [CrossRef]
- Zheng, Z.; Wu, Y.; Qian, D.; Lian, J. Off-label use of chloroquine, hydroxychloroquine, azithromycin and lopinavir/ritonavir in COVID-19 risks prolonging the Q.T. interval by targeting the hERG channel. Eur. J. Pharmacol. 2021, 893, 173813. [Google Scholar]
- Axfors, C.; Schmitt, A.M.; Janiaud, P.; Hooft, J.V.; Abd-Elsalam, S.; Abdo, E.F.; Abella, B.S.; Akram, J.; Amaravadi, R.K.; Angus, D.C.; et al. Mortality outcomes with hydroxychloroquine and chloroquine in COVID-19 from an international collaborative meta-analysis of randomized trials. Nat. Commun. 2021, 12, 2349. [Google Scholar] [CrossRef] [PubMed]
- Lester, M.; Sahin, A.; Pasyar, A. The use of dexamethasone in the treatment of COVID-19. Ann. Med. Surg. 2020, 56, 218–219. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.A.; Pranata, R. Worrying situation regarding the use of dexamethasone for COVID-19. Ther. Adv. Respir. Dis. 2020, 14, 1753466620942131. [Google Scholar] [CrossRef]
- Brotherton, H.; Usuf, E.; Nadjm, B.; Forrest, K.; Bojang, K.; Samateh, A.L.; Bittaye, M.; Roberts, C.A.; D’Alessandro, U.; Roca, A. Dexamethasone for COVID-19: Data needed from randomised clinical trials in Africa. Lancet Glob. Health 2020, 8, e1125–e1126. [Google Scholar] [CrossRef]
- Ahmed, M.H.; Hassan, A. Dexamethasone for the Treatment of Coronavirus Disease (COVID-19): A Review. SN Compr. Clin. Med. 2020, 2, 2637–2646. [Google Scholar] [CrossRef] [PubMed]
- Shaman, J.; Galanti, M. Will SARS-CoV-2 become endemic? Science 2020, 370, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Fontanet, A.; Autran, B.; Lina, B.; Kieny, M.P.; Karim, S.S.A.; Sridhar, D. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 2021, 397, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Variant: S:N501. CoVariants. Available online: https://covariants.org/variants/S.N501 (accessed on 26 April 2021).
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2017, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.S.; Mogensen, T.H.; Agergaard, J.; Schiøttz-Christensen, B.; Østergaard, L.; Vibholm, L.K.; Leth, S. High-dose coenzyme Q10 therapy versus placebo in patients with post COVID-19 condition: A randomized, phase 2, crossover trial. Lancet Reg. Health Eur. 2023, 24, 100539. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, Y.; Tian, Z.; Zhu, M.; Zhang, B.; Du, S.; Li, Y.; Liu, Z.; Hou, S.; Yang, Y. Coenzyme Q10 Attenuates Human Platelet Aggregation Induced by SARS-CoV-2 Spike Protein via Re-ducing Oxidative Stress In Vitro. Int. J. Mol. Sci. 2022, 23, 12345. [Google Scholar] [CrossRef]
- wwPDB Consortium. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Gulotta, M.R.; Lombino, J.; Perricone, U.; De Simone, G.; Mekni, N.; De Rosa, M.; Diana, P.; Padova, A. Targeting SARS-CoV-2 RBD interface: A supervised computational data-driven approach to identify po-tential modulators. ChemMedChem 2020, 15, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640. [Google Scholar] [CrossRef]
- Wu, N.C.; Yuan, M.; Liu, H.; Lee, C.-C.D.; Zhu, X.; Bangaru, S.; Torres, J.L.; Caniels, T.G.; Brouwer, P.J.M.; van Gils, M.J.; et al. An alternative binding mode of IGHV3-53 antibodies to the SARS-CoV-2 receptor binding domain. Cell Rep. 2020, 33, 108274. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Baxter, C.A.; Murray, C.W.; Clark, D.E.; Westhead, D.R.; Eldridge, M.D. Flexible docking using Tabu search and an em-pirical estimate of binding affinity. Proteins Struct. Funct. Bioinform. 1998, 33, 367–382. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2019, 59, 895–913. [Google Scholar] [CrossRef]
- Palacio-Rodríguez, K.; Lans, I.; Cavasotto, C.N.; Cossio, P. Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 2019, 9, 5142. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2018, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Svensson, F.; Zoufir, A.; Bender, A. eMolTox: Prediction of molecular toxicity with confidence. Bioinformatics 2018, 34, 2508–2509. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.; Sabbadin, D.; Cuzzolin, A.; Martínez-Rosell, G.; Gora, J.; Manchester, J.; Duca, J.S.; De Fabritiis, G. PathwayMap: Molecular Pathway Association with Self-Normalizing Neural Networks. J. Chem. Inf. Model. 2018, 59, 1172–1181. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Protein interfaces, surfaces and assemblies service P.I.S.A. at European Bioinformatics Institute. J Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Detection of protein assemblies in crystals. In International Symposium on Computational Life Science; Springer: Berlin/Heidelberg, Germany, 2005; pp. 163–174. [Google Scholar]
- Krissinel, E. Crystal contacts as nature’s docking solutions. J. Comput. Chem. 2010, 31, 133–143. [Google Scholar] [CrossRef]
- Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey, M. A consistent description of HYdrogen bond and DEhydration en-ergies in protein–ligand complexes: Methods behind the HYDE scoring function. J. Comput. Aided Mol. Des. 2013, 27, 15–29. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M.J.J. Contact-based prediction of binding affinity in protein-protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Fu, W.; Chen, Y.; Wang, K.; Hettinghouse, A.; Hu, W.; Wang, J.-Q.; Lei, Z.-N.; Chen, Z.-S.; Stapleford, K.A.; Liu, C.-J. Repurposing FDA-approved drugs for SARS-CoV-2 through an ELISA-based screening for the inhibition of RBD/ACE2 interaction. Protein Cell 2021, 12, 586–591. [Google Scholar] [CrossRef]
- Bojadzic, D.; Alcazar, O.; Buchwald, P. Methylene Blue Inhibits the SARS-CoV-2 spike–ACE2 Protein-Protein Interaction–a Mechanism that can Contribute to its Antiviral Activity Against COVID-19. Front. Pharmacol. 2021, 11, 600372. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Li, Y.; Zhang, Y.; Liu, Z.; Mu, X.; Gu, C.; Liu, J.; Li, Y.; Li, G.; Chen, J. Ceftazidime Is a Potential Drug to Inhibit SARS-CoV-2 Infection In Vitro by Blocking Spike Protein-ACE2 Interaction. Available online: https://biorxiv.org/content/10.1101/2020.09.14.295956v1.full (accessed on 20 February 2021).
- Mulgaonkar, N.S.; Wang, H.; Mallawarachchi, S.; Růžek, D.; Martina, B.; Fernando, S. Bcr-Abl Tyrosine Kinase Inhibitor Imatinib as a Potential Drug for COVID-19. Available online: https://biorxiv.org/content/10.1101/2020.06.18.158196v2 (accessed on 20 February 2021).
- Bojadzic, D.; Alcazar, O.; Chen, J.; Buchwald, P. Small-Molecule In Vitro Inhibitors of the Coronavirus Spike-ACE2 Protein-Protein Interaction as Blockers of Viral Attachment and Entry for SARS-CoV-2. Available online: https://biorxiv.org/content/10.1101/2020.10.22.351056v1 (accessed on 20 February 2021).
- Virtanen, S.I.; Niinivehmas, S.P.; Pentikäinen, O.T. Case-specific performance of MM-PBSA, MM-GBSA, and SIE in virtual screening. J. Mol. Graph. Model. 2015, 62, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RBD | Ligand | Control | Natural | Synthetic | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1st | 2nd | 3rd | 1st | 2nd | 3rd | 1st | 2nd | 3rd | ||
| N501 | Index | c19 | c12 | c11 | n115 | n67 | n29 | s131 | s443 | s422 |
| ECR | 0.107 | 0.104 | 0.0973 | 0.0240 | 0.0234 | 0.0231 | 0.00612 | 0.00609 | 0.00606 | |
| Chemscore | 30.10 | 31.87 | 31.96 | 37.21 | 27.23 | 29.58 | 38.67 | 33.38 | 36.49 | |
| ΔG [kJ mol−1] | −33.24 | −34.45 | −35.26 | −39.71 | −28.81 | −33.64 | −44.46 | −39.35 | −40.69 | |
| ChemPLP | 55.53 | 43.77 | 33.32 | 70.83 | 59.58 | 48.56 | 67.95 | 66.45 | 56.45 | |
| N501Y | Index | c11 | c19 | c12 | n115 | n29 | n96 | s131 | s443 | s422 |
| ECR | 0.0747 | 0.0617 | 0.0612 | 0.0240 | 0.0232 | 0.0229 | 0.00612 | 0.00609 | 0.00608 | |
| Chemscore | 33.68 | 31.04 | 32.52 | 38.07 | 31.43 | 29.03 | 43.12 | 36.70 | 36.77 | |
| ΔG [kJ mol−1] | −37.59 | −32.77 | −35.66 | −40.44 | −33.36 | −29.79 | −48.71 | −42.65 | −40.06 | |
| ChemPLP | 57.09 | 59.52 | 51.51 | 73.89 | 54.81 | 58.59 | 95.09 | 82.10 | 62.77 | |
| Ligand + PDB Structure | MM-GBSA |
|---|---|
| n115 + N501 7JMP | −20.60 |
| n115 + Y501 7JMP | −21.10 |
| s131 + N501 6XE1 | −24.34 |
| s131 + Y501 6XE1 | −15.59 |
| s443 + N501 7JMP | −28.62 |
| s443 + Y501 7JMP | −29.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, C.F.A.; Camalhonte, S.O.; de Oliveira Almeida, M.; Araujo, S.C.; Sannomiya, M.; Lago, J.H.G.; Honorio, K.M. Effect of the N501Y Mutation on Ligands Complexed with SARS-CoV-2 RBD: Insights on Potential Drug Candidates for COVID-19. Drugs Drug Candidates 2023, 2, 433-458. https://doi.org/10.3390/ddc2020022
da Silva CFA, Camalhonte SO, de Oliveira Almeida M, Araujo SC, Sannomiya M, Lago JHG, Honorio KM. Effect of the N501Y Mutation on Ligands Complexed with SARS-CoV-2 RBD: Insights on Potential Drug Candidates for COVID-19. Drugs and Drug Candidates. 2023; 2(2):433-458. https://doi.org/10.3390/ddc2020022
Chicago/Turabian Styleda Silva, Camila Fonseca Amorim, Samanta Omae Camalhonte, Michell de Oliveira Almeida, Sheila Cruz Araujo, Miriam Sannomiya, João Henrique Ghilardi Lago, and Kathia Maria Honorio. 2023. "Effect of the N501Y Mutation on Ligands Complexed with SARS-CoV-2 RBD: Insights on Potential Drug Candidates for COVID-19" Drugs and Drug Candidates 2, no. 2: 433-458. https://doi.org/10.3390/ddc2020022
APA Styleda Silva, C. F. A., Camalhonte, S. O., de Oliveira Almeida, M., Araujo, S. C., Sannomiya, M., Lago, J. H. G., & Honorio, K. M. (2023). Effect of the N501Y Mutation on Ligands Complexed with SARS-CoV-2 RBD: Insights on Potential Drug Candidates for COVID-19. Drugs and Drug Candidates, 2(2), 433-458. https://doi.org/10.3390/ddc2020022