

Optimizing Protein Production in Therapeutic Phages against a Bacterial Pathogen, Mycobacterium abscessus


Abstract
1. Introduction
1.1. General Criteria for Optimizing Therapeutic Phages against Pathogenic Bacteria
1.2. Principles of Optimizing Phage mRNA
1.3. A Case Study: Optimizing Phages against M. abscessus
2. Results
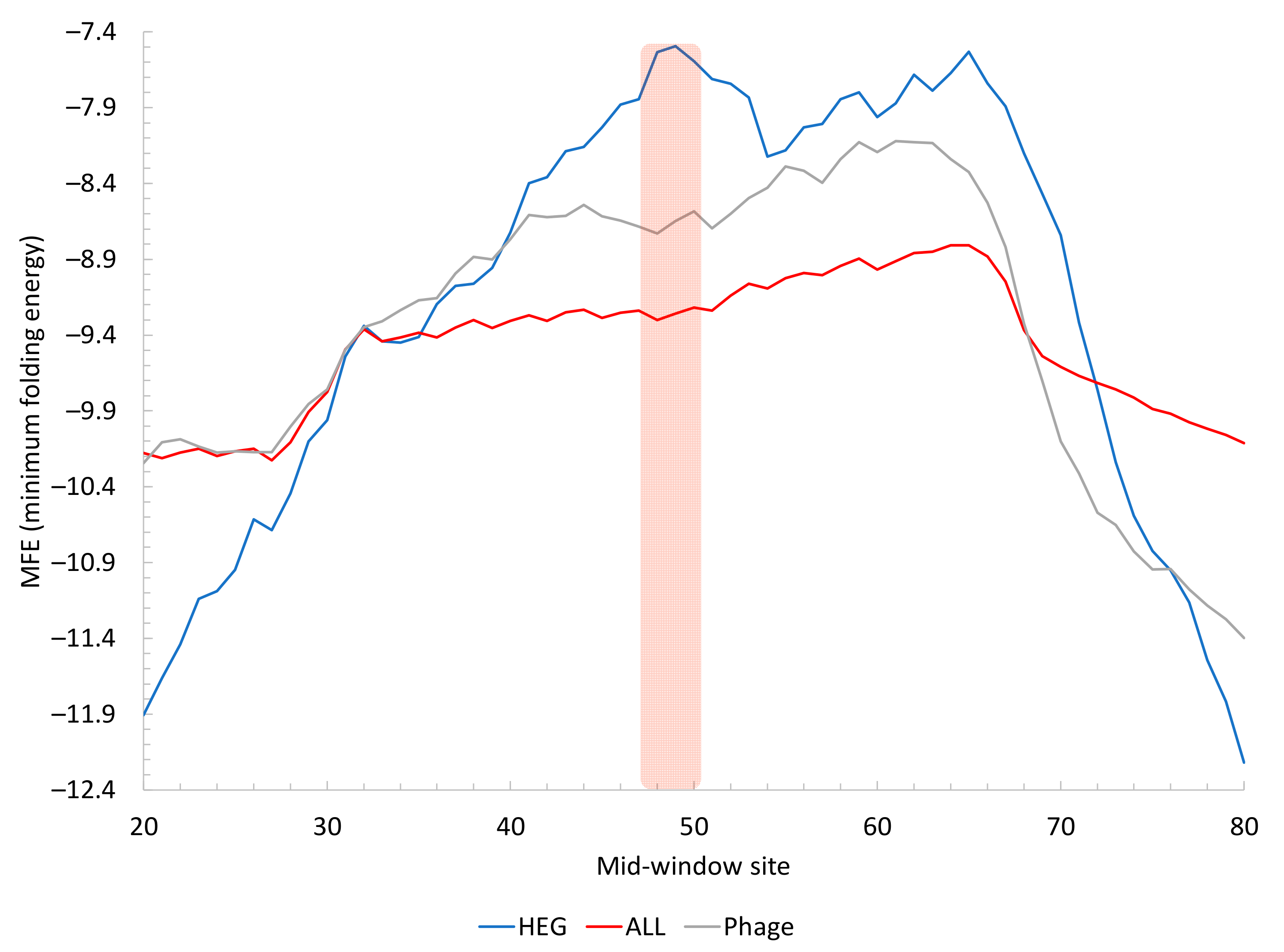
2.1. Translation Initiation
2.1.1. Start Codon Usage
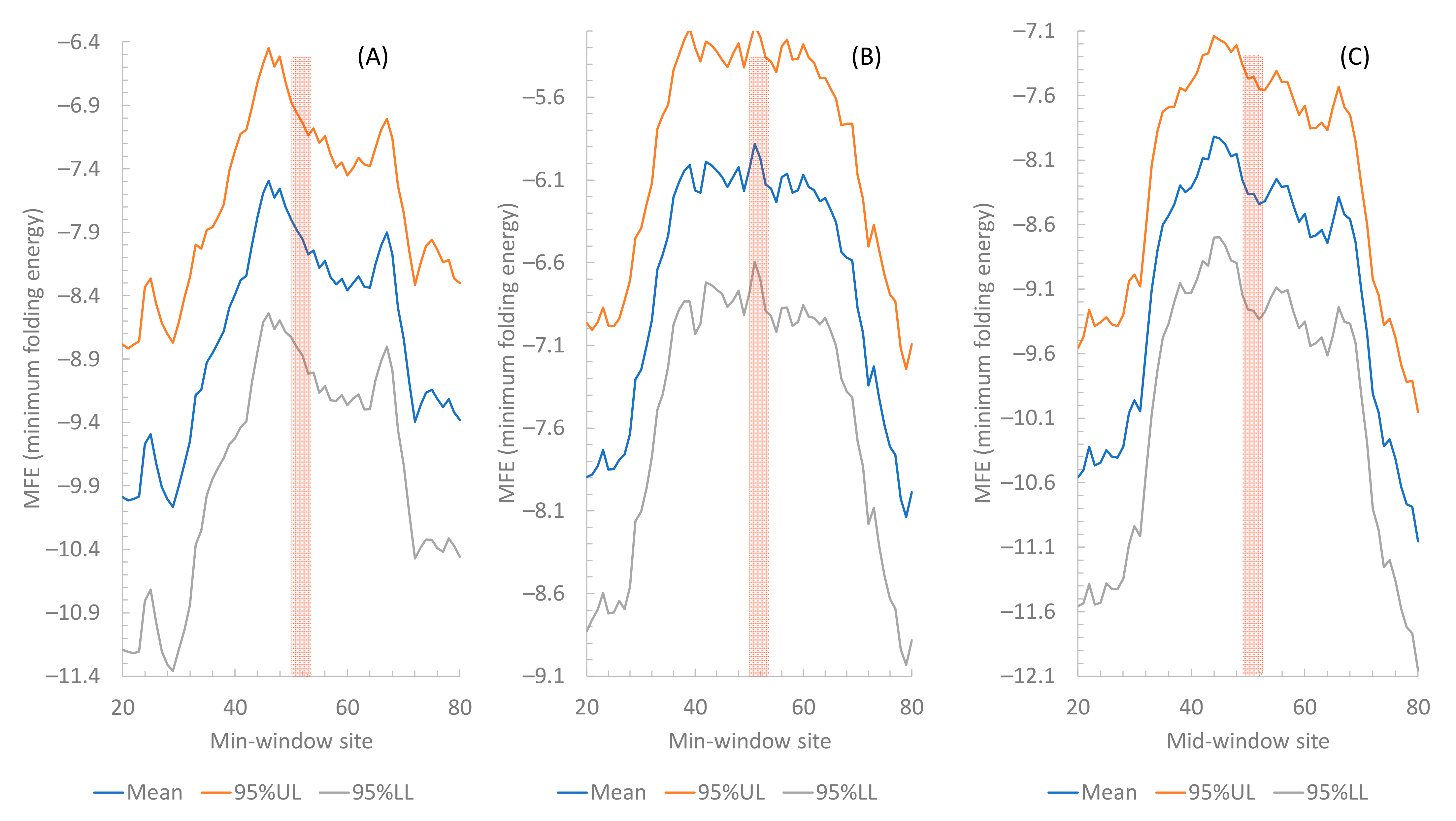
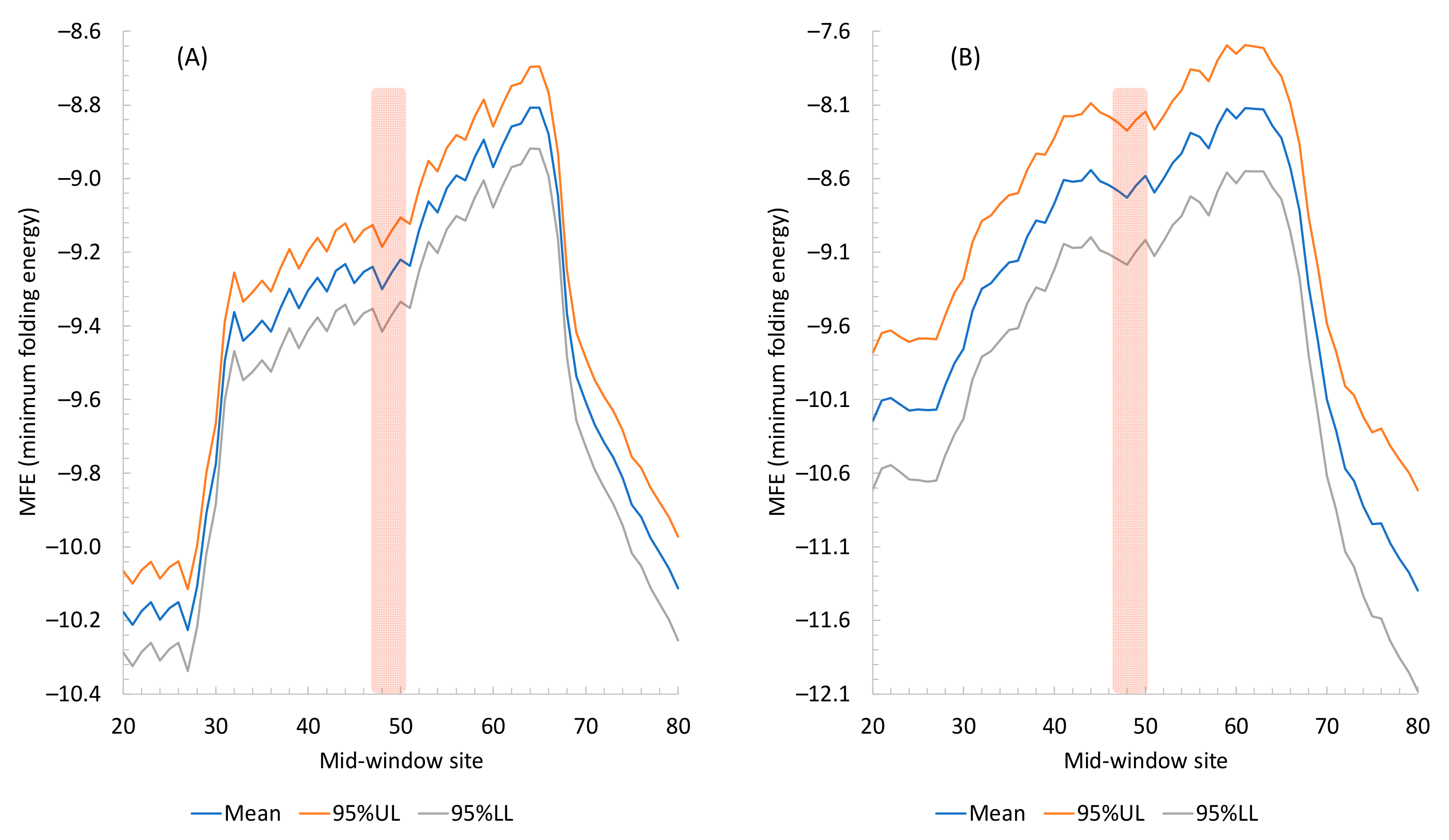
2.1.2. Secondary Structure Flanking the Start Codon
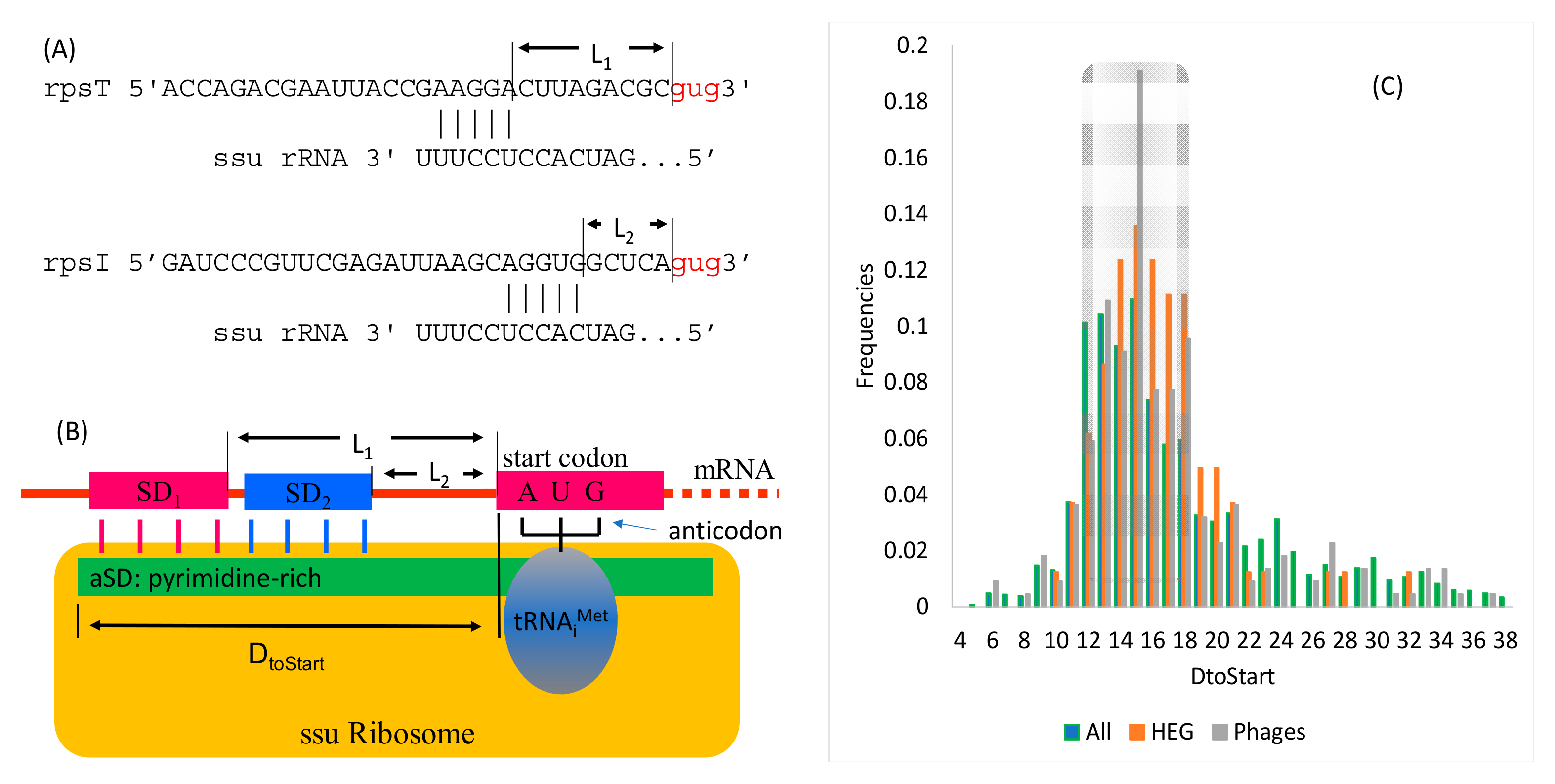
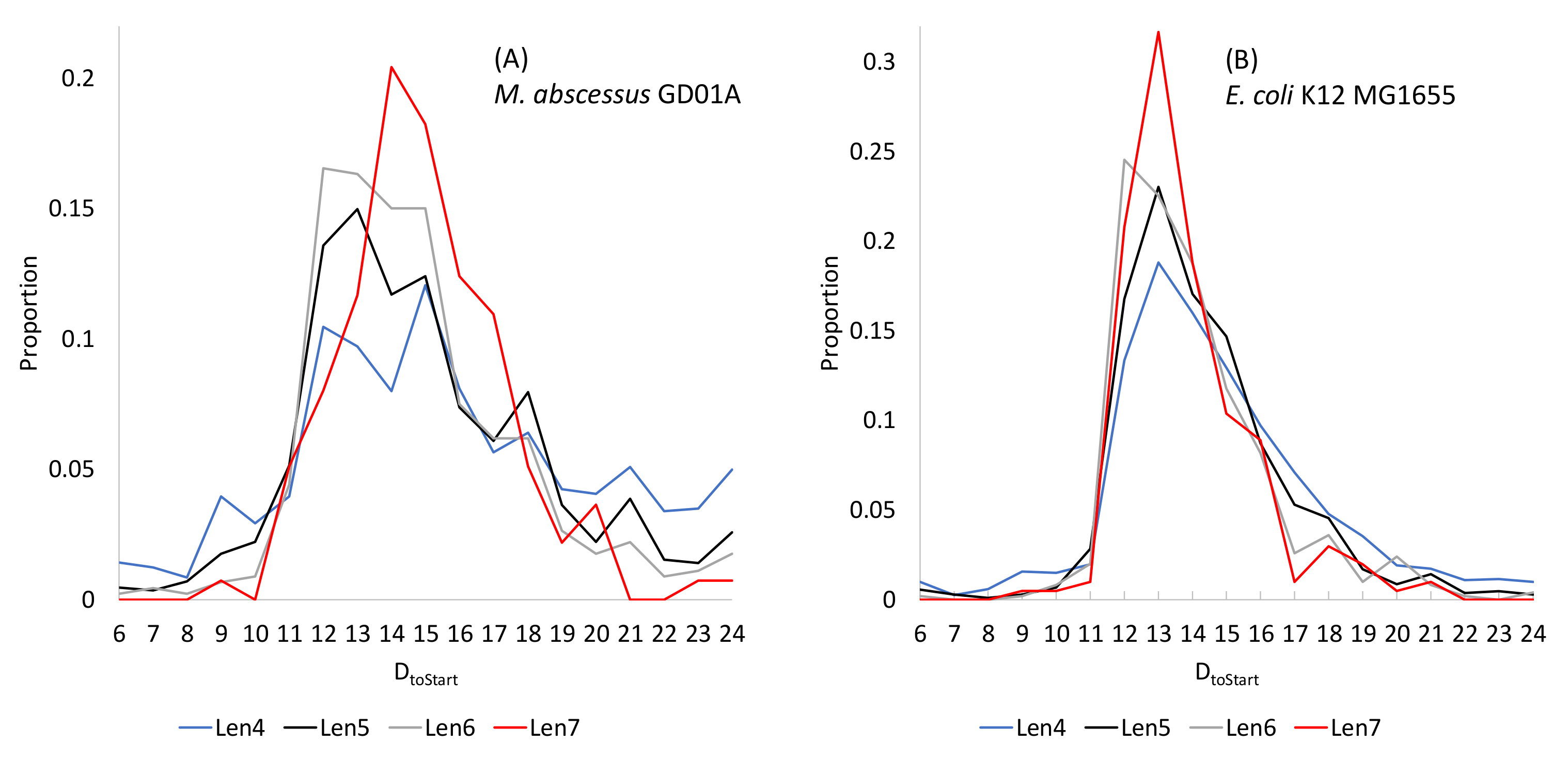
2.1.3. Shine-Dalgarno Paring
2.2. Translation Elongation and Codon-Anticodon Adaptation
2.3. Termination Signals
2.3.1. Identification of Optimal Stop Signals
2.3.2. Differential Nucleotide Preference at the +4 Site
2.3.3. Reduction in Secondary Structure Stability in Sequences Flanking the Stop Codon
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Capparelli, R.; Parlato, M.; Borriello, G.; Salvatore, P.; Iannelli, D. Experimental phage therapy against Staphylococcus aureus in mice. Antimicrob. Agents Chemother. 2007, 51, 2765–2773. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Brives, C.; Pourraz, J. Phage therapy as a potential solution in the fight against AMR: Obstacles and possible futures. Palgrave Commun. 2020, 6, 100. [Google Scholar] [CrossRef]
- Gurney, J.; Pradier, L.; Griffin, J.S.; Gougat-Barbera, C.; Chan, B.K.; Turner, P.E.; Kaltz, O.; Hochberg, M.E. Phage steering of antibiotic-resistance evolution in the bacterial pathogen, Pseudomonas aeruginosa. Evol. Med. Public Health 2020, 2020, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.P.; Chain, E. An enzyme from bacteria able to destroy penicillin. Rev. Infect. Dis. 1940, 10, 677–678. [Google Scholar] [CrossRef]
- Abraham, E.P.; Chain, E.; Fletcher, C.M.; Florey, H.W.; Gardner, A.D.; Heatley, N.G.; Jennings, M.A. Further observations on penicillin. Lancet 1941, 238, 177–189. [Google Scholar] [CrossRef]
- Jernigan, J.A.; Hatfield, K.M.; Wolford, H.; Nelson, R.E.; Olubajo, B.; Reddy, S.C.; McCarthy, N.; Paul, P.; McDonald, L.C.; Kallen, A.; et al. Multidrug-Resistant Bacterial Infections in U.S. Hospitalized Patients, 2012–2017. N. Engl. J. Med. 2020, 382, 1309–1319. [Google Scholar] [CrossRef]
- Meile, S.; Du, J.; Dunne, M.; Kilcher, S.; Loessner, M.J. Engineering therapeutic phages for enhanced antibacterial efficacy. Curr. Opin. Virol. 2022, 52, 182–191. [Google Scholar] [CrossRef]
- Ferry, T.; Kolenda, C.; Briot, T.; Souche, A.; Lustig, S.; Josse, J.; Batailler, C.; Pirot, F.; Medina, M.; Leboucher, G.; et al. Past and Future of Phage Therapy and Phage-Derived Proteins in Patients with Bone and Joint Infection. Viruses 2021, 13, 2414. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.D.; Alcaraz, M.; Dedrick, R.M.; Roquet-Banères, F.; Hamela, C.; Hatfull, G.F.; Kremer, L. Mycobacteriophage–antibiotic therapy promotes enhanced clearance of drug-resistant Mycobacterium abscessus. Dis. Model. Mech. 2021, 14, dmm049159. [Google Scholar] [CrossRef]
- Knezevic, P.; Aleksic Sabo, V. Combining bacteriophages with other antibacterial agents to combat bacteria. In Phage Therapy: A Practical Approach; Górski, A., Międzybrodzki, R., Borysowski, J., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 257–293. [Google Scholar]
- Gabard, J.; Jault, P. How to Achieve a Good Phage Therapy Clinical Trial? In Phage Therapy: A Practical Approach; Górski, A., Międzybrodzki, R., Borysowski, J., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 147–168. [Google Scholar]
- Tzipilevich, E.; Pollak-Fiyaksel, O.; Shraiteh, B.; Ben-Yehuda, S. Bacteria elicit a phage tolerance response subsequent to infection of their neighbors. EMBO J. 2022, 41, e109247. [Google Scholar] [CrossRef] [PubMed]
- García, R.; Latz, S.; Romero, J.; Higuera, G.; García, K.; Bastías, R. Bacteriophage Production Models: An Overview. Front. Microbiol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Krut, O.; Bekeredjian-Ding, I. Contribution of the Immune Response to Phage Therapy. J. Immunol. 2018, 200, 3037–3044. [Google Scholar] [CrossRef] [PubMed]
- Loh, B.; Gondil, V.S.; Manohar, P.; Khan, F.M.; Yang, H.; Leptihn, S. Encapsulation and Delivery of Therapeutic Phages. Appl. Environ. Microbiol. 2020, 87, e01979-20. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.D.; Silva, A.L.N.; Ribeiro, J.M.; Mascarenhas, D.P.A.; Quirino, G.F.S.; Santos, L.L.; Flavell, R.A.; Zamboni, D.S. AIM2 Engages Active but Unprocessed Caspase-1 to Induce Noncanonical Activation of the NLRP3 Inflammasome. Cell Rep. 2017, 20, 794–805. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Weaver, R.F. Molecular Biology, 5th ed.; McGraw-Hill: Boston, MA, USA, 2012. [Google Scholar]
- Xia, X. How optimized is the translational machinery in Escherichia coli, Salmonella typhimurium and Saccharomyces cerevisiae? Genetics 1998, 149, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Bioinformatics and translation initiation. In Bioinformatics and the Cell: Modern Computational Approaches in Genomics, Proteomics and Transcriptomics; Springer: Cham, Switzerland, 2018; pp. 173–195. [Google Scholar]
- Xia, X. Bioinformatics and translation termination in bacteria. In Bioinformatics and the Cell: Modern Computational Approaches in Genomics, Proteomics and Transcriptomics; Springer: Cham, Switzerland, 2018; pp. 239–254. [Google Scholar]
- Chithambaram, S.; Prabhakaran, R.; Xia, X. Differential Codon Adaptation between dsDNA and ssDNA Phages in Escherichia coli. Mol. Biol. Evol. 2014, 31, 1606–1617. [Google Scholar] [CrossRef] [PubMed]
- Chithambaram, S.; Prabhakaran, R.; Xia, X. The Effect of Mutation and Selection on Codon Adaptation in Escherichia coli Bacteriophage. Genetics 2014, 197, 301–315. [Google Scholar] [CrossRef]
- Prabhakaran, R.; Chithambaram, S.; Xia, X. Aeromonas phages encode tRNAs for their overused codons. Int. J. Comput. Biol. Drug Des. 2014, 7, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, R.; Chithambaram, S.; Xia, X. Escherichia coli and Staphylococcus phages: Effect of translation initiation efficiency on differential codon adaptation mediated by virulent and temperate lifestyles. J. Gen. Virol. 2015, 96, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Sezonov, G.; Joseleau-Petit, D.; D’Ari, R. Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 2007, 189, 8746–8749. [Google Scholar] [CrossRef]
- Cortes, M.A.; Nessar, R.; Singh, A.K. Laboratory maintenance of Mycobacterium abscessus. Curr. Protoc. Microbiol. 2010, 18, 10D.1.1–10D.1.12. [Google Scholar] [CrossRef]
- Van Weringh, A.; Ragonnet-Cronin, M.; Pranckeviciene, E.; Pavon-Eternod, M.; Kleiman, L.; Xia, X. HIV-1 Modulates the tRNA Pool to Improve Translation Efficiency. Mol. Biol. Evol. 2011, 28, 1827–1834. [Google Scholar] [CrossRef]
- Xia, X. A Major Controversy in Codon-Anticodon Adaptation Resolved by a New Codon Usage Index. Genetics 2015, 199, 573–579. [Google Scholar] [CrossRef]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-Sequence Determinants of Gene Expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef]
- Tuller, T.; Waldman, Y.Y.; Kupiec, M.; Ruppin, E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. USA 2010, 107, 3645–3650. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Optimizing Phage Translation Initiation. OBM Genet. 2019, 3, 16. [Google Scholar] [CrossRef]
- Gualerzi, C.O.; Pon, C.L. Initiation of mRNA translation in bacteria: Structural and dynamic aspects. Cell. Mol. Life Sci. CMLS 2015, 72, 4341–4367. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Kurland, C.G. Ram ribosomes are defective proofreaders. Mol. Gen. Genet. 1983, 191, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Bulmer, M. The effect of context on synonymous codon usage in genes with low codon usage bias. Nucleic Acids Res. 1990, 18, 2869–2873. [Google Scholar] [CrossRef]
- Bulmer, M. The selection-mutation-drift theory of synonymous codon usage. Genetics 1991, 129, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Liljenstrom, H.; von Heijne, G. Translation rate modification by preferential codon usage: Intragenic position effects. J. Theor. Biol. 1987, 124, 43–55. [Google Scholar] [CrossRef]
- Shine, J.; Dalgarno, L. The 3’-terminal sequence of Escherichia coli 16S ribosomal RNA: Complementarity to nonsense triplets and ribosome binding sites. Proc. Natl. Acad. Sci. USA 1974, 71, 1342–1346. [Google Scholar] [CrossRef]
- Shine, J.; Dalgarno, L. Identical 3’-terminal octanucleotide sequence in 18S ribosomal ribonucleic acid from different eukaryotes. A proposed role for this sequence in the recognition of terminator codons. Biochem. J. 1974, 141, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Shine, J.; Dalgarno, L. Determinant of cistron specificity in bacterial ribosomes. Nature 1975, 254, 34–38. [Google Scholar] [CrossRef]
- Hui, A.; de Boer, H.A. Specialized ribosome system: Preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli. Proc. Natl. Acad. Sci. USA 1987, 84, 4762–4766. [Google Scholar] [CrossRef] [PubMed]
- Steitz, J.A.; Jakes, K. How ribosomes select initiator regions in mRNA: Base pair formation between the 3’ terminus of 16S rRNA and the mRNA during initiation of protein synthesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 1975, 72, 4734–4738. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Weissmann, C. Inhibition of Qbeta RNA 70S ribosome initiation complex formation by an oligonucleotide complementary to the 3’ terminal region of E. coli 16S ribosomal RNA. Nature 1978, 275, 770–772. [Google Scholar] [CrossRef]
- Nakamoto, T. A unified view of the initiation of protein synthesis. Biochem. Biophys. Res. Commun. 2006, 341, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 1981, 146, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Bioinformatics and translation elongation. In Bioinformatics and the Cell: Modern Computational Approaches in Genomics, Proteomics and Transcriptomics; Springer: Cham, Switzerland, 2018; pp. 197–238. [Google Scholar]
- Ikemura, T. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J. Mol. Biol. 1982, 158, 573–597. [Google Scholar] [PubMed]
- Gouy, M.; Gautier, C. Codon usage in bacteria: Correlation with gene expressivity. Nucleic Acids Res. 1982, 10, 7055–7064. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.R. UGA nonsense mutations in Salmonella typhimurium. J. Bacteriol. 1970, 102, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.F.; Fan, D.P.; Brenner, S. A strong suppressor specific for UGA. Nature 1967, 214, 452–453. [Google Scholar] [CrossRef]
- Strigini, P.; Brickman, E. Analysis of specific misreading in Escherichia coli. J. Mol. Biol. 1973, 75, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Jones, D.S.; Khorana, H.G. A further study of misreading of codons induced by streptomycin and neomycin using ribopolynucleotides containing two nucleotides in alternating sequence as templates. J. Mol. Biol. 1966, 18, 48–57. [Google Scholar] [CrossRef]
- Ryden, S.M.; Isaksson, L.A. A temperature-sensitive mutant of Escherichia coli that shows enhanced misreading of UAG/A and increased efficiency for some tRNA nonsense suppressors. Mol. Gen. Genet. 1984, 193, 38–45. [Google Scholar] [CrossRef]
- Bossi, L. Context effects: Translation of UAG codon by suppressor tRNA is affected by the sequence following UAG in the message. J. Mol. Biol. 1983, 164, 73–87. [Google Scholar] [CrossRef]
- Bossi, L.; Roth, J.R. The influence of codon context on genetic code translation. Nature 1980, 286, 123. [Google Scholar] [CrossRef]
- Miller, J.H.; Albertini, A.M. Effects of surrounding sequence on the suppression of nonsense codons. J. Mol. Biol. 1983, 164, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, J.; Xia, X. Coevolution between Stop Codon Usage and Release Factors in Bacterial Species. Mol. Biol. Evol. 2016, 33, 2357–2367. [Google Scholar] [CrossRef]
- Geller, A.I.; Rich, A. A UGA termination suppression tRNATrp active in rabbit reticulocytes. Nature 1980, 283, 41–46. [Google Scholar] [CrossRef]
- Jorgensen, F.; Adamski, F.M.; Tate, W.P.; Kurland, C.G. Release factor-dependent false stops are infrequent in Escherichia coli. J. Mol. Biol. 1993, 230, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Parker, J. Errors and alternatives in reading the universal genetic code. Microbiol. Rev. 1989, 53, 273–298. [Google Scholar] [CrossRef]
- Tate, W.P.; Mansell, J.B.; Mannering, S.A.; Irvine, J.H.; Major, L.L.; Wilson, D.N. UGA: A dual signal for ‘stop’ and for recoding in protein synthesis. Biochemistry 1999, 64, 1342–1353. [Google Scholar]
- Cesar Sanchez, J.; Padron, G.; Santana, H.; Herrera, L. Elimination of an HuIFN alpha 2b readthrough species, produced in Escherichia coli, by replacing its natural translational stop signal. J. Biotechnol. 1998, 63, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.Y.; Hui, J.O.; Haniu, M.; Tsai, L.B. Analysis of translational termination of recombinant human methionyl-neurotrophin 3 in Escherichia coli. Biochem. Biophys. Res. Commun. 1995, 211, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Tate, W.P.; Brown, C.M. Translational termination: “Stop” for protein synthesis or “pause” for regulation of gene expression. Biochemistry 1992, 31, 2443–2450. [Google Scholar] [CrossRef]
- Tate, W.P.; Mannering, S.A. Three, four or more: The translational stop signal at length. Mol. Microbiol. 1996, 21, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xia, X. The Role of +4U as an Extended Translation Termination Signal in Bacteria. Genetics 2017, 205, 539–549. [Google Scholar] [CrossRef]
- Konecki, D.S.; Aune, K.C.; Tate, W.; Caskey, C.T. Characterization of reticulocyte release factor. J. Biol. Chem. 1977, 252, 4514–4520. [Google Scholar] [CrossRef] [PubMed]
- McCaughan, K.K.; Brown, C.M.; Dalphin, M.E.; Berry, M.J.; Tate, W.P. Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc. Natl. Acad. Sci. USA 1995, 92, 5431–5435. [Google Scholar] [CrossRef]
- Brown, C.M.; Stockwell, P.A.; Trotman, C.N.; Tate, W.P. Sequence analysis suggests that tetra-nucleotides signal the termination of protein synthesis in eukaryotes. Nucleic Acids Res. 1990, 18, 6339–6345. [Google Scholar] [CrossRef] [PubMed]
- Craigen, W.J.; Caskey, C.T. Expression of peptide chain release factor 2 requires high-efficiency frameshift. Nature 1986, 322, 273–275. [Google Scholar] [CrossRef]
- Craigen, W.J.; Cook, R.G.; Tate, W.P.; Caskey, C.T. Bacterial peptide chain release factors: Conserved primary structure and possible frameshift regulation of release factor 2. Proc. Natl. Acad. Sci. USA 1985, 82, 3616–3620. [Google Scholar] [CrossRef]
- Curran, J.F.; Yarus, M. Use of tRNA suppressors to probe regulation of Escherichia coli release factor 2. J. Mol. Biol. 1988, 203, 75–83. [Google Scholar] [CrossRef]
- Arisaka, F. Stoichiometry of protein interactions in bacteriophage tail assembly. In Stoichiometry and Research—The Importance of Quantity in Biomedicine; Innocenti, A., Ed.; IntechOpen: London, UK, 2012; pp. 243–258. [Google Scholar]
- Griffith, D.E.; Daley, C.L. Treatment of Mycobacterium abscessus Pulmonary Disease. Chest 2022, 161, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Andersson, G.E.; Sharp, P.M. Codon usage in the Mycobacterium tuberculosis complex. Microbiology 1996, 142, 915–925. [Google Scholar] [CrossRef]
- Belinky, F.; Rogozin, I.B.; Koonin, E.V. Selection on start codons in prokaryotes and potential compensatory nucleotide substitutions. Sci. Rep. 2017, 7, 12422. [Google Scholar] [CrossRef] [PubMed]
- Hartz, D.; McPheeters, D.S.; Gold, L. Influence of mRNA determinants on translation initiation in Escherichia coli. J. Mol. Biol. 1991, 218, 83–97. [Google Scholar] [CrossRef]
- Hecht, A.; Glasgow, J.; Jaschke, P.R.; Bawazer, L.A.; Munson, M.S.; Cochran, J.R.; Endy, D.; Salit, M. Measurements of translation initiation from all 64 codons in E. coli. Nucleic Acids Res. 2017, 45, 3615–3626. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.E.; Hauser, P.M.; Sharpe, R.G.; Errington, J. Bacillus subtilis cell cycle as studied by fluorescence microscopy: Constancy of cell length at initiation of DNA replication and evidence for active nucleoid partitioning. J. Bacteriol. 1998, 180, 547–555. [Google Scholar] [CrossRef]
- Shinedling, S.; Gayle, M.; Pribnow, D.; Gold, L. Mutations affecting translation of the bacteriophage T4 rIIB gene cloned in Escherichia coli. Mol. Gen. Genet. 1987, 207, 224–232. [Google Scholar] [CrossRef]
- Sun, S.; Kondabagil, K.; Gentz, P.M.; Rossmann, M.G.; Rao, V.B. The Structure of the ATPase that Powers DNA Packaging into Bacteriophage T4 Procapsids. Mol. Cell 2007, 25, 943–949. [Google Scholar] [CrossRef]
- Jofre, J.; Muniesa, M. Minimal Methods of Processing|Potential Use of Phages and Lysins. In Encyclopedia of Food Microbiology, 2nd ed.; Batt, C.A., Tortorello, M.L., Eds.; Academic Press: Oxford, UK, 2014; pp. 752–758. [Google Scholar]
- Vázquez, R.; García, E.; García, P. Phage Lysins for Fighting Bacterial Respiratory Infections: A New Generation of Antimicrobials. Front. Immunol. 2018, 9, 2252. [Google Scholar] [CrossRef] [PubMed]
- De Smit, M.H.; van Duin, J. Control of prokaryotic translational initiation by mRNA secondary structure. Prog. Nucleic Acid Res. Mol. Biol. 1990, 38, 1–35. [Google Scholar] [PubMed]
- Ikemura, T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: A proposal for a synonymous codon choice that is optimal for the E coli translational system. J. Mol. Biol. 1981, 151, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. An Improved Implementation of Codon Adaptation Index. Evol. Bioinform. 2007, 3, 53–58. [Google Scholar] [CrossRef]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35 Pt 4, 1550–1552. [Google Scholar] [CrossRef]
- Goz, E.; Mioduser, O.; Diament, A.; Tuller, T. Evidence of translation efficiency adaptation of the coding regions of the bacteriophage lambda. DNA Res. 2017, 24, 333–342. [Google Scholar] [CrossRef]
- Poole, E.S.; Brown, C.M.; Tate, W.P. The identity of the base following the stop codon determines the efficiency of in vivo translational termination in Escherichia coli. Embo. J. 1995, 14, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Beznoskova, P.; Gunisova, S.; Valasek, L.S. Rules of UGA-N decoding by near-cognate tRNAs and analysis of readthrough on short uORFs in yeast. RNA 2016, 22, 456–466. [Google Scholar] [CrossRef]
- Jungreis, I.; Chan, C.S.; Waterhouse, R.M.; Fields, G.; Lin, M.F.; Kellis, M. Evolutionary Dynamics of Abundant Stop Codon Readthrough. Mol. Biol. Evol. 2016, 33, 3108–3132. [Google Scholar] [CrossRef] [PubMed]
- Jungreis, I.; Lin, M.F.; Spokony, R.; Chan, C.S.; Negre, N.; Victorsen, A.; White, K.P.; Kellis, M. Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 2011, 21, 2096–2113. [Google Scholar] [CrossRef] [PubMed]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Translational readthrough potential of natural termination codons in eucaryotes--The impact of RNA sequence. RNA Biol. 2015, 12, 950–958. [Google Scholar] [CrossRef]
- Xia, X. Detailed Dissection and Critical Evaluation of the Pfizer/BioNTech and Moderna mRNA Vaccines. Vaccines 2021, 9, 734. [Google Scholar] [CrossRef]
- Brown, C.M.; Tate, W.P. Direct recognition of mRNA stop signals by Escherichia coli polypeptide chain release factor two. J. Biol. Chem. 1994, 269, 33164–33170. [Google Scholar] [CrossRef]
- Brown, C.M.; Stockwell, P.A.; Trotman, C.N.; Tate, W.P. The signal for the termination of protein synthesis in procaryotes. Nucleic Acids Res. 1990, 18, 2079–2086. [Google Scholar] [CrossRef]
- Pirnay, J.-P.; De Vos, D.; Verbeken, G.; Merabishvili, M.; Chanishvili, N.; Vaneechoutte, M.; Zizi, M.; Laire, G.; Lavigne, R.; Huys, I.; et al. The Phage Therapy Paradigm: Prêt-à-Porter or Sur-mesure? Pharm. Res. 2011, 28, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J. A bacteriophages journey through the human body. Immunol. Rev. 2017, 279, 106–122. [Google Scholar] [CrossRef] [PubMed]
- De Sordi, L.; Khanna, V.; Debarbieux, L. The Gut Microbiota Facilitates Drifts in the Genetic Diversity and Infectivity of Bacterial Viruses. Cell Host Microbe 2017, 22, 801–808.e803. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The "Known Unknown" of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Dills, M.; Young, M.J. The Human Gut Phage Community and Its Implications for Health and Disease. Viruses 2017, 9, 141. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Kaur, S.; Harjai, K.; Chhibber, S. Bacteriophage-aided intracellular killing of engulfed methicillin-resistant Staphylococcus aureus (MRSA) by murine macrophages. Appl. Microbiol. Biotechnol. 2014, 98, 4653–4661. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sun, L.; Wei, R.; Gao, Q.; He, T.; Xu, C.; Liu, X.; Wang, R. Intracellular Staphylococcus aureus Control by Virulent Bacteriophages within MAC-T Bovine Mammary Epithelial Cells. Antimicrob. Agents Chemother. 2017, 61, e01990-16. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Baker, K.; Padman, B.S.; Patwa, R.; Dunstan, R.A.; Weston, T.A.; Schlosser, K.; Bailey, B.; Lithgow, T.; Lazarou, M.; et al. Bacteriophage Transcytosis Provides a Mechanism To Cross Epithelial Cell Layers. mBio 2017, 8, e01874-17. [Google Scholar] [CrossRef] [PubMed]
- Tetz, G.; Tetz, V. Bacteriophages as New Human Viral Pathogens. Microorganisms 2018, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.S.; Chauhan, D.P.; Carrier, E. In utero detection of T7 phage after systemic administration to pregnant mice. Biotechniques 2004, 37, 81–83. [Google Scholar] [CrossRef]
- Tétart, F.; Repoila, F.; Monod, C.; Krisch, H.M. Bacteriophage T4 host range is expanded by duplications of a small domain of the tail fiber adhesin. J. Mol. Biol. 1996, 258, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, I.E.H.; Lee, H.; Allison, A.B.; Lopez-Astacio, R.; Goodman, L.B.; Oyesola, O.O.; Omobowale, O.; Fagbohun, O.; Dubovi, E.J.; Hafenstein, S.L.; et al. Limited Intrahost Diversity and Background Evolution Accompany 40 Years of Canine Parvovirus Host Adaptation and Spread. J. Virol. 2019, 94, 108760. [Google Scholar] [CrossRef] [PubMed]
- Flint, S.J.; Enquist, L.W.; Racaniello, V.R.; Skalka, A.M. Principles of Virology: Molecular Biology, Pathogenesis, and Control of Animal Viruses, 2nd ed.; ASM Press: Washington, DC, USA, 2004; p. 918. [Google Scholar]
- Gibson, D.G.; Glass, J.I.; Lartigue, C.; Noskov, V.N.; Chuang, R.Y.; Algire, M.A.; Benders, G.A.; Montague, M.G.; Ma, L.; Moodie, M.M.; et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010, 329, 52–56. [Google Scholar] [CrossRef]
- Becker, M.M.; Graham, R.L.; Donaldson, E.F.; Rockx, B.; Sims, A.C.; Sheahan, T.; Pickles, R.J.; Corti, D.; Johnston, R.E.; Baric, R.S.; et al. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 19944–19949. [Google Scholar] [CrossRef] [PubMed]
- Yount, B.; Curtis, K.M.; Fritz, E.A.; Hensley, L.E.; Jahrling, P.B.; Prentice, E.; Denison, M.R.; Geisbert, T.W.; Baric, R.S. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2003, 100, 12995–13000. [Google Scholar] [CrossRef] [PubMed]
- Scobey, T.; Yount, B.L.; Sims, A.C.; Donaldson, E.F.; Agnihothram, S.S.; Menachery, V.D.; Graham, R.L.; Swanstrom, J.; Bove, P.F.; Kim, J.D.; et al. Reverse genetics with a full-length infectious cDNA of the Middle East respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 16157–16162. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L., Jr.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Thiel, K. Old dogma, new tricks—21st Century phage therapy. Nat. Biotechnol. 2004, 22, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Mrazek, J.; Campbell, A.; Kaiser, D. Characterizations of highly expressed genes of four fast-growing bacteria. J. Bacteriol. 2001, 183, 5025–5040. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable (1) | E. coli | B. subtilis | M. abscessus | |||
|---|---|---|---|---|---|---|
| HEG (2) | REST (2) | HEG (2) | REST (2) | HEG (2) | REST (2) | |
| NAUG | 64 | 3848 | 58 | 3188 | 55 | 3078 |
| NNonAUG | 3 | 428 | 7 | 922 | 18 | 1556 |
| PAUG | 0.9552 | 0.8999 | 0.8923 | 0.7757 | 0.7534 | 0.6642 |
| PNonAUG | 0.0448 | 0.1001 | 0.1077 | 0.2243 | 0.2466 | 0.3358 |
| Start | HEG (1) | REST (2) | Muddy | BPs | ZoeJ |
|---|---|---|---|---|---|
| AUG | 55 (75.34) | 3078 (66.42) | 49 (69.01) | 55 (87.30) | 61 (66.30) |
| GUG | 16 (21.92) | 1388 (29.95) | 18 (25.35) | 8 (12.70) | 27 (29.35) |
| UUG | 2 (2.74) | 120 (2.59) | 4 (5.63) | 0 | 4 (4.35) |
| CUG | 0 | 22 (0.47) | 0 | 0 | 0 |
| AUA | 0 | 4 (0.09) | 0 | 0 | 0 |
| AUC | 0 | 16 (0.35) | 0 | 0 | 0 |
| AUU | 0 | 6 (0.13) | 0 | 0 | 0 |
| Sum | 73 | 4634 | 71 | 63 | 92 |
| Species (1) | Sequence | NNuc | A | C | G | T |
|---|---|---|---|---|---|---|
| Ma GD01A | Between | 367,972 | 0.2015 | 0.2987 | 0.2984 | 0.2013 |
| CS3 | 1,509,600 | 0.0919 | 0.417 | 0.3702 | 0.121 | |
| HEG-CS3 | 14,379 | 0.0438 | 0.4875 | 0.3656 | 0.1031 | |
| Ms MC2 155 | Between | 513,911 | 0.1928 | 0.3085 | 0.3071 | 0.1916 |
| CS3 | 2,148,887 | 0.0667 | 0.4747 | 0.385 | 0.0736 | |
| HEG-CS3 | 12,342 | 0.0488 | 0.50081 | 0.36072 | 0.0897 |
| Group | n | Mean | SE |
|---|---|---|---|
| HEG | 73 | 0.767922 | 0.006768 |
| REST | 4634 | 0.642809 | 0.000799 |
| Pseudogene | 63 | 0.547333 | 0.008181 |
| Phage BPs | 63 | 0.651445 | 0.007063 |
| Phage Muddy | 71 | 0.612486 | 0.009194 |
| Phage ZoeJ | 92 | 0.675267 | 0.006514 |
| Stop | HEG (1) | REST (2) | Muddy | BPs | ZoeJ |
|---|---|---|---|---|---|
| UAA | 16 (21.9) | 722 (15.6) | 17 (23.94) | 12 (19.05) | 17 (18.48) |
| UAG | 27 (37.0) | 1640 (35.4) | 14 (19.72) | 9 (14.29) | 14 (15.22) |
| UGA | 30 (41.1) | 2272 (49.0) | 40 (56.34) | 42 (66.67) | 61 (66.30) |
| Sum | 73 | 4634 | 71 | 63 | 92 |
| Genes | A | C | G | T | Sum |
|---|---|---|---|---|---|
| ALL (1) | |||||
| UAA | 126 (17.1) | 323 (43.8) | 165 (22.4) | 124 (16.8) | 738 |
| UAG | 295 (17.7) | 673 (40.4) | 413 (24.8) | 286 (17.2) | 1667 |
| UGA | 299 (13.0) | 840 (36.5) | 644 (28.0) | 519 (22.5) | 2302 |
| HEG (2) | A | C | G | T | |
| UAA | 3 (18.8) | 2 (12.5) | 7 (43.8) | 4 (25) | 16 |
| UAG | 0 (0) | 7 (25.9) | 9 (33.3) | 11 (40.7) | 27 |
| UGA | 1 (3.3) | 11 (36.7) | 4 (13.3) | 14 (46.7) | 30 |
| Phage (3) | A | C | G | T | |
| UAA | 5(10.9) | 17(37) | 14(30.4) | 10(21.7) | 46 |
| UAG | 5(13.5) | 13(35.1) | 16(43.2) | 3(8.1) | 37 |
| UGA | 13(9.1) | 50(35) | 43(30.1) | 37(25.9) | 143 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, X. Optimizing Protein Production in Therapeutic Phages against a Bacterial Pathogen, Mycobacterium abscessus. Drugs Drug Candidates 2023, 2, 189-209. https://doi.org/10.3390/ddc2010012
Xia X. Optimizing Protein Production in Therapeutic Phages against a Bacterial Pathogen, Mycobacterium abscessus. Drugs and Drug Candidates. 2023; 2(1):189-209. https://doi.org/10.3390/ddc2010012
Chicago/Turabian StyleXia, Xuhua. 2023. "Optimizing Protein Production in Therapeutic Phages against a Bacterial Pathogen, Mycobacterium abscessus" Drugs and Drug Candidates 2, no. 1: 189-209. https://doi.org/10.3390/ddc2010012
APA StyleXia, X. (2023). Optimizing Protein Production in Therapeutic Phages against a Bacterial Pathogen, Mycobacterium abscessus. Drugs and Drug Candidates, 2(1), 189-209. https://doi.org/10.3390/ddc2010012