Abstract
This review focuses on the pivotal roles of nuclear receptors (NRs) in driving endocrine resistance in prostate and breast cancers. In prostate cancer (PCa), androgen receptor (AR) amplification, mutations, and altered coactivator interactions sustain tumor growth under androgen deprivation therapy (ADT), leading to castration-resistant prostate cancer (CRPC). Orphan NRs like RORβ, TLX, and COUP-TFII further contribute to CRPC by regulating stemness and therapeutic resistance mechanisms. In breast cancer, NRs, including estrogen receptor alpha (ERα), androgen receptor (AR), glucocorticoid receptor (GR), and liver receptor homolog-1 (LRH-1), modulate estrogen signaling pathways and alternative survival mechanisms like PI3K/AKT/mTOR and NFκB, promoting resistance to endocrine therapies such as tamoxifen. Understanding these NR-mediated mechanisms is critical for developing targeted therapies to overcome endocrine resistance and improve patient outcomes in hormone-dependent cancers.
1. Introduction
Nuclear receptors (NRs) have emerged as pivotal players in the resistance to endocrine therapies in prostate and breast cancers. In prostate cancer (PCa), particularly in castration-resistant forms, the elevated expression levels of orphan nuclear receptors such as retinoic acid receptor-related orphan receptor β (RORβ), tailless X (TLX), chicken ovalbumin upstream promoter transcription factor II (COUP-TFII), nuclear receptor-related factor 1 (NURR1), and liver receptor homolog-1 (LRH-1) drives cancer stem cell properties and androgen insensitivity [1]. These receptors enhance cell proliferation and survival and activate alternative pathways to sustain tumor growth despite therapy, underscoring their potential as therapeutic targets in managing resistance [2,3,4]. Recent research highlights the role of specific orphan nuclear receptors in castration-resistant prostate cancer (CRPC). Notably, some nuclear receptors promote androgen biosynthesis and directly activate the androgen receptor (AR), enabling CRPC cells to thrive in low-androgen environments [5]. Similarly, in breast cancer, resistance to endocrine therapies targeting the estrogen receptor alpha (ERα) involves intricate mechanisms, including altered expression of nuclear receptors, such as the glucocorticoid receptor (GR), NURR1, LRH-1 and, COUP-TFII, and are positively associated with endocrine resistance in breast cancer [6,7,8,9]. This understanding of the roles of NRs in therapy resistance in both prostate and breast cancers emphasizes the need for further research to develop targeted therapies that can overcome resistance and improve patient outcomes.
2. Nuclear Receptors in Prostate Cancer and Castration Resistance
In humans, there are 48 transcription factors (TFs) within the nuclear receptor (NR) superfamily, which includes intracellular receptors that regulate a wide range of physiological functions [10]. Several endogenous mechanisms influence NR actions, such as ligand binding, post-translational modification, protein dimerization, nuclear translocation, and protein–protein interactions with activators and repressors [11,12]. NRs are classified into seven subfamilies based on sequence similarity: thyroid hormone receptors, retinoid X receptors, steroid receptors, nerve growth factor IB-like receptors, steroidogenic factors, germ cell nuclear factors, and various other receptors, including DSS-AHC Critical Region On The X Chromosome Protein 1 (DAX1) and Small Heterodimer Partner (SHP) [13]. NRs have become attractive targets for the development of pharmacological therapies due to the presence of distinctive ligand-binding sites. Endogenous ligands have not been described for a subgroup of NRs known as “orphans” and have recently been linked to various aspects with a wide range of diseases, such as rheumatoid arthritis (RA), inflammatory bowel disease (IBD) and cancer [4,14,15].
PCa is the second most commonly diagnosed cancer in men. The primary treatment for advanced tumors is androgen deprivation therapy (ADT), which aims to reduce serum testosterone levels, preventing androgen receptor (AR) activation and thereby inducing tumor regression [16]. Huggins and Hodges were the first to introduce the concept of PCa as an androgen-dependent disease; since continued androgen stimulation is necessary to maintain prostate function, the absence of these hormones causes the gland to become affected [17]. After an initial response to ADT, the tumor progresses in a large number of patients, leading to CRPC, a condition in which cancerous cells survive and proliferate despite ADT. Previously, it was believed that the disease was independent of the AR signaling pathway, leading to its classification as androgen-independent cancer [18,19]. However, recent years have shown that most cases of CRPC rely on the AR signaling pathway. The main mechanisms that the AR uses to resist castration include AR amplification, AR mutations (promiscuity), splicing variants, and activation [2,17]. The use of AR antagonists has led to an increase in more aggressive variants of PCa, such as those with low AR expression or lacking AR altogether. Additionally, certain AR-independent tumors, which are unresponsive to ADT, may arise due to the clonal selection of cells with low or absent AR expression, or through phenotypic transformation into neuroendocrine-like cells [20].
Several studies have demonstrated that AR gene amplification occurs in approximately 30% of PCa patients treated with hormone suppression therapy, and significant amplification of AR has been found in CRPC cell lines, ranging from 30% to 80%. Amplification of the AR gene can increase AR levels, enhancing transcription, mRNA stability, protein stability, and nuclear localization despite ADT and low androgen levels persist in the body [21,22]. Specific PCa cells adapt to become sensitive to these reduced androgen levels in this environment. Mutations in the AR gene allow the receptor to be activated by non-androgen molecules [23]. For example, substitutions such as threonine to alanine at codon 877 (T877A) and histidine to tyrosine at codon 874 (H874Y) enable AR activation by various steroids like progesterone and hydrocortisone [24]. Mutations in AR have also demonstrated the importance of noncanonical Wnt signaling in regulating androgen-dependent tumor growth in PCa. In a study involving mice with a point mutation (ARpe-T877A/Y), the animals developed hypertrophic prostates that responded to both an androgen antagonist and estrogen, although they did not develop prostatic tumors. However, in transgenic mouse models of PCa, the introduction of the AR T877A mutation significantly increased both the onset of prostatic tumorigenesis and tumor growth, suggesting that this pathway stimulates the development of prostatic tumors with AR hyperfunction [25]. Also, AR mutation where valine is replaced by leucine at codon 89 (V89L) leads to increased 5α-reductase levels, raising DHT levels despite low testosterone [26].
More than 150 different co-activators and co-repressor molecules regulate AR transcriptional activity in PCa [27]. FKBP51 is an AR target gene and coactivator found to be upregulated in recurrent LAPC-4 tumors in castrated mice. This co-activator promotes the formation of a superchaperone complex by regulating the recruitment of p23 to ATP-bound Hsp90. This helps maintain AR in a ligand-binding conformation, thus enhancing transcriptional activity and androgen-stimulated growth [28]. Other co-activators related to AR possess acetyltransferase activity, enhancing AR-induced transcription by promoting the assembly of complexes between AR-associated enhancers/promoters and transcription start sites of AR target genes that include steroid receptor co-activators such as SRC-1, SRC-2 (TIF-2, GRIP-1, NcoA2), and SRC-3 [3]. The next generation of antiandrogens approved by the FDA, such as enzalutamide, apalutamide, and darolutamide, has been used to treat CRPC and improve the prognosis of patients with advanced PCa [29]. Enzalutamide, darolutamide, and apalutamide exhibit robust antagonistic effects against the AR [30]. Their common mechanism of action involves inhibiting AR nuclear translocation, AR-mediated transcription, and coactivator recruitment [31,32,33]. The result of impeding the influence of androgens is a decrease in PCa cell proliferation and tumor size [34].
Despite these advances, drug resistance still occurs. Current evidence shows that the AR missense mutation F877L is linked to apalutamide resistance in preclinical models and apalutamide-treated patients [35]. Additionally, chronic treatment with apalutamide in C4-2B cells has been shown to activate the steroid hormone biosynthesis pathway and increase AKR1C3 expression [36]. Recently, a shared resistance mechanism for apalutamide, enzalutamide, and darolutamide has been identified, which is related to the AKR1C3/AR-V7 axis [36].
AR-V7 is described as an AR variant that can activate distinct oncogenic transcriptional programs and confer resistance to enzalutamide. On the other hand, AKR1C3 is known to catalyze androgen synthesis, which is activated in enzalutamide-resistant PCa cells [37]. Previous studies have demonstrated that AKR1C3 can bind with AR-V7 to confer resistance to enzalutamide in advanced PCa [38]. However, the knockdown of the AKR1C3/AR-V7 axis has the opposite effect, resensitizing cells to apalutamide and darolutamide treatment [36].
Other Nuclear Receptors Associated with Prostate Cancer Castration Resistance
In recent decades, nuclear receptors have been demonstrated to drive resistance to castration. Notably, models of CRPC xenografts and spheroids exhibit distinct patterns of nuclear receptor expression. Analysis by Zhu Wang and colleagues identified five key orphan nuclear receptors—RORβ, TLX, COUP-TFII, NURR1, and LRH-1—with positively regulated mRNA and protein levels in both models. Overexpression of these NRs increased cancer stem cell markers and spheroid formation, suggesting their involvement in CRPC growth (Figure 1) [1].
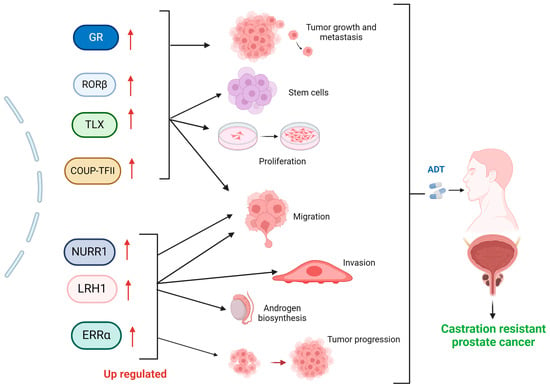
Figure 1.
Impact of elevated nuclear receptor expression on the Progression of Castration-Resistant Prostate Cancer (CRPC). Upregulation (up red arrows) of nuclear receptors and their role in CRPC development. These receptors contribute to key processes such as tumor growth and metastasis (GR), maintenance of stem cell populations, cellular proliferation and migration (RORβ, TLX, COUP-TFII), migration, invasion, androgen biosynthesis (NURR1, LRH1), and tumor progression (ERRα), ultimately leading to resistance against androgen deprivation therapy (ADT). Each receptor activates (black arrows) specific pathways that enhance tumor aggressiveness and resistance mechanisms, highlighting their potential as therapeutic targets for CRPC. Created with BioRender.com (accessed on 1 June 2024).
The retinoic acid receptor-related orphan receptors (RORs) are a subfamily of the thyroid hormone receptor within the nuclear receptors (NR) superfamily. Comprising RORα (NR1F1), RORβ (NR1F2), and RORγ (NR1F3), RORs are homologous to retinoic acid receptors (RARs) and retinoid X receptors (RXRs) [39,40]. RORβ is mainly involved in circadian rhythms, stem cell proliferation, and differentiation [41,42]. RORβ was found to be upregulated in clinical PCa samples compared to normal prostate gland tissues. This upregulation was also observed in PCa cell line-derived prostatospheroids and castration-relapse VCaP-CRPC xenografts, as confirmed by qRT-PCR and immunoblot analyses [1]. Conversely, TLX (NR2E1) is another orphan NR belonging to the nuclear receptor superfamily. TLX is essentially expressed in adult neural stem cells to regulate their proliferation. Removal of TLX from the adult mouse brain decreases stem cell proliferation [43]. Concerning CPRC, models such as xenografts and VCaP and LNCaP cell lines demonstrate that TLX is upregulated in CRPC and contributes to androgen insensitivity by promoting cancer stem cell-like properties and enhancing resistance to androgen deprivation therapy. TLX achieves this through regulating stem cell proliferation and maintaining undifferentiated states [44]. COUP-TFs are highly conserved nuclear orphan receptors expressed in the mouse embryonic stomach mesenchyme and play a significant role in endothelial cell growth and regulation of the Notch signaling pathway [45,46]. COUP-TFII shows a notable upregulation in both PCa cell line-derived prostatospheroids and VCaP-CRPC xenografts. These 3D-cultured prostatospheroids, derived from both AR-positive or AR-negative PCa cell lines and post-castration VCaP-CRPC xenografts, contained increased populations of prostate cancer stem cells (PCSCs), specifically CD44+/CD133+ cells. The common upregulation of COUP-TFII in these models underscores its potential role in promoting the survival and proliferation of PCSCs in CRPC [1].
Another NR intimately related to stem cells is NURR1, a member of the nuclear receptor superfamily of transcription factors, which is primarily expressed in the central nervous system, particularly in developing and mature dopaminergic neurons. It has been found to influence proliferation, migration, invasion, and apoptosis in human PCa cells [47,48]. NURR1 promotes stemness and epithelial-mesenchymal transition in PCa by targeting the Wnt/β-catenin signaling pathway. NURR1 was upregulated in PCa cell lines, stem-like cells, and CRPC xenografts. It promotes cancer stemness and epithelial-mesenchymal transition (EMT) by activating the Wnt/β-catenin signaling pathway. The research highlights that targeting NURR1 could be a potential therapeutic strategy for advanced PCa [49].
In CRPC, the retinoic acid receptor-related orphan receptor gamma (RORγ, also known as RORC) plays a crucial role in regulating the expression of the AR and its variant AR-V7. This regulation is significant as AR and AR-V7 are key drivers in the progression of CRPC, which is resistant to conventional hormone therapies targeting androgen signaling. Research has shown that targeting RORγ can modulate AR signaling pathways, thus offering a potential therapeutic approach for managing CRPC [50]. Additionally, the development of specific ligands that suppress CRPC through the inhibition of RORγ highlights a promising area for therapeutic intervention [51].
LRH-1, belonging to the nuclear receptor NR5A (Ftz-F1) subfamily, is found in tissues originating from the endoderm [52]. It is an orphan nuclear receptor that shows increased expression in various cancers like breast, colon, and pancreas [53,54,55]. In PCa, LRH-1 serves as a potent transcription factor, aiding in the development of CRPC [56]. The mechanisms underlying LRH-1 resistance to castration are intertwined with the upregulation of enzymes such as CYP11A1 and CYP17A1, which is pivotal for androgen biosynthesis. Additionally, LRH-1 directly activates the AR, enabling CRPC cells to thrive and proliferate without external androgens (Figure 1) [5].
Another receptor implicated in CRPC resistance, such as estrogen-related receptor alpha (ERRα), is a nuclear receptor encoded by the ESRRA gene in humans. ERRα belongs to a group of 48 structurally related, ligand-activated transcription factors and shares significant structural similarities with the ligand-dependent estrogen receptors (ERα and ERβ). Still, it does not bind estrogens [57,58,59]. In PCa, ERRα is upregulated and is implicated as a negative prognostic marker for the disease [60] (Figure 1). Interestingly, in xenograft models of CRPC, ERRα expression is upregulated in metastatic CRPC. The mechanism described involves two androgen synthesis enzymes, CYP11A1 and AKR1C3, which enhance the intraprostatic production of dihydrotestosterone (DHT) and activation of AR signaling in PCa cells [61]. Additionally, in VCaP PCa cell lines, ERRα has been reported to induce promoter–enhancer DNA looping and activate TMPRSS2:ERG fusion genes regulated by the AR, promoting castration resistance through the AKT pathway [62].
The glucocorticoid receptors (GR), encoded by the NR3C1 gene, belong to the subfamily of steroid hormone receptors and serve as the receptor for cortisol and other glucocorticoids [63]. The analysis of gene expression in a mouse model with patient-derived xenografts (PDX) of CRPC and samples from human PCa metastases reveals that the increase in GR activation is accompanied by a significant rise in AMPc/PKA pathway activation [64]. Interestingly, it has been demonstrated that CRPC has higher expression of the GR, along with AMPc/PKA activation [65,66]. Further research has indicated that in LAPC4 and CWR-22Rv1 PCa cells, GR activation through an MDV3100 agonist increased the secretion of prostate-specific antigen (PSA) and induced the expression of SGKI and MKP1/DUSP genes. Conversely, cell viability decreased with treatment using the GR antagonist CORT 122928 (Corcept Therapeutics, Merlo Park, CA, USA). In xenografts, the use of GR siRNAs (Open Biosystems, Huntsville, AL, USA) delayed the formation of castration-resistant tumors, thereby substantiating the role of GR in GR-mediated CRPC progression (Figure 1) [67].
3. Role of Nuclear Receptors in Endocrine Resistance of Breast Cancer
Approximately 70% of breast carcinomas express the α-estrogen receptor (ERα), classifying them as luminal breast cancers. Endocrine therapy is the primary treatment for most ERα patients, typically involving anti-estrogens like tamoxifen (TAM), which blocks ERα transcriptional activity by directly binding to it [68]. However, endocrine therapy resistance is a significant challenge, driven by various factors such as alterations in ERa signaling, interactions between the growth factor receptor (GFR) network, activation of the PI3K/AKT/mTOR pathway (including PTEN inactivation), and induction of NFκB signaling [69].
The vitamin D receptor (VDR) is a nuclear receptor that regulates gene expression, and it plays a crucial role in various cellular processes, including cell proliferation, differentiation, apoptosis, and immune response modulation. Impaired vitamin D and VDR signaling may lead to abnormal mammary gland development, potentially contributing to early-onset breast cancer. The VDR signaling can protect against breast cancer by regulating long noncoding RNAs, which are involved in gene expression regulation [70]. A study of 718 invasive breast tumors found that high VDR expression is associated with favorable prognostic factors and lower breast cancer mortality, making it a positive prognostic factor [71].
Peroxisome proliferator-activated receptors (PPARs) regulate cellular functions such as differentiation, proliferation, metabolism, and apoptosis, all of which are involved in tumor formation. The role of PPARs in breast cancer is complex, with some studies suggesting they may act as either tumor promoters or suppressors [72]. Specifically, PPARγ is highlighted as a potential tumor suppressor, with preclinical studies showing that natural and synthetic PPARγ ligands can modulate tumor cell signaling and inhibit breast tumor growth in both in vitro and in vivo models [73].
The AR is a crucial driver of tumor growth in PCa, and recent research highlights its significant role in breast cancer development. Although its role is still controversial, it has been found that AR expression is associated with improved breast cancer-specific survival and predicted response to endocrine therapy [74,75]. In breast cancer, NR associated with CRPC shows dual effects in endocrine therapy resistance.
3.1. Variable Role of the Glucocorticoid Receptor in Breast Cancer Endocrine Resistance
The role of the GR varies depending on the molecular subtype of breast cancer. In luminal breast cancer, GR expression is linked to a better prognosis and reduced proliferation. Conversely, in triple-negative breast cancer (TNBC), GR expression promotes migration, chemotherapy resistance, and cell survival [76]. The expression of GR according to the molecular subtypes PAM50 of breast cancer exhibits high levels of GR in luminal A breast cancer and is associated with favorable patient outcomes. Interestingly, Dexamethasone, a synthetic glucocorticoid, reduced viability in models derived from luminal A tumors. At the genomic level, the use of chip-seq data revealed that MCF7 cells treated with dexamethasone show an overlap of ER and GR in different genes. Specifically, it was observed that the ZBTB16 gene has bindings for ER and GR at intronic sites and was identified as a potential driver of Dexa-induced growth suppression [77]. ZBTB1 functions as a tumor suppressor that is significantly downregulated in breast tumors compared to normal tissues. Conversely, the overexpression of ZBTB1 sensitizes cells resistant to tamoxifen through HER2 suppression in MCF-7/TamR cells [78]. In another study, the application of GR agonists in MCF7 cells demonstrated that the co-activation of ER and GR reduces the activation of proliferative genes, particularly the expression of cyclin-dependent kinases (CDKs), CDK1, CDK2, and CDK6 genes. Additionally, the use of GR agonists also reduced the growth of xenograft tumors from MCF7 cells [79].
Importantly, the overactivation of CDKs, such as CDK6, contributes to tumor progression by promoting cell cycle progression through the inhibition of the retinoblastoma (Rb) protein [80]. Since CDKs play a crucial role in initiating the cell cycle in cancers, CDK4/6 inhibitors have been developed as important cancer therapeutics for hormone receptor-expressing breast cancer. These inhibitors are designed to halt or slow cancer cell growth by arresting the cell cycle in the G1 phase [81]. INX-315, a highly selective CDK2 inhibitor, has shown promise in patient-derived xenografts and murine models, where it induces cell cycle arrest and cellular senescence-like states [82]. Coincidentally, resistance to CDK4/6 inhibitors also promotes resistance to ER inhibitors. For example, chronic exposure of MCF7 cells to a CDK4/6 inhibitor (LY2835219) induces resistance to fulvestrant. Additionally, MCF7 cells resistant to CDK inhibitors also exhibit elevated IC50 concentrations in relation to 4-hydroxytamoxifen, with an IC50 of 2030 nM compared to an IC50 of 113.2 nM [83].
In endocrine resistance, a novel model of endoxifen-resistant luminal breast cancer cells, MCF7 and T47D, GR activation via Dexamethasone significantly reduces the proliferation rates by 50–60% [84]. Furthermore, dexamethasone demonstrated a synergistic effect with endoxifen in suppressing cell proliferation, suggesting that this combined treatment could be an effective therapy for endocrine-resistant disease [6] (Figure 2A).
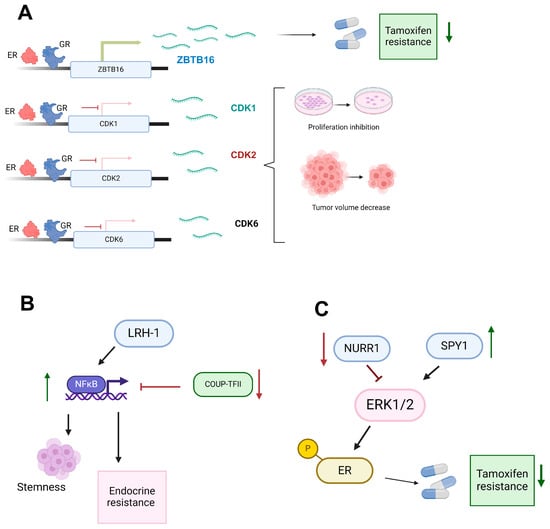
Figure 2.
Modulation of nuclear receptor functions in breast cancer endocrine resistance. (A) The glucocorticoid receptor (GR) and estrogen receptor (ER) synergistically modulate gene expression, enhancing ZBTB16 expression (green arrow) and contributing to a reduction in tamoxifen resistance (down green arrow). In contrast, GR and ER negatively regulate CDKs (CDK1, CDK2, and CDK6) (red inhibition arrow), leading to inhibition of proliferation and a decrease in tumor volume. (B) NFκB activation (up green arrow) by LRH-1 and inactivation (red inhibition arrow) by COUP-TFII promote stemness and endocrine resistance, suggesting a regulatory role in mechanisms of endocrine resistance in breast cancer. (C) NURR1 downregulation (down red arrow) and SPY1 upregulation (result in the activation of the ERK1/2 signaling pathway, which in turn activates (black arrow) the estrogen receptor (ER). The activation of ER is also associated with a decrease in tamoxifen resistance (down green arrow). Created with BioRender.com (accessed on 1 June 2024).
3.2. UP-TFII’s Dual Role Modulating Angiogenesis, Metastasis, and Endocrine Therapy Resistance through NFκB Pathway Regulation in Cancer
Evidence shows that COUP-TFII plays dual roles as an oncogene by regulating angiogenesis and lymphangiogenesis. In contrast, as a suppressor, it is related to inhibiting tumor metastasis and leads to favorable therapeutic outcomes in cancer therapy [85]. In therapy resistance, COUP-TFII is capable of regulating the NFκB family of dimeric transcription factors that play crucial roles in immune and inflammatory responses, and it has recently been found to promote endocrine resistance [9]. Regarding NFκB, it has been shown that in MCF-7, T47D, ZR75-1, and BT474 cells, exposure to tamoxifen, both in vitro and in vivo, results in a tamoxifen-tolerant population with increased NFκB pathway activity and enhanced stemness characteristics [86]. Specifically, endocrine-resistant LCC9 cells with low endogenous COUP-TFII show approximately five times higher basal NFκB activity than parental endocrine-sensitive MCF-7 breast cancer cells. Overexpression of COUP-TFII in LCC9 cells reduces NFκB expression and NFκB target gene expression. An inverse correlation between COUP-TFII and NFκB was confirmed in breast cancer patient samples [9]. Notably, the low expression of COUP-TFII in cells with endocrine therapy resistance is associated with higher methylation in the first exon in LCC2 and LCC9 endocrine-resistant breast cancer cells compared to MCF-7 cells. Additionally, treatment of LCC2 and LCC9 with 5-aza-2′-deoxycytidine (AZA), a cytidine analog that disrupts the interaction between DNA and DNA methyltransferase (DNMT), increased COUP-TFII expression, and the epigenetic changes mediated the decrease in COUP-TFII [87] (Figure 2B).
3.3. The Oncogenic Role of LRH-1 in Endocrine-Sensitive and Endocrine-Resistant Breast Cancer: Mechanisms and Therapeutic Implications
LRH-1 plays a critical role in both endocrine-sensitive and endocrine-resistant breast cancer. LRH-1 is a nuclear receptor involved in regulating genes associated with cell proliferation, metabolism, and steroidogenesis [88]. Its expression promotes the transcriptional activity of ER target genes, thereby enhancing estrogen signaling pathways. In endocrine-resistant breast cancer, LRH-1 maintains cell proliferation and survival by activating alternative signaling pathways that compensate for the lack of estrogen, such as the PI3K/AKT and NFκB pathways [89]. Interestingly, the nuclear receptors LRH-1 exhibit oncogenic behavior similar to that observed in PCa. LRH-1 plays a critical role in both endocrine-sensitive and endocrine-resistant breast cancer. LRH-1 expression is upregulated in endocrine-resistant breast cancer cell MCF-7/LCC9 and castration resistance xenografts. Its expression promotes the transcriptional activity of ER target genes, thereby enhancing estrogen signaling pathways [8]. ChIP-seq analysis demonstrated that LRH-1 binds to estrogen response elements in ERα-target genes, such as the TFF1/pS2 gene. Overexpression of LRH-1 enhances ERα recruitment to these binding sites, whereas knockdown reduces it. This indicates that LRH-1 is essential in regulating ERα target genes in breast cancer cells. Furthermore, the study reveals that LRH-1 and ERα co-binding to estrogen response elements is a critical mechanism for controlling the expression of estrogen-responsive genes [90] (Figure 2B).
3.4. Divergent Roles of NURR1 in Breast Cancer Progression: Implications for Endocrine Therapy and ERK Signaling Modulation
On the other hand, the NURR1 receptor appears to have divergent roles in cancer progression. In breast biopsy specimens, NURR1 showed high expression in non-cancerous breast epithelium. However, when comparing its expression among molecular subtypes, basal-like cancer displayed lower levels of NURR1 mRNA expression, while luminal A cancer exhibited the highest expression compared to all other subtypes. Additionally, findings unveiled a positive association between NURR1 expression and enhanced 5-year relapse-free survival among patients treated with endocrine therapy within the luminal A breast cancer subgroup [7] (Figure 2C).
Interestingly, the alteration of signaling pathways such as ERK contributed to endocrine resistance, specifically through the protein SPY1. In MCF7 cells, SPY1-mediated activation leads to the phosphorylation of ERK1/2, and subsequently, ERK1/2 can phosphorylate ER at S118, changing the sensitivity to tamoxifen [91]. Within the ERK signaling mechanism, the NURR1 receptor can promote its inhibition [92]. Specifically, western blot assays revealed that NURR1 improved responsiveness to tamoxifen by suppressing ERK signaling, specifically the p-SRC, p-MEK1/2, and p-ERK1/2 proteins in estrogen receptor-positive (ER+) breast cancer, suggesting that the NURR1/ERK signaling axis modulates tamoxifen resistance. Also, NURR1 gene expression is repressed in tamoxifen-resistant MCF7 (TamR) cells compared to MCF7 cells. Interestingly, overexpression of NURR1 in TamR cells promotes the restoration of sensitivity to tamoxifen and suppresses cell proliferation, migration, and invasion [92] (Figure 2C).
4. Conclusions
The intricate roles of NRs in the therapy resistance of prostate and breast cancers highlight their multifaceted contributions to disease progression and therapeutic challenges. This review explored how NRs influence cancer cell behavior and treatment outcomes, emphasizing their potential as therapeutic targets and prognostic indicators. PCa remains a leading cause of cancer-related mortality in men, with ADT being the cornerstone of treatment for advanced disease [16,93]. Despite initial responses, many patients progress to CRPC, necessitating deeper exploration into the molecular mechanisms driving resistance. NRs play pivotal roles in CRPC through mechanisms to sustain AR signaling under low androgen conditions, promoting tumor survival and growth [5,56].
Additionally, orphan NRs like RORβ, TLX, LRH-1, and COUP-TFII have emerged as potential regulators of CRPC, underscoring their potential as novel therapeutic targets [1,44,56]. In luminal breast cancers, endocrine therapy is mediated by complex interactions involving NRs such as GR, NURR1, COUP-TFII, and LRH-1 [6,7,8,87]. These NRs modulate estrogen signaling pathways and promote alternative survival pathways like PI3K/AKT/mTOR and NFkB to contribute to endocrine resistance [9]. Understanding the crosstalk between NRs and these pathways is crucial for developing effective therapeutic strategies that can overcome or prevent endocrine resistance in breast cancer.
Author Contributions
Conception of the manuscript, M.B.S.-C., S.I.N.-O., R.H.-B., and J.P.-R.; literature review, M.B.S.-C., S.I.N.-O., R.H.-B., E.M.C.-M., M.E.A.-S., and J.P.-R.; draft of the manuscript, M.B.S.-C., S.I.N.-O., R.H.-B., and J.P.-R.; reviewing and editing, M.B.S.-C., S.I.N.-O., R.H.-B., E.M.C.-M., M.E.A.-S., and J.P.-R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We thank the División de Investigación of the Hospital Juárez de México and the Laboratorio de Patogenesis Celular y Molecular Humana y Veterinaria of Posgrado en Ciencias Genómicas of the UACM for the facilities provided. This article is a part of the objectives of Institutional project HJM 009-23-I.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wang, Z.; Wu, D.; Ng, C.F.; Teoh, J.Y.; Yu, S.; Wang, Y.; Chan, F.L. Nuclear receptor profiling in prostatospheroids and castration-resistant prostate cancer. Endocr. Relat. Cancer 2018, 25, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, E.; Bergh, A.; Wikström, P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef]
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol. Cell 2005, 19, 631–642. [Google Scholar] [CrossRef]
- Sakellakis, M. Orphan receptors in prostate cancer. Prostate 2022, 82, 1016–1024. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, Y.; Xu, K.; Hu, H.; Xu, Z.; Wu, D.; Wang, Z.; You, W.; Ng, C.F.; Yu, S.; et al. Nuclear Receptor LRH-1 Functions to Promote Castration-Resistant Growth of Prostate Cancer via Its Promotion of Intratumoral Androgen Biosynthesis. Cancer Res. 2018, 78, 2205–2218. [Google Scholar] [CrossRef]
- Jones, C.J.; Goetz, M.P.; Ingle, J.N.; Hawse, J.R. Glucocorticoid receptor activation inhibits proliferation of endoxifen-resistant breast cancer cells and resensitizes cells to hormonal therapy [abstract]. In: Proceedings of the 2018 San Antonio Breast Cancer Symposium; 2018 Dec 4–8; San Antonio, TX. Philadelphia (PA): AACR. Cancer Res. 2019, 79 (Suppl. 4), P5-04-05. [Google Scholar] [CrossRef]
- Shaik, S.; Campbell, H.; Williams, C. NURR1 Is Differentially Expressed in Breast Cancer According to Patient Racial Identity and Tumor Subtype. BioMedInformatics 2022, 2, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Bianco, S.; Brunelle, M.; Jangal, M.; Magnani, L.; Gévry, N. LRH-1 governs vital transcriptional programs in endocrine-sensitive and -resistant breast cancer cells. Cancer Res. 2014, 74, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, L.M.; Appana, S.N.; Datta, S.; Klinge, C.M. COUP-TFII inhibits NFkappaB activation in endocrine-resistant breast cancer cells. Mol. Cell Endocrinol. 2014, 382, 358–367. [Google Scholar] [CrossRef][Green Version]
- Burris, T.P.; de Vera, I.M.S.; Cote, I.; Flaveny, C.A.; Wanninayake, U.S.; Chatterjee, A.; Walker, J.K.; Steinauer, N.; Zhang, J.; Coons, L.A.; et al. International Union of Basic and Clinical Pharmacology CXIII: Nuclear Receptor Superfamily-Update 2023. Pharmacol. Rev. 2023, 75, 1233–1318. [Google Scholar] [CrossRef]
- Giguère, V. Orphan nuclear receptors: From gene to function. Endocr. Rev. 1999, 20, 689–725. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Glass, C.K. Nuclear receptors versus inflammation: Mechanisms of transrepression. Trends Endocrinol. Metab. 2006, 17, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, A.; Schmutz, C.; Askari, A.; Kuiper, J.H.; Middleton, J. Orphan receptor GPR15/BOB is up-regulated in rheumatoid arthritis. Cytokine 2014, 67, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Klepsch, V.; Moschen, A.R.; Tilg, H.; Baier, G.; Hermann-Kleiter, N. Nuclear Receptors Regulate Intestinal Inflammation in the Context of IBD. Front. Immunol. 2019, 10, 1070. [Google Scholar] [CrossRef]
- Le, T.K.; Duong, Q.H.; Baylot, V.; Fargette, C.; Baboudjian, M.; Colleaux, L.; Taïeb, D.; Rocchi, P. Castration-Resistant Prostate Cancer: From Uncovered Resistance Mechanisms to Current Treatments. Cancers 2023, 15, 5047. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Shah, R.B.; Mehra, R.; Chinnaiyan, A.M.; Shen, R.; Ghosh, D.; Zhou, M.; Macvicar, G.R.; Varambally, S.; Harwood, J.; Bismar, T.A.; et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004, 64, 9209–9216. [Google Scholar] [CrossRef]
- Hu, C.D.; Choo, R.; Huang, J. Neuroendocrine differentiation in prostate cancer: A mechanism of radioresistance and treatment failure. Front. Oncol. 2015, 5, 90. [Google Scholar] [CrossRef]
- Liu, W.; Xie, C.C.; Zhu, Y.; Li, T.; Sun, J.; Cheng, Y.; Ewing, C.M.; Dalrymple, S.; Turner, A.R.; Isaacs, J.T.; et al. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia 2008, 10, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ibaragi, S.; Hu, G.F. Angiogenin as a molecular target for the treatment of prostate cancer. Curr. Cancer Ther. Rev. 2011, 7, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Duff, J.; McEwan, I.J. Mutation of histidine 874 in the androgen receptor ligand-binding domain leads to promiscuous ligand activation and altered p160 coactivator interactions. Mol. Endocrinol. 2005, 19, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Watanabe, T.; Okada, M.; Inoue, K.; Ueda, T.; Takada, I.; Watabe, T.; Yamamoto, Y.; Fukuda, T.; Nakamura, T.; et al. Noncanonical Wnt signaling mediates androgen-dependent tumor growth in a mouse model of prostate cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 4938–4943. [Google Scholar] [CrossRef]
- Giwercman, Y.L.; Abrahamsson, P.A.; Giwercman, A.; Gadaleanu, V.; Ahlgren, G. The 5alpha-reductase type II A49T and V89L high-activity allelic variants are more common in men with prostate cancer compared with the general population. Eur. Urol. 2005, 48, 679–685. [Google Scholar] [CrossRef]
- Crowley, F.; Sterpi, M.; Buckley, C.; Margetich, L.; Handa, S.; Dovey, Z. A Review of the Pathophysiological Mechanisms Underlying Castration-resistant Prostate Cancer. Res. Rep. Urol. 2021, 13, 457–472. [Google Scholar] [CrossRef]
- Ni, L.; Yang, C.S.; Gioeli, D.; Frierson, H.; Toft, D.O.; Paschal, B.M. FKBP51 promotes assembly of the Hsp90 chaperone complex and regulates androgen receptor signaling in prostate cancer cells. Mol. Cell Biol. 2010, 30, 1243–1253. [Google Scholar] [CrossRef]
- Lavaud, P.; Dumont, C.; Thibault, C.; Albiges, L.; Baciarello, G.; Colomba, E.; Flippot, R.; Fuerea, A.; Loriot, Y.; Fizazi, K. Next-generation androgen receptor inhibitors in non-metastatic castration-resistant prostate cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920978134. [Google Scholar] [CrossRef]
- Podgoršek, E.; Mehra, N.; van Oort, I.M.; Somford, D.M.; Boerrigter, E.; van Erp, N.P. Clinical Pharmacokinetics and Pharmacodynamics of the Next Generation Androgen Receptor Inhibitor-Darolutamide. Clin. Pharmacokinet. 2023, 62, 1049–1061. [Google Scholar] [CrossRef]
- Sugawara, T.; Baumgart, S.J.; Nevedomskaya, E.; Reichert, K.; Steuber, H.; Lejeune, P.; Mumberg, D.; Haendler, B. Darolutamide is a potent androgen receptor antagonist with strong efficacy in prostate cancer models. Int. J. Cancer 2019, 145, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Hoffman-Censits, J.; Kelly, W.K. Enzalutamide: A novel antiandrogen for patients with castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 1335–1339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhao, L.J.; Wang, Y.T. Synthesis and clinical application of small-molecule drugs approved to treat prostatic cancer. Eur. J. Med. Chem. 2023, 262, 115925. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, P.; Hu, M.; Yang, L.; Yan, G.; Xu, R.; Deng, Y.; Li, X.; Chen, Y. Discovery and biological evaluation of darolutamide derivatives as inhibitors and down-regulators of wild-type AR and the mutants. Eur. J. Med. Chem. 2019, 182, 111608. [Google Scholar] [CrossRef]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef]
- Zhao, J.; Ning, S.; Lou, W.; Yang, J.C.; Armstrong, C.M.; Lombard, A.P.; D’Abronzo, L.S.; Evans, C.P.; Gao, A.C.; Liu, C. Cross-Resistance among Next-Generation Antiandrogen Drugs through the AKR1C3/AR-V7 Axis in Advanced Prostate Cancer. Mol. Cancer Ther. 2020, 19, 1708–1718. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Zhu, Y.; Yang, J.C.; Nadiminty, N.; Gaikwad, N.W.; Evans, C.P.; Gao, A.C. Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer Res. 2015, 75, 1413–1422. [Google Scholar] [CrossRef]
- Liu, C.; Yang, J.C.; Armstrong, C.M.; Lou, W.; Liu, L.; Qiu, X.; Zou, B.; Lombard, A.P.; D’Abronzo, L.S.; Evans, C.P.; et al. AKR1C3 Promotes AR-V7 Protein Stabilization and Confers Resistance to AR-Targeted Therapies in Advanced Prostate Cancer. Mol. Cancer Ther. 2019, 18, 1875–1886. [Google Scholar] [CrossRef]
- Giguère, V.; Tini, M.; Flock, G.; Ong, E.; Evans, R.M.; Otulakowski, G. Isoform-specific amino-terminal domains dictate DNA-binding properties of ROR alpha, a novel family of orphan hormone nuclear receptors. Genes. Dev. 1994, 8, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Jetten, A.M.; Takeda, Y.; Slominski, A.; Kang, H.S. Retinoic acid-related Orphan Receptor γ (RORγ): Connecting sterol metabolism to regulation of the immune system and autoimmune disease. Curr. Opin. Toxicol. 2018, 8, 66–80. [Google Scholar] [CrossRef]
- Chang, M.R.; Griffin, P.R. RORβ modulates a gene program that is protective against articular cartilage damage. PLoS ONE 2022, 17, e0268663. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Chen, M.; Guo, W.; Guo, K.; Du, P.; Fang, Y.; Gao, M.; Wang, Q. RORβ suppresses the stemness of gastric cancer cells by downregulating the activity of the Wnt signaling pathway. Oncol. Rep. 2021, 46, 180. [Google Scholar] [CrossRef]
- Zhang, C.L.; Zou, Y.; He, W.; Gage, F.H.; Evans, R.M. A role for adult TLX-positive neural stem cells in learning and behaviour. Nature 2008, 451, 1004–1007. [Google Scholar] [CrossRef]
- Jia, L.; Wu, D.; Wang, Y.; You, W.; Wang, Z.; Xiao, L.; Cai, G.; Xu, Z.; Zou, C.; Wang, F.; et al. Orphan nuclear receptor TLX contributes to androgen insensitivity in castration-resistant prostate cancer via its repression of androgen receptor transcription. Oncogene 2018, 37, 3340–3355. [Google Scholar] [CrossRef]
- Chen, X.; Qin, J.; Cheng, C.M.; Tsai, M.J.; Tsai, S.Y. COUP-TFII is a major regulator of cell cycle and Notch signaling pathways. Mol. Endocrinol. 2012, 26, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Koibuchi, N. COUP-TFII in Kidneys, from Embryos to Sick Adults. Diagnostics 2022, 12, 1181. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, J.; Zou, Y.; Huang, G.L.; He, Z.W. Orphan nuclear receptor nurr1 as a potential novel marker for progression in human prostate cancer. Asian Pac. J. Cancer Prev. 2013, 14, 2023–2028. [Google Scholar] [CrossRef]
- Saucedo-Cardenas, O.; Quintana-Hau, J.D.; Le, W.D.; Smidt, M.P.; Cox, J.J.; De Mayo, F.; Burbach, J.P.; Conneely, O.M. Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 4013–4018. [Google Scholar] [CrossRef]
- Zhang, X.; Li, H.; Wang, Y.; Zhao, H.; Wang, Z.; Chan, F.L. Nuclear receptor NURR1 functions to promote stemness and epithelial-mesenchymal transition in prostate cancer via its targeting of Wnt/β-catenin signaling pathway. Cell Death Dis. 2024, 15, 234. [Google Scholar] [CrossRef]
- Wang, J.; Zou, J.X.; Xue, X.; Cai, D.; Zhang, Y.; Duan, Z.; Xiang, Q.; Yang, J.C.; Louie, M.C.; Borowsky, A.D.; et al. ROR-γ drives androgen receptor expression and represents a therapeutic target in castration-resistant prostate cancer. Nat. Med. 2016, 22, 488–496. [Google Scholar] [CrossRef]
- Fang, W.; Zheng, J.; Deng, L.; An, Y.; Rong, D.; Wei, J.; Xiong, X.F.; Wang, J.; Wang, Y. Discovery of the First-in-Class RORγ Covalent Inhibitors for Treatment of Castration-Resistant Prostate Cancer. J. Med. Chem. 2024, 67, 1481–1499. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Goodwin, B.; Chung, A.C.; Xu, X.; Wheeler, D.A.; Price, R.R.; Galardi, C.; Peng, L.; Latour, A.M.; Koller, B.H.; et al. Orphan nuclear receptor LRH-1 is required to maintain Oct4 expression at the epiblast stage of embryonic development. Mol. Cell Biol. 2005, 25, 3492–3505. [Google Scholar] [CrossRef] [PubMed]
- Bianco, S.; Jangal, M.; Garneau, D.; Gévry, N. LRH-1 controls proliferation in breast tumor cells by regulating CDKN1A gene expression. Oncogene 2015, 34, 4509–4518. [Google Scholar] [CrossRef] [PubMed]
- Kramer, H.B.; Lai, C.F.; Patel, H.; Periyasamy, M.; Lin, M.L.; Feller, S.M.; Fuller-Pace, F.V.; Meek, D.W.; Ali, S.; Buluwela, L. LRH-1 drives colon cancer cell growth by repressing the expression of the CDKN1A gene in a p53-dependent manner. Nucleic Acids Res. 2016, 44, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Aihara, A.; Chung, W.; Li, Y.; Chen, X.; Huang, Z.; Weng, S.; Carlson, R.I.; Nadolny, C.; Wands, J.R.; et al. LRH1 promotes pancreatic cancer metastasis. Cancer Lett. 2014, 350, 15–24. [Google Scholar] [CrossRef]
- You, W.; Wang, Y.; Chan, L. Functional cross-talk between nuclear receptor LRH-1 and androgen receptor signaling in prostate cancer [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2018; 2018 Apr 14–18; Chicago, IL. Philadelphia (PA): AACR. Cancer Res. 2018, 78 (Suppl. 13), nr 3740. [Google Scholar] [CrossRef]
- Alaynick, W.A. Nuclear receptors, mitochondria and lipid metabolism. Mitochondrion 2008, 8, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.M.; Giguère, V. The NR3B subgroup: An ovERRview. Nucl. Recept. Signal 2007, 5, e009. [Google Scholar] [CrossRef]
- Duellman, S.J.; Calaoagan, J.M.; Sato, B.G.; Fine, R.; Klebansky, B.; Chao, W.R.; Hobbs, P.; Collins, N.; Sambucetti, L.; Laderoute, K.R. A novel steroidal inhibitor of estrogen-related receptor alpha (ERR alpha). Biochem. Pharmacol. 2010, 80, 819–826. [Google Scholar] [CrossRef]
- Cheung, C.P.; Yu, S.; Wong, K.B.; Chan, L.W.; Lai, F.M.; Wang, X.; Suetsugi, M.; Chen, S.; Chan, F.L. Expression and functional study of estrogen receptor-related receptors in human prostatic cells and tissues. J. Clin. Endocrinol. Metab. 2005, 90, 1830–1844. [Google Scholar] [CrossRef]
- Xu, Z.; Ma, T.; Zhou, J.; Gao, W.; Li, Y.; Yu, S.; Wang, Y.; Chan, F.L. Nuclear receptor ERRα contributes to castration-resistant growth of prostate cancer via its regulation of intratumoral androgen biosynthesis. Theranostics 2020, 10, 4201–4216. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Y.; Xiao, Z.G.; Zou, C.; Zhang, X.; Wang, Z.; Wu, D.; Yu, S.; Chan, F.L. Nuclear receptor ERRα and transcription factor ERG form a reciprocal loop in the regulation of TMPRSS2:ERG fusion gene in prostate cancer. Oncogene 2018, 37, 6259–6274. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Wardell, S.E.; Burnstein, K.L.; Defranco, D.; Fuller, P.J.; Giguere, V.; Hochberg, R.B.; McKay, L.; Renoir, J.M.; Weigel, N.L.; et al. International Union of Pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: Glucocorticoid, mineralocorticoid, progesterone, and androgen receptors. Pharmacol. Rev. 2006, 58, 782–797. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.; Jaiswal, P.K.; Harkless, R.V.; Long, T.M.; Gao, N.; Vandenburg, B.; Selman, P.; Durdana, I.; Lastra, R.R.; Vander Griend, D.; et al. Glucocorticoid Receptor (GR) Activation Is Associated with Increased cAMP/PKA Signaling in Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2024, 23, 552–563. [Google Scholar] [CrossRef]
- Pollack, A.; Bae, K.; Khor, L.Y.; Al-Saleem, T.; Hammond, M.E.; Venkatesan, V.; Byhardt, R.W.; Asbell, S.O.; Shipley, W.U.; Sandler, H.M. The importance of protein kinase A in prostate cancer: Relationship to patient outcome in Radiation Therapy Oncology Group trial 92-02. Clin. Cancer Res. 2009, 15, 5478–5484. [Google Scholar] [CrossRef]
- Li, J.; Alyamani, M.; Zhang, A.; Chang, K.H.; Berk, M.; Li, Z.; Zhu, Z.; Petro, M.; Magi-Galluzzi, C.; Taplin, M.E.; et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. eLife 2017, 6, e20183. [Google Scholar] [CrossRef]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Vander Griend, D.J.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef]
- Ali, S.; Rasool, M.; Chaoudhry, H.; Pushparaj, P.; Jha, P.; Hafiz, A.; Mahfooz, M.; Abdus Sami, G.; Azhar Kamal, M.; Bashir, S.; et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation 2016, 12, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Rondón-Lagos, M.; Villegas, V.E.; Rangel, N.; Sánchez, M.C.; Zaphiropoulos, P.G. Tamoxifen Resistance: Emerging Molecular Targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar] [CrossRef]
- Blasiak, J.; Chojnacki, J.; Pawlowska, E.; Jablkowska, A.; Chojnacki, C. Vitamin D May Protect against Breast Cancer through the Regulation of Long Noncoding RNAs by VDR Signaling. Int. J. Mol. Sci. 2022, 23, 3189. [Google Scholar] [CrossRef]
- Huss, L.; Butt, S.T.; Borgquist, S.; Elebro, K.; Sandsveden, M.; Rosendahl, A.; Manjer, J. Vitamin D receptor expression in invasive breast tumors and breast cancer survival. Breast Cancer Res. 2019, 21, 84. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Xin, Z.; Ren, P.; Wu, H. The Role of PPARs in Breast Cancer. Cells 2022, 12, 130. [Google Scholar] [CrossRef] [PubMed]
- Augimeri, G.; Giordano, C.; Gelsomino, L.; Plastina, P.; Barone, I.; Catalano, S.; Andò, S.; Bonofiglio, D. The Role of PPARγ Ligands in Breast Cancer: From Basic Research to Clinical Studies. Cancers 2020, 12, 2623. [Google Scholar] [CrossRef] [PubMed]
- Kensler, K.H.; Poole, E.M.; Heng, Y.J.; Collins, L.C.; Glass, B.; Beck, A.H.; Hazra, A.; Rosner, B.A.; Eliassen, A.H.; Hankinson, S.E.; et al. Androgen Receptor Expression and Breast Cancer Survival: Results From the Nurses’ Health Studies. J. Natl. Cancer Inst. 2019, 111, 700–708. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Palla, S.L.; Carey, M.; Agarwal, R.; Meric-Berstam, F.; Traina, T.A.; Hudis, C.; Hortobagyi, G.N.; Gerald, W.L.; et al. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clin. Cancer Res. 2009, 15, 2472–2478. [Google Scholar] [CrossRef]
- Abigail, B.C.; Suzanne, D.C. Glucocorticoid receptor-mediated oncogenic activity is dependent on breast cancer subtype. J. Steroid Biochem. Mol. Biol. 2024, 243, 106518. [Google Scholar] [CrossRef]
- Prekovic, S.; Chalkiadakis, T.; Roest, M.; Roden, D.; Lutz, C.; Schuurman, K.; Opdam, M.; Hoekman, L.; Abbott, N.; Tesselaar, T.; et al. Luminal breast cancer identity is determined by loss of glucocorticoid receptor activity. EMBO Mol. Med. 2023, 15, e17737. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, Y.; Qian, K.; Li, L.; Zhang, C.; Fu, X.; Zhang, X.; Chen, H.; Liu, Q.; Cao, S.; et al. A novel tumor suppressor ZBTB1 regulates tamoxifen resistance and aerobic glycolysis through suppressing. J. Biol. Chem. 2020, 295, 14140–14152. [Google Scholar] [CrossRef] [PubMed]
- Tonsing-Carter, E.; Hernandez, K.M.; Kim, C.R.; Harkless, R.V.; Oh, A.; Bowie, K.R.; West-Szymanski, D.C.; Betancourt-Ponce, M.A.; Green, B.D.; Lastra, R.R.; et al. Glucocorticoid receptor modulation decreases ER-positive breast cancer cell proliferation and suppresses wild-type and mutant ER chromatin association. Breast Cancer Res. 2019, 21, 82. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Łukasik, P.; Baranowska-Bosiacka, I.; Kulczycka, K.; Gutowska, I. Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action. Int. J. Mol. Sci. 2021, 22, 2806. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.; Trub, A.; Ahn, A.; Taylor, M.; Ambani, K.; Chan, K.T.; Lu, K.H.; Mahendra, C.A.; Blyth, C.; Coulson, R.; et al. INX-315, a Selective CDK2 Inhibitor, Induces Cell Cycle Arrest and Senescence in Solid Tumors. Cancer Discov. 2024, 14, 446–467. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.J.; Acero-Bedoya, S.; Mykytyn, A.V.; Goetz, M.P.; Hawse, J.R. Glucocorticoid receptor signaling elicits anti-cancer effects in endocrine-resistant breast cancer via induction of AZGP1 [abstract]. In: Proceedings of the 2019 San Antonio Breast Cancer Symposium; 2019 Dec 10–14; San Antonio, TX. Philadelphia (PA): AACR. Cancer Res. 2020, 80 (Suppl. 4), P6-04-16. [Google Scholar] [CrossRef]
- Litchfield, L.M.; Klinge, C.M. Multiple roles of COUP-TFII in cancer initiation and progression. J. Mol. Endocrinol. 2012, 49, R135–R148. [Google Scholar] [CrossRef]
- Kastrati, I.; Joosten, S.E.P.; Semina, S.E.; Alejo, L.H.; Brovkovych, S.D.; Stender, J.D.; Horlings, H.M.; Kok, M.; Alarid, E.T.; Greene, G.L.; et al. The NF-κB Pathway Promotes Tamoxifen Tolerance and Disease Recurrence in Estrogen Receptor-Positive Breast Cancers. Mol. Cancer Res. 2020, 18, 1018–1027. [Google Scholar] [CrossRef]
- Al-Rayyan, N.; Litchfield, L.M.; Ivanova, M.M.; Radde, B.N.; Cheng, A.; Elbedewy, A.; Klinge, C.M. 5-Aza-2-deoxycytidine and trichostatin A increase COUP-TFII expression in antiestrogen-resistant breast cancer cell lines. Cancer Lett. 2014, 347, 139–150. [Google Scholar] [CrossRef]
- Fayard, E.; Auwerx, J.; Schoonjans, K. LRH-1: An orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004, 14, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Deng, K.; Huang, J.; Zeng, R.; Zuo, J. Progress in the Understanding of the Mechanism of Tamoxifen Resistance in Breast Cancer. Front. Pharmacol. 2020, 11, 592912. [Google Scholar] [CrossRef]
- Lai, C.F.; Flach, K.D.; Alexi, X.; Fox, S.P.; Ottaviani, S.; Thiruchelvam, P.T.; Kyle, F.J.; Thomas, R.S.; Launchbury, R.; Hua, H.; et al. Co-regulated gene expression by oestrogen receptor α and liver receptor homolog-1 is a feature of the oestrogen response in breast cancer cells. Nucleic Acids Res. 2013, 41, 10228–10240. [Google Scholar] [CrossRef]
- Ferraiuolo, R.M.; Tubman, J.; Sinha, I.; Hamm, C.; Porter, L.A. The cyclin-like protein, SPY1, regulates the ERα and ERK1/2 pathways promoting tamoxifen resistance. Oncotarget 2017, 8, 23337–23352. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Kim, C.Y.; Oh, J.H.; Kim, M.H. NR4A1 Regulates Tamoxifen Resistance by Suppressing ERK Signaling in ER-Positive Breast Cancer. Cells 2021, 10, 1633. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Ross, A.E.; Parikh, R.B.; Nohria, A.; Morgans, A.K. How to Treat Prostate Cancer With Androgen Deprivation and Minimize Cardiovascular Risk: A Therapeutic Tightrope. JACC CardioOncol. 2021, 3, 737–741. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).