
Peripheral Artery Disease: Atherosclerosis, Decreased Nitric Oxide, and Vascular Arterial Stiffening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction

2. Peripheral Arterial Disease (PAD)
2.1. Atherosclerosis in PAD
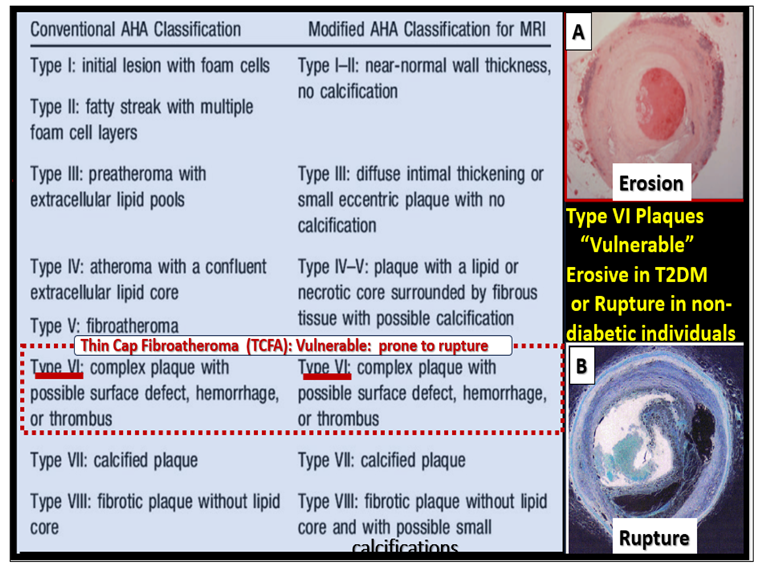
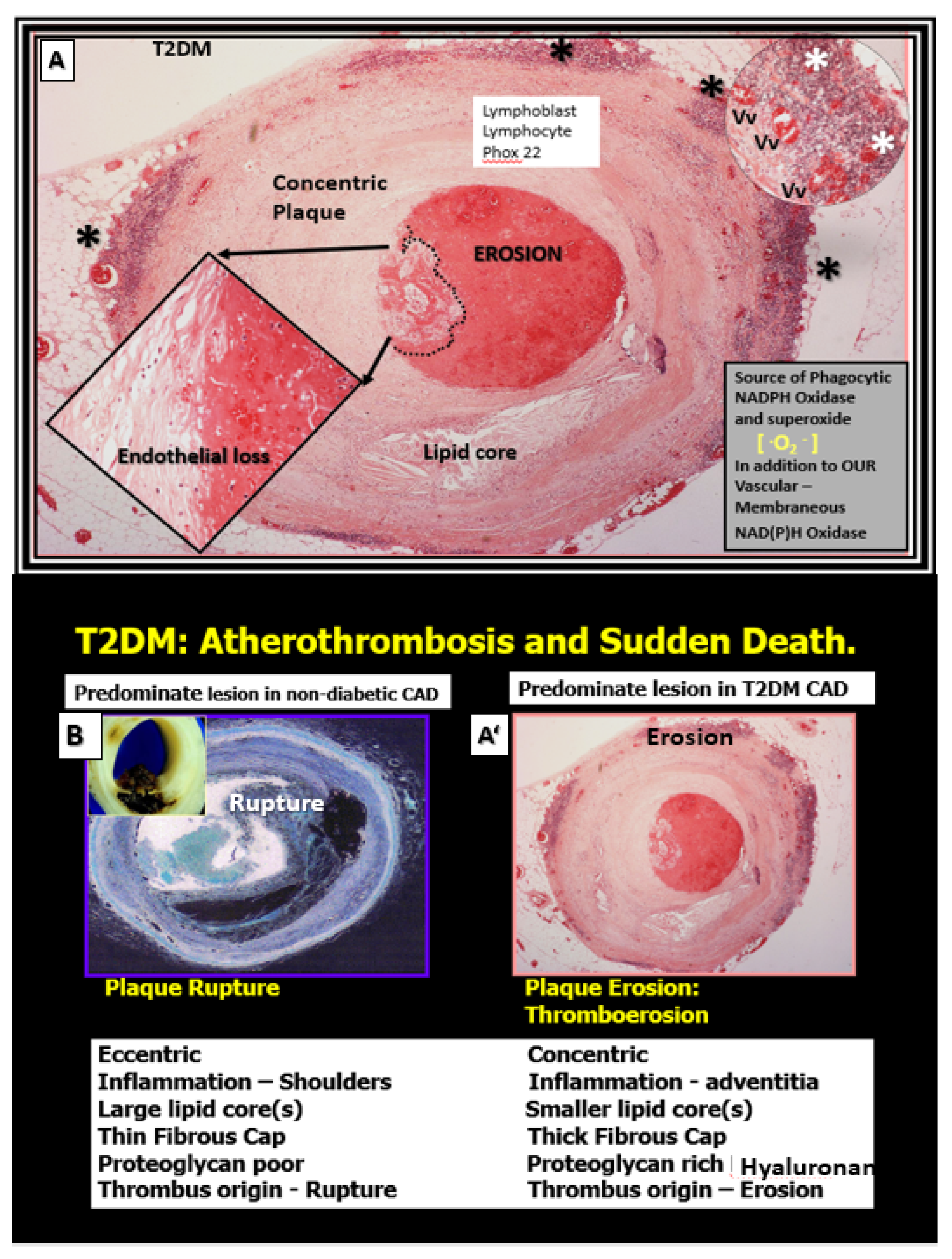
2.2. Advancing Age and Essential Hypertension (eHTN-HTN) in PAD
2.3. The Metabolic Syndrome (MetS) and Type 2 Diabetes Mellitus (T2DM) in PAD
2.4. Tobacco Smoking, Oxidative Redox Stress (OxRS), and Accelerated Atherosclerosis in PAD
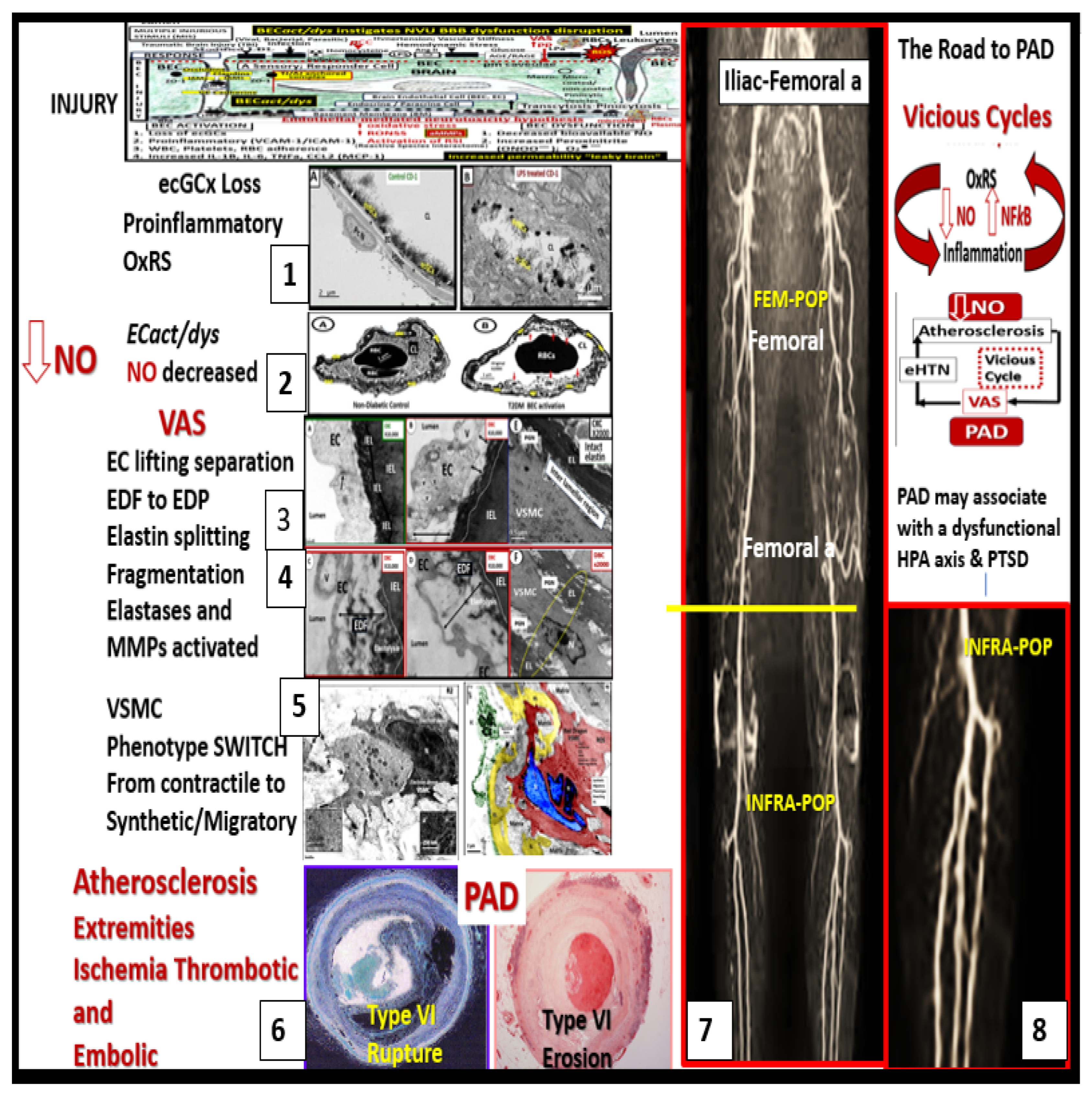
3. Decreased Bioavailability of Endothelial Cell-Derived Nitric Oxide in PAD
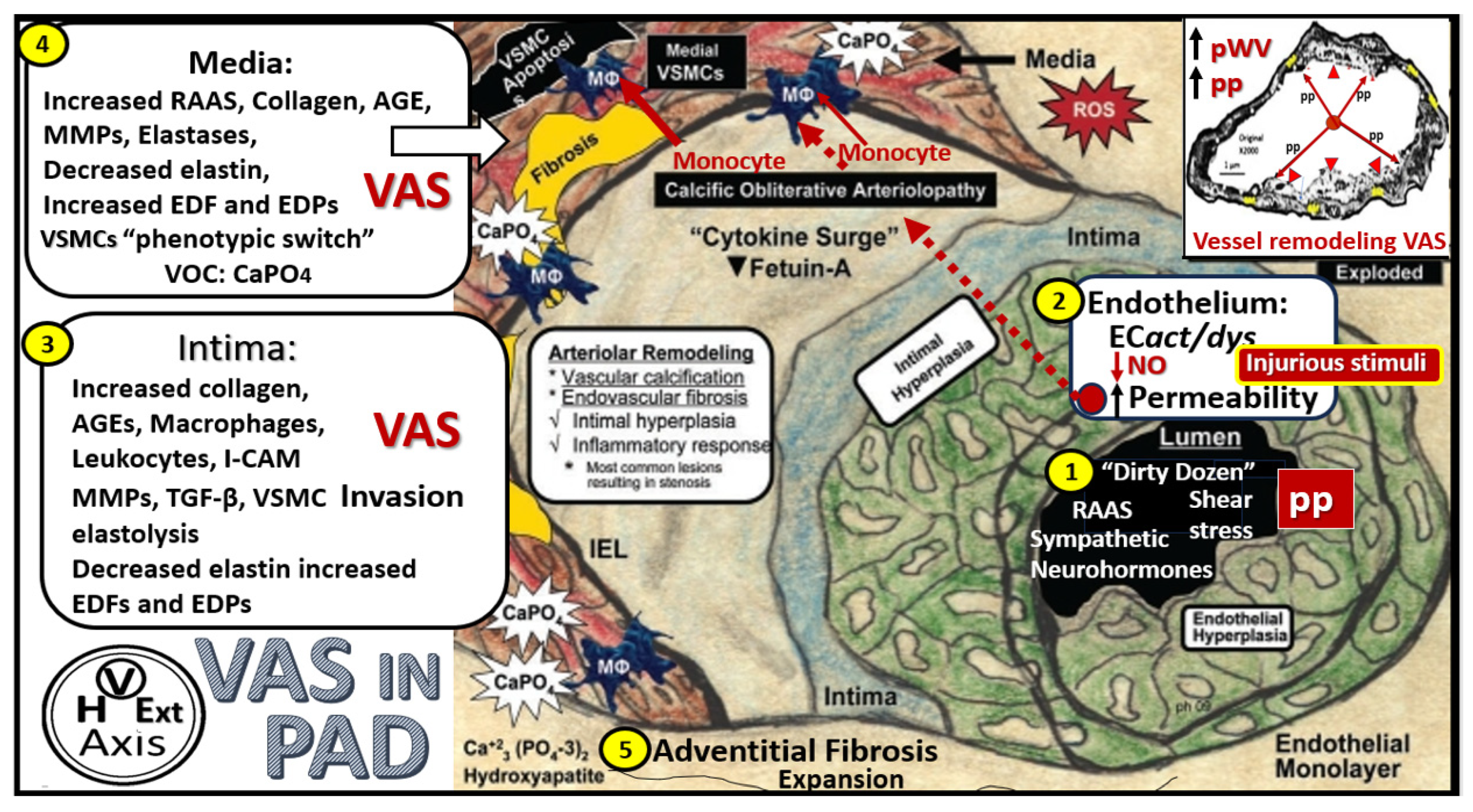
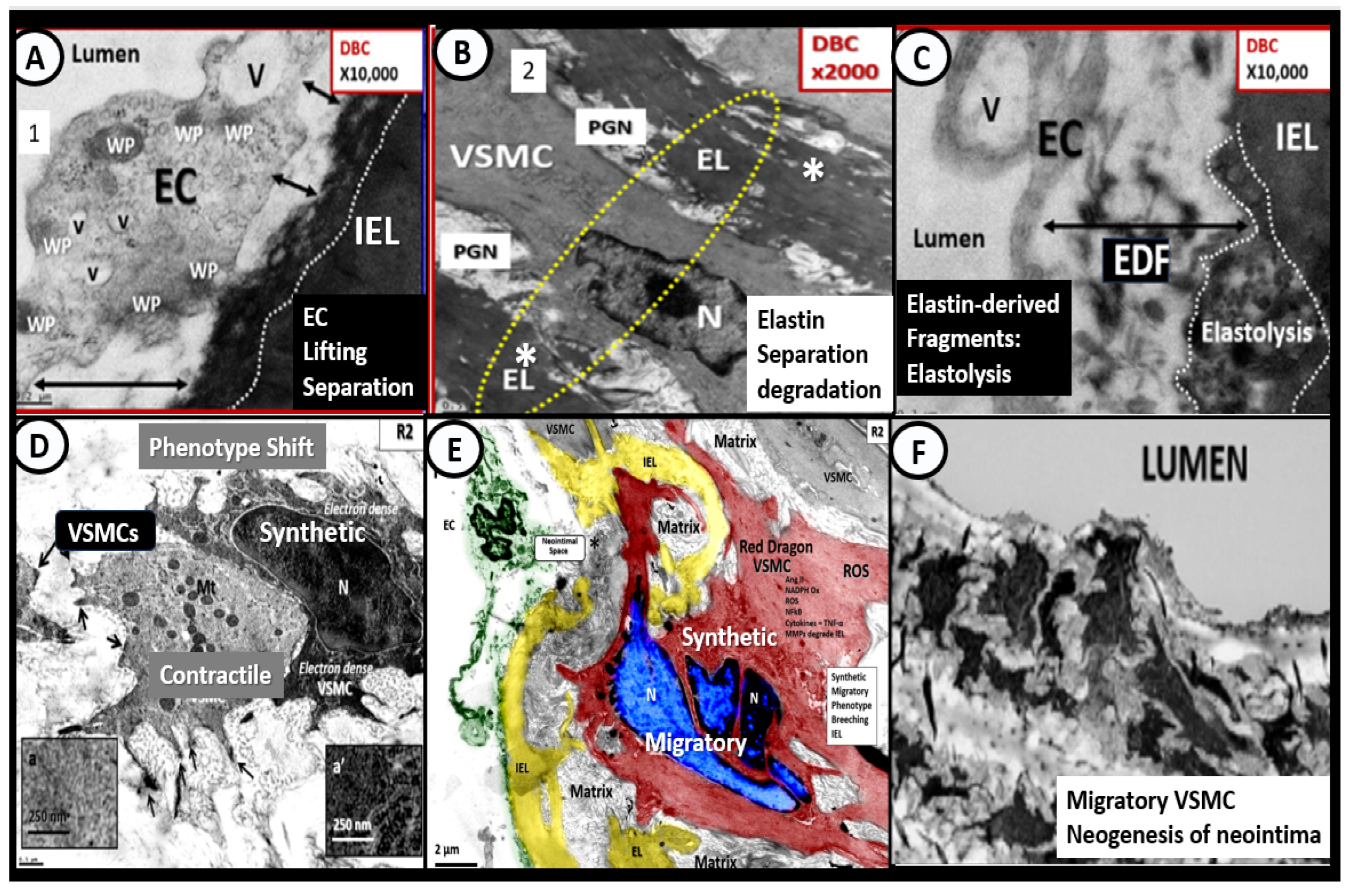
4. Vascular Arterial Stiffening (VAS) in PAD
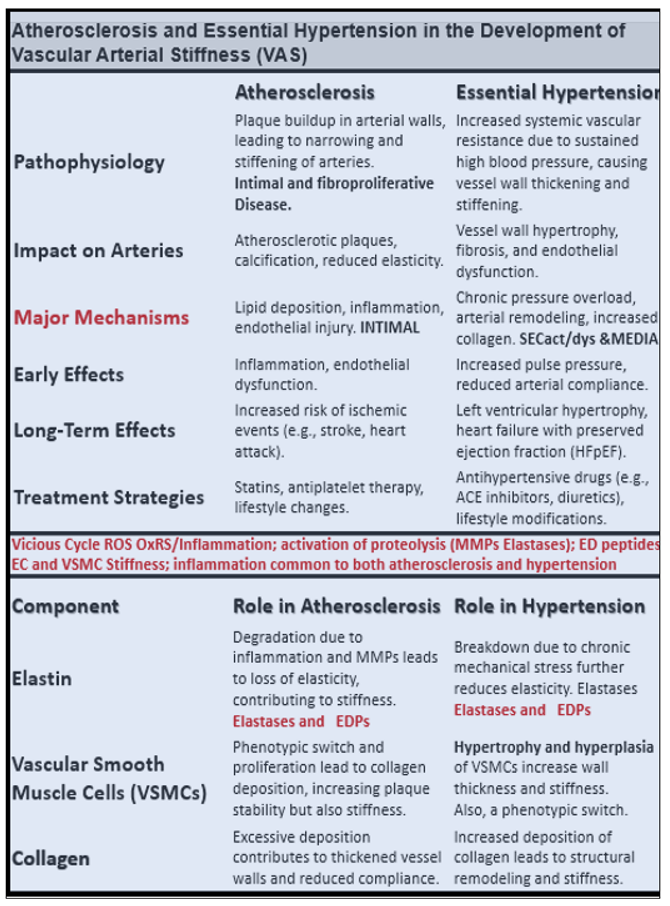
4.1. Bioactive Elastin-Derived Peptides (EDPs) and Transglutaminase 2 (TG2)
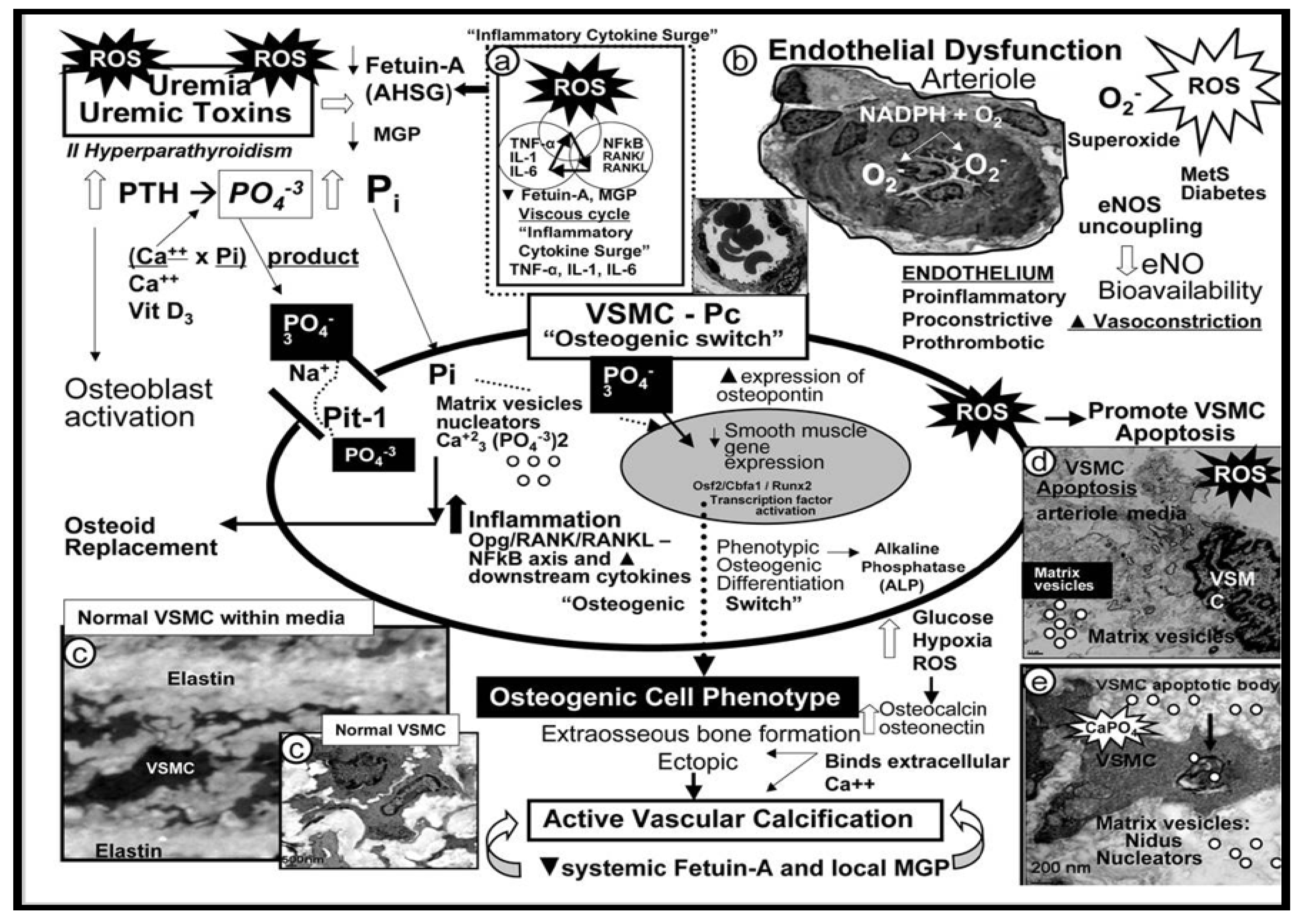
4.2. Vascular Ossification Calcification (VOC) in Type VII Plaques
4.3. PAD Involves Arterial Vessels Large and Small: Microvessel (Mv) Remodeling in Skeletal Muscle (SkM)
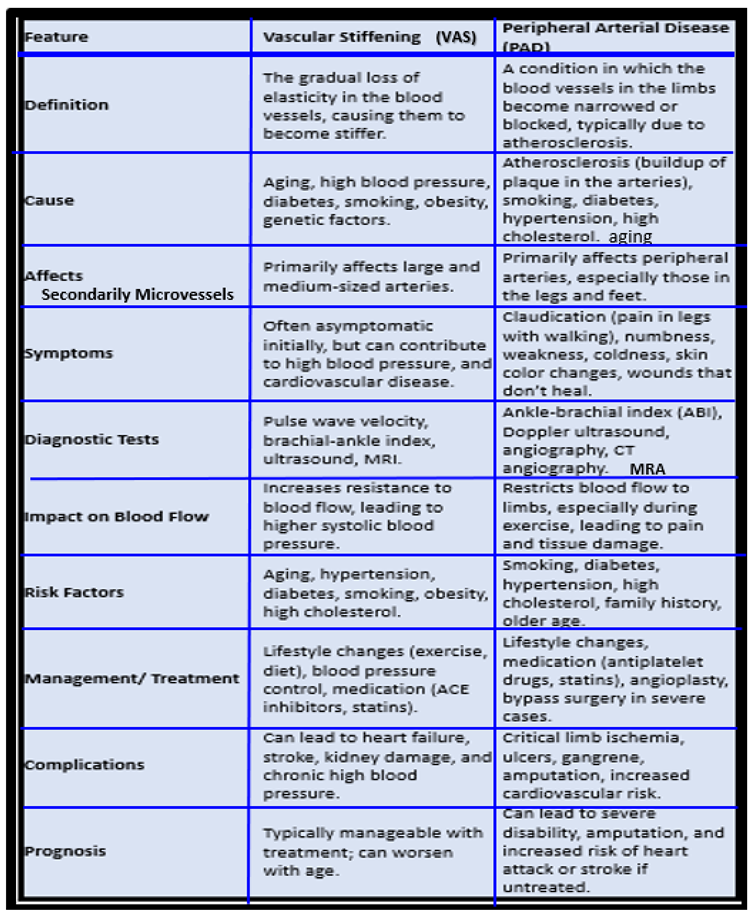
5. Ischemia and Ischemia/Reperfusion (IR) Injury, Microvessel (Mv) Remodeling, Ischemic Neuropathy, Ischemic Skeletal Muscle (SkM) Remodeling with Myopathy and Myalgias, and Pain Perception
6. Conclusions and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hayden, M.R.; Tyagi, S.C. Intimal redox stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovasc. Diabetol. 2002, 1, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Abdelfattah, A.; Allam, A.H.; Wann, S.; Thompson, R.C.; Abdel-Maksoud, G.; Badr, I.; Amer, H.A.R.; El-Din, A.E.-H.N.; Finch, C.E.; Miyamoto, M.I.; et al. Atherosclerotic crdiovasculay disease in Eqyptian women: 1570BCE-2011 CE. Int. J. Cardiol. 2013, 167, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Allam, A.H.; Thompson, R.C.; Wann, L.S.; Miyamoto, M.I.; El-Din, A.E.-H.N.; El-Maksoud, G.A.; Soliman, M.A.-T.; Badr, I.; Amer, H.A.E.-R.; Sutherland, M.L.; et al. Atherosclerosis in ancient Egyptian mummies: The Horus study. JACC Cardiovasc. Imaging 2011, 4, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Fowkes, F.G.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.A.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Song, P.; Rudan, D.; Zhu, Y.; Fowkes, F.J.I.; Rahimi, K.; Fowkes, F.G.R.; Rudan, I. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: An updated systematic review and analysis. Lancet Glob. Health 2019, 7, e1020–e1030. [Google Scholar] [CrossRef]
- The Diabetes Prevention Program Research Group; Crandall, J.; Schade, D.; Ma, Y.; Fujimoto, W.Y.; Barrett-Connor, E.; Fowler, S.; Dagogo-Jack, S.; Andres, R. The Influence of Age on the Effects of Lifestyle Modification and Metformin in Prevention of Diabetes. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 1075–1081. [Google Scholar]
- United Nations Department of Economics and Social Affairs Populations Division. Population Ageing and Sustainable Development. Available online: https://www.un.org/en/development/desa/population/publications/pdf/popfacts/PopFacts_2017-1.pdf (accessed on 14 January 2025).
- Hayden, M.R. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262. [Google Scholar] [CrossRef]
- Macabrey, D.; Longchamp, A.; MacArthur, M.R.; Lambelet, M.; Urfer, S.; Deglise, S.; Allagnat, F. Sodium thiosulfate, a source of hydrogen sulfide, stimulates endothelial cell proliferation and neovascularization. EBioMedicine 2022, 78, 103954. [Google Scholar] [CrossRef]
- Eraso, L.H.; Fukaya, E.; Mohler, E.R.; Xie, D.; Sha, D.; Berger, J.S. Peripheral arterial disease, prevalence and cumulative risk factor profile analysis. Eur. J. Prev. Cardiol. 2014, 21, 704–711. [Google Scholar] [CrossRef]
- Parwani, D.; Ahmed, M.A.; Mahawar, A.; Gorantla, V.R. Peripheral Arterial Disease: A Narrative Review. Cureus 2023, 15, e40267. [Google Scholar] [CrossRef]
- Grenon, S.M.; Vittinghoff, E.; Owens, C.D.; Conte, M.S.; Whooley, M.; E Cohen, B. Peripheral artery disease and risk of cardiovascular events in patients with coronary artery disease: Insights from the Heart and Soul Study. Vasc. Med. 2013, 18, 176–184. [Google Scholar] [CrossRef]
- Liao, J.K.; Shin, W.S.; Lee, W.Y.; Clark, S.L. Oxidized low-density lipoprotein decreases the expression of endothelial nitric oxide synthase. J. Biol. Chem. 1995, 270, 319–324. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Endothelial activation and dysfunction in metabolic syndrome, type 2 diabetes mellitus, and coronavirus disease 2019. J. Int. Med. Res. 2020, 48, 0300060520939746. [Google Scholar] [CrossRef]
- Hayden, M.R. Brain Endothelial Cells Play a Central Role in the Development of Enlarged Perivascular Spaces in the Metabolic Syndrome. Medicina 2023, 59, 1124. [Google Scholar] [CrossRef]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. The Brain Endothelial Cell Glycocalyx Plays a Crucial Role in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Life 2023, 13, 1955. [Google Scholar] [CrossRef]
- Hayden, M.R. Brain endothelial cell activation and dysfunction associate with and contribute to the development of enlarged perivascular spaces and cerebral small vessel disease. Histol. Histopathol. 2024, 39, 1565–1586. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, N. Extracranial Vascular Arterial Stiffness Contributes to Cerebral Small Vessel Disease, Stroke, and Late-Onset Alzheimer’s Disease. Preprints. arXiv 2025, arXiv:2025010993. [Google Scholar] [CrossRef]
- Refaat, A.; Abdou, M.; Ismael, A.; Alhelali, I. Aortic stiffness and microalbuminuria in patients with chronic obstructive pulmonary disease. Egypt. J. Chest Dis. Tuberc. 2015, 64, 541–549. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef]
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Allen, M.F.; Pekas, E.J.; Park, S.-Y. Arterial Stiffness as a Prognostic Marker for Peripheral Artery Disease Risk: Clinical Relevance and Considerations. JACC Asia 2023, 3, 298–300. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Vasa vasorum in plaque angiogenesis, metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: A malignant transformation. Cardiovasc. Diabetol. 2002, 22, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47 (Suppl. 8), C7–C12. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Hayden, M.R.; Reidy, M. Many roads lead to atheroma. Nat. Med. 1995, 1, 22–23. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Xie, B.; Shi, X.; Xing, Y.; Tang, Y. Association between atherosclerosis and Alzheimer’s disease: A systematic review and meta-analysis. Brain Behav. 2020, 10, e01601. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. Composition and classification of human atherosclerotic lesions. Virchows Arch. A 1992, 421, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Blankenhorn, D.H.; Chandler, A.B.; Glagov, S.; Insull, W.; Richardson, M.; E Rosenfeld, M.; A Schaffer, S.; Schwartz, C.J.; Wagner, W.D. A definition of the intima of human arteries and of its atherosclerosis-prone regions. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1992, 85, 391–405. [Google Scholar] [CrossRef]
- Rizzo, M.; Berneis, K. Lipid triad or atherogenic lipoprotein phenotype: A role in cardiovascular prevention? J. Atheroscler Thromb. 2005, 12, 237–239. [Google Scholar] [CrossRef]
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam Cells in Atherosclerosis: Novel Insights into Its Origins, Consequences, and Molecular Mechanisms. Front. Cardiovasc. Med. 2022, 9, 845942. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhang, X.J.; Cao, L.J.; Liu, X.H.; Liu, Z.H.; Wang, X.Q.; Chen, Q.J.; Lu, L.; Shen, W.F.; Liu, Y. Toll-like receptor 4 mediates inflammatory cytokine secretion in smooth muscle cells induced by oxidized low-density lipoprotein. PLoS ONE 2014, 9, e95935. [Google Scholar] [CrossRef]
- Chen, Z.; Xue, Q.; Cao, L.; Wang, Y.; Chen, Y.; Zhang, X.; Xiao, F.; Yang, Y.; Hayden, M.R.; Liu, Y.; et al. Toll-Like Receptor 4 Mediated Oxidized Low-Density Lipoprotein-Induced Foam Cell Formation in Vascular Smooth Muscle Cells via Src and Sirt1/3 Pathway. Mediat. Inflamm. 2021, 2021, 6639252. [Google Scholar] [CrossRef]
- Cai, J.-M.; Hatsukami, T.S.; Ferguson, M.S.; Small, R.; Polissar, N.L.; Yuan, C. Classification of Human Carotid Atherosclerotic Lesions With In Vivo Multicontrast Magnetic Resonance Imaging. Circulation 2002, 106, 1368–1373. [Google Scholar] [CrossRef]
- Costopoulos, C.; Huang, Y.; Brown, A.J.; Calvert, P.A.; Hoole, S.P.; West, N.E.; Gillard, J.H.; Teng, Z.; Bennett, M.R. Plaque Rupture in Coronary Atherosclerosis Is Associated With Increased Plaque Structural Stress. JACC Cardiovasc. Imaging 2017, 10, 1472–1483. [Google Scholar] [CrossRef]
- Nasir, K. Cainzos-Achirica M. Role of coronary artery calcium score in the primary prevention of cardiovascular disease. BMJ 2021, 373, n776. [Google Scholar] [CrossRef]
- Stehouwer, C.D.A.; Henry, R.M.A.; Ferreira, I. Arterial stiffness in diabetes and the metabolic syndrome: A pathway to cardiovascular disease. Diabetologia 2008, 51, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Karuparthi, P.R.; Chowdhury, N.A.; Govindarajan, G.; Habibi, J.; Ortmann, R.A. Autoimmune Vasculitis and Plaque Erosion in the Cardiometabolic Syndrome and Type 2 Diabetes mellitus. J. Cardiometab. Syn. 2006, 1, 228–232. [Google Scholar] [CrossRef]
- Lorentzen, K.A.; Chai, S.; Chen, H.; Danielsen, C.C.; Simonsen, U.; Wogensen, L. Mechanisms involved in extracellular matrix remodeling and arterial stiffness induced by hyaluronan accumulation. Atherosclerosis 2016, 244, 195–203. [Google Scholar] [CrossRef]
- Barger, A.C.; Beeuwkes, R.; Lainey, L.L.; Silverman, K.J. Hypothesis: Vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis. N. Engl. J. Med. 1984, 310, 175–177. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Gold, H.K.; Burke, A.P.; Fowler, D.R.; Kruth, H.S.; Weber, D.K.; Farb, A.; Guerrero, J.L.; Hayase, M.; Kutys, R.; et al. Intraplaque Hemorrhage and Progression of Coronary Atheroma. N. Engl. J. Med. 2003, 349, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Stefanadis, C.; Toutouzas, K.; Tsiamis, E.; Stratos, C.; Vavuranakis, M.; Kallikazaros, I.; Panagiotakos, D.; Toutouzas, P. Increased local temperature in human coronary atherosclerotic plaques: An independent predictor of clinical outcome in patients undergoing a percutaneous coronary intervention. J. Am. Coll. Cardiol. 2001, 37, 1277–1283. [Google Scholar] [CrossRef]
- Narula, N.; Dannenberg, A.J.; Olin, J.W.; Bhatt, D.L.; Johnson, K.W.; Nadkarni, G.; Min, J.; Torii, S.; Poojary, P.; Anand, S.S.; et al. Pathology of Peripheral Artery Disease in Patients With Critical Limb Ischemia. J. Am. Coll. Cardiol. 2018, 72, 2152–2163. [Google Scholar] [CrossRef]
- Narula, N.; Olin, J.W.; Narula, N. Pathologic Disparities Between Peripheral Artery Disease and Coronary Artery Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Savji, N.; Rockman, C.B.; Skolnick, A.H.; Guo, Y.; Adelman, M.A.; Riles, T.; Berger, J.S. Association Between Advanced Age and Vascular Disease in Different Arterial Territories: A Population Database of Over 3.6 Million Subjects. J. Am. Coll. Cardiol. 2013, 61, 1736–1743. [Google Scholar] [CrossRef]
- Hooglugt, A.; Klatt, O.; Huveneers, S. Vascular stiffening and endothelial dysfunction in atherosclerosis. Curr. Opin. Lipidol. 2022, 33, 353–363. [Google Scholar] [CrossRef]
- Chirinos, J.A.; Segers, P.; Hughes, T.; Townsend, R. Large Artery Stiffness in Health and Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 1237–1263. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.F. Arterial Stiffness and Hypertension: Chicken or Egg? Hypertension 2014, 64, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, S.E. Ageing of the conduit arteries. J. Pathol. 2007, 211, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.T.; Mojaddedi, S.; Loseke, I.H.; Bray, C. Hypertension in Patients with Peripheral Artery Disease: An Updated Literature Review. Cureus 2024, 16, e62246. [Google Scholar] [CrossRef]
- Hurtubise, J.; McLellan, K.; Durr, K.; Onasanya, O.; Nwabuko, D.; Ndisang, J.F. The different facets of dyslipidemia and hypertension in atherosclerosis. Curr. Atheroscler. Rep. 2016, 18, 82. [Google Scholar] [CrossRef]
- Hayden, M.R. Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease. Medicina 2023, 59, 561. [Google Scholar] [CrossRef]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Heart disease and stroke statistics--2014 update: A report from the american heart association. Circulation 2014, 129, e28–e292. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Hypertension as a risk factor for atherosclerosis: Cardiovascular risk assessment. Front. Cardiovasc. Med. 2022, 9, 959285. [Google Scholar] [CrossRef]
- Scuteri, A.; Najjar, S.S.; Muller, D.C.; Andres, R.; Hougaku, H.; Metter, E.; Lakatta, E.G. Metabolic syndrome amplifies the age-associated increases in vascular thickness and stiffness. J. Am. Coll. Cardiol. 2004, 43, 1388–1395. [Google Scholar] [CrossRef]
- Schillaci, G.; Pirro, M.; Vaudo, G.; Mannarino, M.R.; Savarese, G.; Pucci, G.; Franklin, S.S.; Mannarino, E. Metabolic Syndrome Is Associated With Aortic Stiffness in Untreated Essential Hypertension. Hypertension 2005, 45, 1078–1082. [Google Scholar] [CrossRef]
- Lopes-Vicente, W.R.P.; Rodrigues, S.; Cepeda, F.X.; Jordão, C.P.; Costa-Hong, V.; Dutra-Marques, A.C.B.; Carvalho, J.C.; Alves, M.J.N.N.; Bortolotto, L.A.; Trombetta, I.C. Arterial stiffness and its association with clustering of metabolic syndrome risk factors. Diabetol. Metabol. Syndr. 2017, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhao, W.; Liu, Q.; Qin, M. Association Between Metabolic Syndrome and Peripheral Artery Disease in Elderly Patients with Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 4783–4789. [Google Scholar] [CrossRef] [PubMed]
- Soyoye, D.O.; Abiodun, O.O.; Ikem, R.T.; Kolawole, B.A.; Akintomide, A.O. Diabetes and peripheral artery disease: A review. World J. Diabetes 2021, 12, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.L.; Stafford, L.K.; McLaughlin, S.A.; Boyko, E.J.; Vollset, S.E.; Smith, A.E.; Dalton, B.E.; Duprey, J.; Cruz, J.A.; Hagins, H.; et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. GBD 2021 Diabetes Collaborators. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus–Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef]
- Ye, J.; Li, L.; Wang, M.; Ma, Q.; Tian, Y.; Zhang, Q.; Liu, J.; Li, B.; Zhang, B.; Liu, H.; et al. Diabetes Mellitus Promotes the Development of Atherosclerosis: The Role of NLRP3. Front. Immunol. 2022, 13, 900254. [Google Scholar] [CrossRef]
- Joosten, M.M.; Pai, J.K.; Bertoia, M.L.; Rimm, E.B.; Spiegelman, D.; Mittleman, M.A.; Mukamal, K.J. Associations between conventional cardiovascular risk factors and risk of peripheral artery disease in men. JAMA 2012, 308, 1660–1667. [Google Scholar] [CrossRef]
- Alnima, T.; Meijer, R.I.; Spronk, H.M.; Warlé, M.; Cate, H.T. Diabetes- versus smoking-related thrombo-inflammation in peripheral artery disease. Cardiovasc. Diabetol. 2023, 22, 257. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, T.; Geng, K.; Yuan, G.; Chen, Y.; Xu, Y. Smoking and the Pathophysiology of Peripheral Artery Disease. Front Cardiovasc. Med. 2021, 8, 704106. [Google Scholar] [CrossRef]
- O’Callaghan, P.; Meleady, R.; Fitzgerald, T.; Graham, I.; European COMAC group. Smoking and plasma homocysteine. Eur. Heart J. 2002, 23, 1580–1586. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Homocysteine and reactive oxygen species in metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: The pleiotropic effects of folate supplementation. Nutr. J. 2004, 10, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C. Impaired folate one-carbon metabolism in Type 2 Diabetes, Late-Onset Alzheimer’s Disease and Long COVID. Medicina 2021, 58, 16. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef]
- Ismaeel, A.; Brumberg, R.S.; Kirk, J.S.; Papoutsi, E.; Farmer, P.J.; Bohannon, W.T.; Smith, R.S.; Eidson, J.L.; Sawicki, I.; Koutakis, P. Oxidative Stress and Arterial Dysfunction in Peripheral Artery Disease. Antioxidants 2018, 7, 145. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Ye, Z.-X.; Wang, X.-F.; Chang, J.; Yang, M.-W.; Zhong, H.-H.; Hong, F.-F.; Yang, S.-L. Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed. Pharmacother. 2018, 97, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Epstein, F.H.; Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar] [CrossRef]
- Cooke, J.P.; Dzau, V.J. Nitric oxide synthase: Role in the genesis of vascular disease. Annu. Rev. Med. 1997, 48, 489–509. [Google Scholar] [CrossRef]
- Loscalzo, J.; Jin, R.C. Vascular nitric oxide: Formation and function. J. Blood Med. 2010, 1, 147–162. [Google Scholar] [CrossRef]
- Cooke, J.P. The pivotal role of nitric oxide for vascular health. Can. J. Cardiol. 2004, 20 (Suppl. B), 7B–15B. [Google Scholar]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Allen, J.D.; Giordano, T.; Kevil, C.G. Nitrite and Nitric Oxide Metabolism in Peripheral Artery Disease. Nitric Oxide 2012, 26, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.; Giaginis, C.; Tsigris, C.; Patsouris, E.; Theocharis, S. NF-κB signaling at the crossroads of inflammation and atherogenesis: Searching for new therapeutic links. Expert Opin. Ther. Targets 2014, 18, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Altaany, Z.; Yang, G.; Wang, R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J. Cell Mol. Med. 2013, 17, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Kim, J.; Kwak, S.N.; Lee, K.S.; Lee, D.K.; Ha, K.S.; Won, M.H.; Jeoung, D.; Lee, H.; Kwon, Y.G.; et al. Functional role of NF-κB in expression of human endothelial nitric oxide synthase. Biochem. Biophys. Res. Commun. 2014, 448, 101–107. [Google Scholar] [CrossRef]
- Barbato, J.E.; Tzeng, E. Nitric oxide and arterial disease. J. Vasc. Surg. 2004, 40, 187–193. [Google Scholar] [CrossRef]
- Kamieńska, A.; Danieluk, A.; Niwińska, M.M.; Chlabicz, S. Arterial Stiffness and Ankle-Brachial Index—Cross-Sectional Study of 259 Primary Care Patients ≥ 50 Year-Old. Med. Sci. Monit. 2024, 30, e942718-1. [Google Scholar] [CrossRef]
- Cecelja, M.; Chowienczyk, P. Role of arterial stiffness in cardiovascular disease. JRSM Cardiovasc. Dis. 2012, 1, 1–10. [Google Scholar] [CrossRef]
- Mitchell, G.F.; Parise, H.; Benjamin, E.J.; Larson, M.G.; Keyes, M.J.; Vita, J.A.; Vasan, R.S.; Levy, D. Changes in arterial stiffness and wave reflection with advancing age in the Framingham Heart Study. Hypertension 2004, 43, 1239–1245. [Google Scholar] [CrossRef]
- Maier, J.A.; Andrés, V.; Castiglioni, S.; Giudici, A.; Lau, E.S.; Nemcsik, J.; Seta, F.; Zaninotto, P.; Catalano, M.; Hamburg, N.M. Aging and Vascular Disease: A Multidisciplinary Overview. J. Clin. Med. 2023, 12, 5512. [Google Scholar] [CrossRef]
- Laurent, S.; Boutouyrie, P.; Asmar, R.; Gautier, I.; Laloux, B.; Guize, L.; Ducimetiere, P.; Benetos, A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 2001, 37, 1236–1241. [Google Scholar] [CrossRef]
- Safar, M.E.; Asmar, R.; Benetos, A.; Blacher, J.; Boutouyrie, P.; Lacolley, P.; Laurent, S.; London, G.; Pannier, B.; Protogerou, A.; et al. Interaction Between Hypertension and Arterial Stiffness. Hypertension 2018, 72, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.B.; Mitchell, G.F.; Gill, D.; Burgess, S.; Rahman, M.; Hanff, T.C.; Ramachandran, V.S.; Mutalik, K.M.; Townsend, R.R.; Chirinos, J.A. Arterial Stiffness and Diabetes Risk in Framingham Heart Study and UK Biobank. Circ. Res. 2022, 131, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Huang, J.; Wu, T.; Qi, L. Arterial Stiffness, Genetic Risk, and Type 2 Diabetes: A Prospective Cohort Study. Diabetes Care 2022, 45, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Urbina, E.M.; Gao, Z.; Khoury, P.R.; Martin, L.J.; Dolan, L.M. Insulin resistance and arterial stiffness in healthy adolescents and young adults. Diabetologia 2012, 55, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Safar, M.E.; Czernichow, S.A.A.; Blacher, J. Obesity, arterial stiffness, and cardiovascular risk. J. Am. Soc. Nephrol. 2006, 17 (Suppl. 2), S109–S111. [Google Scholar] [CrossRef]
- Aroor, A.R.; Jia, G.; Sowers, J.R. Cellular mechanisms underlying obesity-induced arterial stiffness. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 314, R387–R398. [Google Scholar] [CrossRef]
- Camplain, R.; Meyer, M.L.; Tanaka, H.; Palta, P.; Agarwal, S.K.; Aguilar, D.; Butler, K.R.; Heiss, G. Smoking Behaviors and Arterial Stiffness Measured by Pulse Wave Velocity in Older Adults: The Atherosclerosis Risk in Communities (ARIC) Study. Am. J. Hypertens. 2016, 29, 1268–1275. [Google Scholar] [CrossRef]
- Saladini, F. Effects of Different Kinds of Physical Activity on Vascular Function. J. Clin. Med. 2023, 13, 152. [Google Scholar] [CrossRef]
- Ramirez-Perez, F.I.; Cabral-Amador, F.J.; Whaley-Connell, A.T.; Aroor, A.R.; Morales-Quinones, M.; Woodford, M.L.; Ghiarone, T.; Ferreira-Santos, L.; Jurrissen, T.J.; Manrique-Acevedo, C.M.; et al. Cystamine reduces vascular stiffness in Western diet-fed female mice. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H167–H180. [Google Scholar] [CrossRef]
- Padilla, J.; Ramirez-Perez, F.I.; Habibi, J.; Bostick, B.; Aroor, A.R.; Hayden, M.R.; Jia, G.; Garro, M.; DeMarco, V.G.; Manrique, C.; et al. Regular Exercise Reduces Endothelial Cortical Stiffness in Western Diet-Fed Female Mice. Hypertension 2016, 68, 1236–1244. [Google Scholar] [CrossRef]
- van Sloten, T.T.; Stehouwer, C.D.A. Carotid Stiffness: A Novel Cerebrovascular Disease Risk Factor. Pulse 2016, 4, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Zieman, S.J.; Melenovsky, V.; Kass, D.A. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arter. Thromb. Vasc. Biol. 2005, 25, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Agbaje, A.O. Arterial stiffness precedes hypertension and metabolic risks in youth: A review. J. Hypertens. 2022, 49, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Shirwany, N.A.; Zou, M.H. Arterial stiffness: A brief review. Acta Pharmacol. Sin. 2010, 31, 1267–1276. [Google Scholar] [CrossRef]
- Sowers, K.M.; Hayden, M.R. Calcific uremic arteriolopathy: Pathophysiology, reactive oxygen species and therapeutic approaches. Oxidative Med. Cell Longev. 2010, 3, 109–121. [Google Scholar] [CrossRef]
- Hayden, M.R. Empagliflozin Ameliorates Tinica Adiposa Expansion and Vascular Stiffening of the Descending Aorta in Female db/db Mice. Adipobiology 2020, 10, 41–54. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R.; DeMarco, V.G. Ultrastructure study of the transgenic REN2 rat aorta—Part 2: Media, external elastic lamina, and adventitia. Biomed. Rev. 2019, 30, 111–123. [Google Scholar] [CrossRef]
- Sowers, J.R.; Whaley-Connell, A.; Hayden, M. The role of overweight and obesity in the cardiorenal syndrome. Cardiorenal. Med. 2011, 1, 5–12. [Google Scholar] [CrossRef]
- Petrie, J.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef]
- Duca, L.; Blaise, S.; Romier, B.; Laffargue, M.; Gayral, S.; El Btaouri, H.; Kawecki, C.; Guillot, A.; Martiny, L.; Debelle, L.; et al. Matrix ageing and vascular impacts: Focus on elastin fragmentation. Cardiovasc Res. 2016, 110, 298–308. [Google Scholar] [CrossRef]
- Le Page, A.; Khalil, A.; Vermette, P.; Frost, E.H.; Larbi, A.; Witkowski, J.M.; Fulop, T. The role of elastin-derived peptides in human physiology and diseases. Matrix Biol. 2019, 84, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Wahart, A.; Hocine, T.; Albrecht, C.; Henry, A.; Sarazin, T.; Martiny, L.; El Btaouri, H.; Maurice, P.; Bennasroune, A.; Romier-Crouzet, B.; et al. Role of elastin peptides and elastin receptor complex in metabolic and cardiovascular diseases. FEBS J. 2019, 286, 2980–2993. [Google Scholar] [CrossRef] [PubMed]
- Babici, D.; Kudej, R.K.; McNulty, T.; Zhang, J.; Oydanich, M.; Berkman, T.; Nishimura, K.; Bishop, S.P.; Vatner, D.E.; Vatner, S.F. Mechanisms of increased vascular stiffness down the aortic tree in aging, premenopausal female monkeys. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H222–H234. [Google Scholar] [CrossRef] [PubMed]
- Gornik, H.L.; Aronow, H.D.; Goodney, P.P.; Arya, S.; Brewster, L.P.; Byrd, L.; Chandra, V.; Drachman, D.E.; Eaves, J.M.; Ehrman, J.K.; et al. 2024ACC/AHA/AACVPR/APMA/ABC/SCAI/SVM/SVN/SVS/SIR/VESS Guideline for the Management of Lower Extremity Peripheral Artery Disease: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1313–e1410. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, Y.; Zhu, Q.; Zhang, H.; Li, Z.; Wang, J.; Pan, H.; Guo, Z.; Zheng, Y.; Li, X.; et al. Combined Evaluation of Arterial Stiffness and Blood Pressure Promotes Risk Stratification of Peripheral Arterial Disease. JACC Asia 2023, 3, 287–297. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Chen, Z.; Zhang, M. Arterial Calcification and Its Association with Stroke: Implication of Risk, Prognosis, Treatment Response, and Prevention. Front. Cell. Neurosci. 2022, 16, 845215. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Mineral exploration: Search for the mechanism of vascular calcification and beyond: The 2003 Jeffrey M. Hoeg Award lecture. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1739–1743. [Google Scholar] [CrossRef]
- Moe, S.M.; Chen, N.X. Pathophysiology of vascular calcification in chronic kidney disease. Circ. Res. 2004, 95, 560–567. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, K.; Xiong, Y.; He, Y.; Zhou, Y.; Hayden, M.R. Phosphate Toxicity and Vascular Calcification in Chronic Kidney Disease: A Closer Look Utilizing Transmission Electron Microscopy. Curr. Protein Pept. Sci. 2023, 24, 621–639. [Google Scholar] [CrossRef]
- Wu, X.H.; Chen, X.-Y.; Wang, L.J.; Wong, K.S. Intracranial Artery Calcification and Its Clinical Significance. J. Clin. Neurol. 2016, 12, 253–261. [Google Scholar] [CrossRef]
- Tesauro, M.; Mauriello, A.; Rovella, V.; Annicchiarico-Petruzzelli, M.; Cardillo, C.; Melino, G.; Di Daniele, N. Arterial ageing: From endothelial dysfunction to vascular calcification. J. Intern. Med. 2017, 281, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular Calcification: An Update on Mechanisms and Challenges in Treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C.; Kolb, L.; Sowers, J.R.; Khanna, R. Vascular ossification-calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis-calcific uremic arteriolopathy: The emerging role of sodium thiosulfate. Cardiovasc. Diabetol. 2005, 4, 1–22. [Google Scholar] [CrossRef]
- Vos, A.; Van Hecke, W.; Spliet, W.G.; Goldschmeding, R.; Isgum, I.; Kockelkoren, R.; Bleys, R.L.; Mali, W.P.; de Jong, P.A.; Vink, A. Predominance of Nonatherosclerotic Internal Elastic Lamina Calcification in the Intracranial Internal Carotid Artery. Stroke 2016, 47, 221–223. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- McEniery, C.M.; McDonnell, B.J.; So, A.; Aitken, S.; Bolton, C.E.; Munnery, M.; Hickson, S.S.; Yasmin; Maki-Petaja, K.M.; Cockcroft, J.R.; et al. Aortic calcification is associated with aortic stiffness and isolated systolic hypertension in healthy individuals. Hypertension 2009, 53, 524–531. [Google Scholar] [CrossRef]
- Bethel, M.; Annex, B.H. Peripheral arterial disease: A small and large vessel problem. Am. Heart J. Plus. Cardiol. Res. Pract. 2023, 28, 100291. [Google Scholar] [CrossRef]
- Mietus, C.J.; Lackner, T.J.; Karvelis, P.S.; Willcockson, G.T.; Shields, C.M.; Lambert, N.G.; Koutakis, P.; Fuglestad, M.A.; Hernandez, H.; Haynatzki, G.R.; et al. Abnormal Microvascular Architecture, Fibrosis, and Pericyte in the Calf Muscle of Peripheral Artery Disease Patients with Claudication and Critical Limb Ischemia. J. Clin. Med. 2020, 9, 2575. [Google Scholar] [CrossRef]
- Duscha, B.D.; E Kraus, W.; Jones, W.S.; Robbins, J.L.; Piner, L.W.; Huffman, K.M.; Allen, J.D.; Annex, B.H. Skeletal muscle capillary density is related to anaerobic threshold and claudication in peripheral artery disease. Vasc. Med. 2020, 25, 411–418. [Google Scholar] [CrossRef]
- Behroozian, A.; Beckman, J.A. Microvascular Disease Increases Amputation in Patients With Peripheral Artery Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 534–540. [Google Scholar] [CrossRef]
- Hayden, M.R.; Yang, Y.; Habibi, J.; Bagree, S.V.; Sowers, J.R. Pericytopathy: Oxidative Stress and Impaired Cellular Longevity in the Pancreas and Skeletal Muscle in Metabolic Syndrome and Type 2 Diabetes. Oxidative Med. Cell. Longevity 2010, 3, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Sfyri, P.; Matsakas, A. Crossroads between peripheral atherosclerosis, western-type diet and skeletal muscle pathophysiology: Emphasis on apolipoprotein E deficiency and peripheral arterial disease. Biomed. Sci. 2017, 24, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, M.; Lee, N.H.; Moon, J.Y.; Han, W.M.; Anderson, S.E.; Choi, J.J.; Shin, E.; Nakhai, S.A.; Tran, T.; Aliya, B.; et al. Critical Limb Ischemia Induces Remodeling of Skeletal Muscle Motor Unit, Myonuclear-, and Mitochondrial-Domains. Sci. Rep. 2019, 9, 9551. [Google Scholar] [CrossRef]
- Shu, J.; Santulli, G. Update on peripheral artery disease: Epidemiology and evidence-based facts. Atherosclerosis 2018, 275, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Paradis, S.; Charles, A.-L.; Meyer, A.; Lejay, A.; Scholey, J.W.; Chakfé, N.; Zoll, J.; Geny, B. Chronology of mitochondrial and cellular events during skeletal muscle ischemia-reperfusion. Am. J. Physiol. Cell Physiol. 2016, 310, C968–C982. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, W.; Zhu, T.; Li, Y.; Cao, P.; Wu, Y.; Zhang, Y. Ischemia-Reperfusion Injury in Peripheral Artery Disease and Traditional Chinese Medicine Treatment. Evid. Based Complement. Altern. Med. 2021, 2021, 4954070. [Google Scholar] [CrossRef]
- England, J.D.; Regensteiner, J.G.; Ringel, S.P.; Carry, M.R.; Hiatt, W.R. Muscle denervation in peripheral arterial disease. Neurology 1992, 42, 994–999. [Google Scholar] [CrossRef]
- Hayden, M.R.; Salam, M.; Sowers, J.R. Reactive Oxygen Species and Diabetic Peripheral Neuropathy—A Closer Look. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 14, pp. 3375–3403. [Google Scholar] [CrossRef]
- Lastra, G.; Habibi, J.; Whaley-Connell, A.T.; Manrique, C.; Hayden, M.R.; Rehmer, J.; Patel, K.; Ferrario, C.; Sowers, J.R. Direct Renin Inhibition Improves Systemic Insulin Resistance and Skeletal Muscle. Endocrinology 2009, 150, 2561–2568. [Google Scholar] [CrossRef]
- Gupta, K.; Harvima, I.T. Mast cell-neural interactions contribute to pain and itch. Immunol. Rev. 2018, 282, 168–187. [Google Scholar] [CrossRef]
- Queme, L.F.; Ross, J.L.; Jankowski, M.P. Peripheral Mechanisms of Ischemic Myalgia. Front. Cell. Neurosci. 2017, 11, 419. [Google Scholar] [CrossRef]
- Smolderen, K.G.; Alabi, O.; Collins, T.C.; Dennis, B.; Goodney, P.P.; Mena-Hurtado, C.; Spertus, J.A.; Decker, C. American Heart Association Council on Peripheral Vascular Disease and Council on Lifestyle and Cardiometabolic Health. Advancing Peripheral Artery Disease Quality of Care and Outcomes Through Patient-Reported Health Status Assessment: A Scientific Statement from the American Heart Association. Circulation 2022, 146, e286–e297. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.G.; McCord, J.L.; Kaufman, M.P. Role played by P2X and P2Y receptors in evoking the muscle chemoreflex. J. Appl. Physiol. 2008, 104, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Ives, S.J.; McDaniel, J.; Witman, M.A.H.; Richardson, R.S. Passive limb movement: Evidence of mechanoreflex sex specificity. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H154–H161. [Google Scholar] [CrossRef] [PubMed]
- Tsilimigras, D.I.; Moris, D.; Karaolanis, G.; Kakkos, S.K.; Filis, K.; Sigala, F. Rivaroxaban versus Clopidogrel for Peripheral Disease: A Clinico-Economic Approach of the COMPASS. Trial. Curr. Pharm. Des. 2018, 24, 4516–4517. [Google Scholar] [CrossRef]
- Hirsch, A.T.; Criqui, M.H.; Treat-Jacobson, D.; Regensteiner, J.G.; Creager, M.A.; Olin, J.W.; Krook, S.H.; Hunninghake, D.B.; Comerota, A.J.; Walsh, M.E.; et al. Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA 2001, 286, 1317–1324. [Google Scholar] [CrossRef]
- Jansen, S.; de Borst, G.J.; Hinchliffe, R.; Teraa, M. Peripheral Artery Disease: Underappreciated Impact and Residual Cardiovascular Risk Despite Revascularization. Clin. Ther. 2023, 45, 1019–1022. [Google Scholar] [CrossRef]
- Olin, J.W.; Sealove, B.A. Peripheral Artery Disease: Current insight Into the Disease and Its Diagnosis and Management. Mayo Clin. Proc. 2010, 85, 678–692. [Google Scholar] [CrossRef]
- Mastracci, T.M.; Anand, S.S.; Aday, A.W. Peripheral Artery Disease: A High-Risk Yet Understudied, Underdiagnosed, and Undertreated Condition-A Call to Action. Can. J. Cardiol. 2022, 38, 553–554. [Google Scholar] [CrossRef]
- McDermott, M.M.; Kerwin, D.R.; Liu, K.; Martin, G.J.; O’brien, E.; Kaplan, H.; Greenland, P. Prevalence and significance of unrecognized lower extremity peripheral arterial disease in general medicine practice. J. Gen. Intern. Med. 2001, 16, 384–390. [Google Scholar] [CrossRef]
- Mohler, E.R.; Treat-Jacobson, D.; Reilly, M.P.; E Cunningham, K.; Miani, M.; Criqui, M.H.; Hiatt, W.R.; Hirsch, A.T. Utility and barriers to performance of the ankle-brachial index in primary care practice. Vasc. Med. 2004, 9, 253–260. [Google Scholar] [CrossRef]
- Arabzadeh, A.; Faghfuri, E.; Soofiyani, S.R.; Abdolahinia, E.D.; Siapush, S.; Nejati-Koshki, K.; Shahrami, B.; Asghariazar, V.; Pahlavan, Y. Current and Novel Emerging Medical Therapies for Peripheral Artery Disease: A Literature Review. Adv. Pharm. Bull. 2022, 13, 259–268. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, N. Sodium Thiosulfate: An Innovative Multi-Target Repurposed Treatment Strategy for Late-Onset Alzheimer’s Disease. Pharmaceuticals 2024, 17, 1741. [Google Scholar] [CrossRef]
- Cicone, J.S.; Petronis, J.B.; Embert, C.D.; Spector, D.A. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am. J. Kidney Dis. 2004, 43, 1104–1108. [Google Scholar] [CrossRef]
- Hayden, M.R.; Kolb, L.G.; Khanna, R. Calciphylaxis and the cardiometabolic syndrome. J. Cardiometab. Syndr. 2006, 1, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Yerram, P.; Saab, G.; Karuparthi, P.R.; Hayden, M.R.; Khanna, R. Nephrogenic systemic fibrosis: A mysterious disease in patients with renal failure—Role of gadolinium-based contrast media in causation and the beneficial effect of intravenous sodium thiosulfate. Clin. J. Am. Soc. Nephrol. 2007, 2, 258–263. [Google Scholar] [CrossRef]
- Rogers, N.M.; Coates, P.T. Calcific uraemic arteriolopathy: An update. Curr. Opin. Nephrol. Hypertens. 2008, 17, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Araya, C.E.; Fennell, R.S.; Neiberger, R.E.; Dharnidharka, V.R. Sodium thiosulfate treatment for calcific uremic arteriolopathy in children and young adults. Clin. J. Am. Soc. Nephrol. 2006, 1, 1161–1166. [Google Scholar] [CrossRef]
- Hayden, M.R.; Goldsmith, D.; Sowers, J.R.; Khanna, R. Calciphylaxis: Calcific uremic arteriolopathy and the emerging role of sodium thiosulfate. Int. Urol. Nephrol. 2008, 40, 443–451. [Google Scholar] [CrossRef]
- Hayden, M.R.; Goldsmith, D.J. New hope for the treatment of calciphylaxis. Semin. Dial. 2010, 23, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Brunelli, S.M.; Meade, D.; Wang, W.; Hymes, J.; Lacson, L., Jr. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin. J. Am. Soc. Nephrol. 2013, 8, 1162–1170. [Google Scholar] [CrossRef]
- Generali, J.A.; Cada, D.J. Sodium Thiosulfate: Calciphylaxis. Hosp. Pharm. 2015, 50, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Pfeifle, C.E.; Howell, S.B.; Felthouse, R.D.; Woliver, T.B.; Andrews, P.A.; Markman, M.; Murphy, M.P. High-dose cisplatin with sodium thiosulfate protection. J. Clin. Oncol. 1985, 3, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Brock, P.R.; Maibach, R.; Childs, M.; Rajput, K.; Roebuck, D.; Sullivan, M.; Laithier, V.; Ronghe, M.; Dall’Igna, P.; Hiyama, E.; et al. Sodium Thiosulfate for Protection from Cisplatin-Induced Hearing Loss. N. Engl. J. Med. 2018, 378, 2376–2385. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Sodium Thiosulfate: Pediatric First Approval. Paediatr. Drugs. 2023, 25, 239–244. [Google Scholar] [CrossRef]
- Sen, U.; Vacek, T.P.; Hughes, W.M.; Kumar, M.; Moshal, K.S.; Tyagi, N.; Metreveli, N.; Hayden, M.R.; Tyagi, S.C. Cardioprotective role of sodium thiosulfate on chronic heart failure by modulating endogenous H2S generation. Pharmacology 2008, 82, 201–213. [Google Scholar] [CrossRef]
- Wen, W.; Portales-Castillo, I.; Seethapathy, R.; Krinsky, S.; Kroshinsky, D.; Kalim, S.; Goverman, J.; Nazarian, R.M.; Chitalia, V.; Malhotra, R.; et al. Intravenous sodium thiosulphate for vascular calcification of hemodialysis patients-a systematic review and meta-analysis. Nephrol. Dial. Transplant. 2023, 38, 733–745. [Google Scholar] [CrossRef]
- Du, J.T.; Li, W.; Yang, J.Y.; Tang, C.S.; Li, Q.; Jin, H.F. Hydrogen sulfide is endogenously generated in rat skeletal muscle and exerts a protective effect against oxidative stress. Chin. Med. J. 2013, 126, 930–936. [Google Scholar] [CrossRef]
- Tokashiki, K.; Ishida, A.; Kouchi, M.; Ishihara, S.; Tomiyama, N.; Kohagura, K.; Iseki, K.; Takishita, S. Successful management of critical limb ischemia with intravenous sodium thiosulfate in a chronic hemodialysis patient. Clin. Nephrol. 2006, 66, 140–143. [Google Scholar] [CrossRef]
- Allen, R.E.; Kirby, K.A. Nocturnal leg cramps. Am. Fam. Physician 2012, 86, 350–355. [Google Scholar]
- Criqui, M.H.; Matsushita, K.; Aboyans, V.; Hess, C.N.; Hicks, C.W.; Kwan, T.W.; McDermott, M.M.; Misra, S.; Ujueta, F. Lower Extremity Peripheral Artery Disease: Contemporary Epidemiology, Management Gaps, and Future Directions: A Scientific Statement from the American Heart Association. Circulation 2021, 144, e171–e191. [Google Scholar] [CrossRef]
- Weber, T.; Meyer, C.C. “Man Is as OLD as His Arteries” Taken Literally: In Search of the Best Metric. Hypertension 2020, 76, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
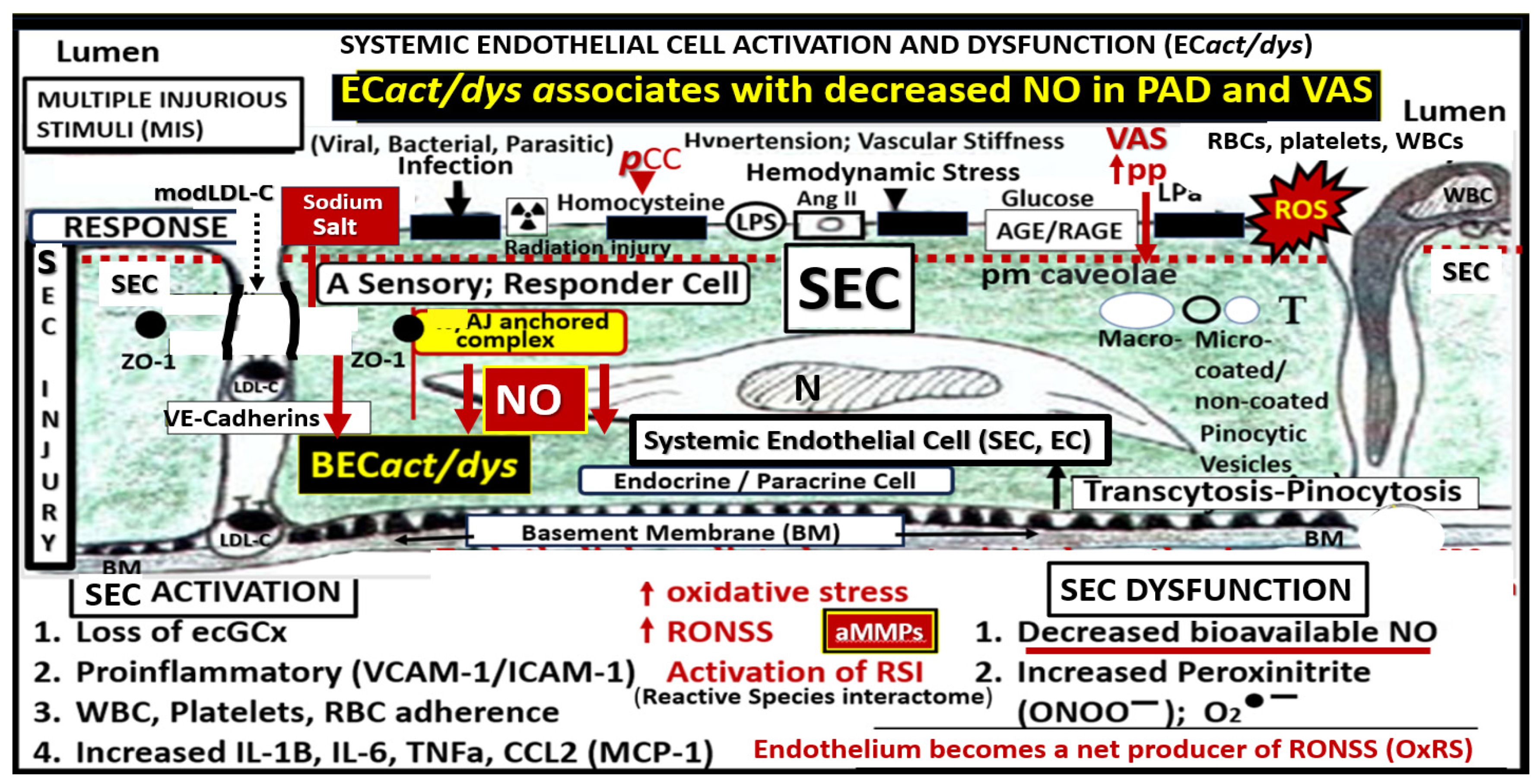

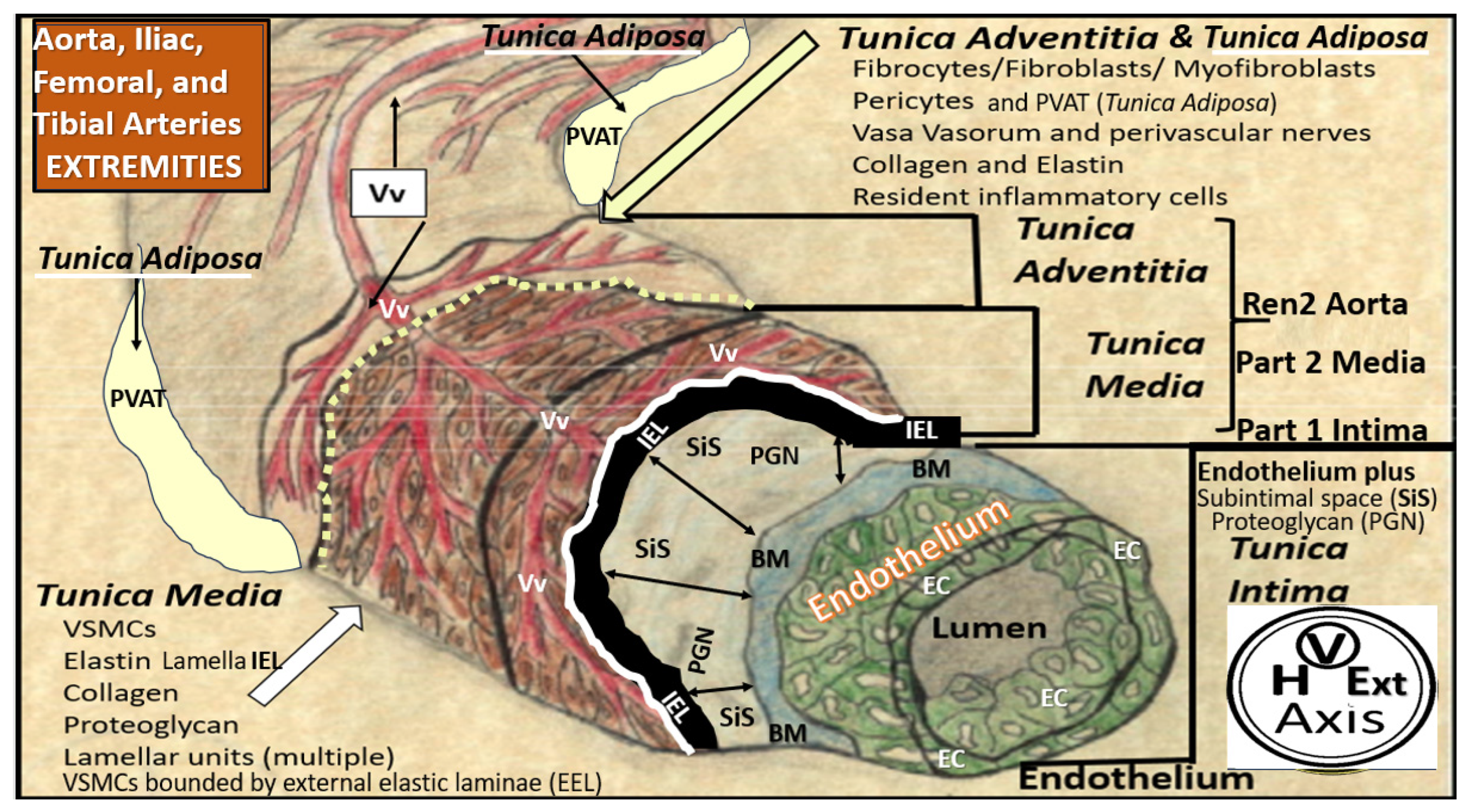
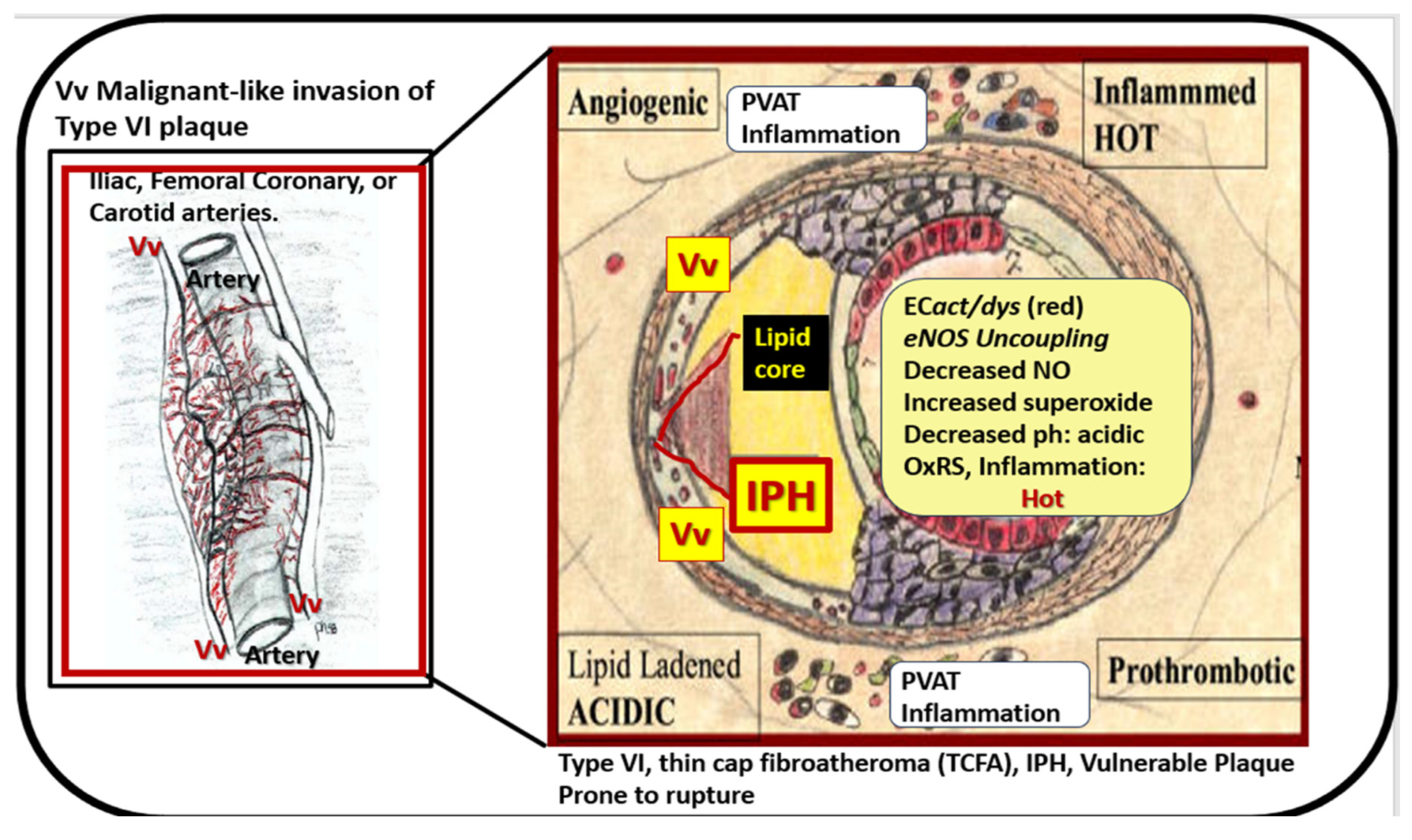
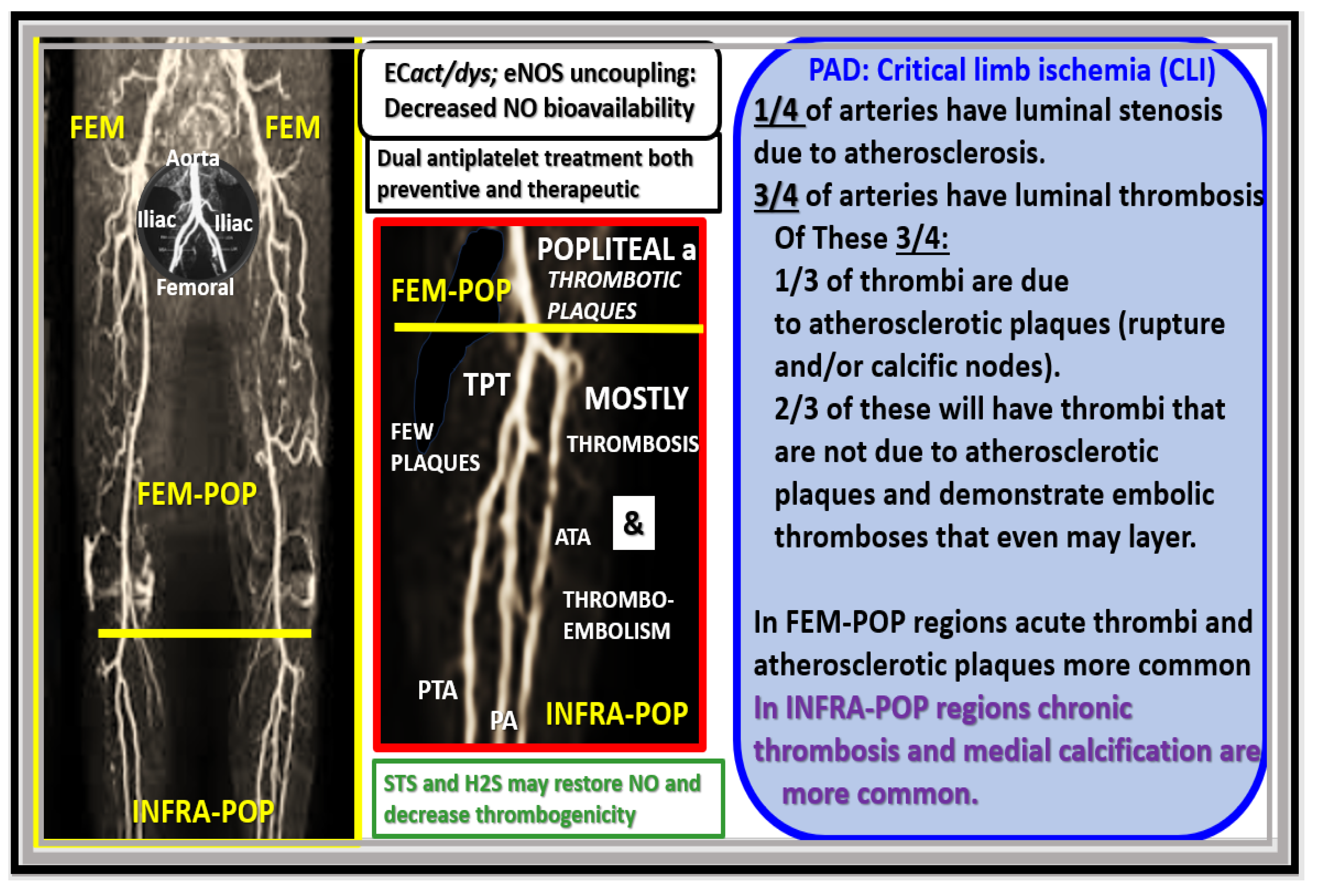
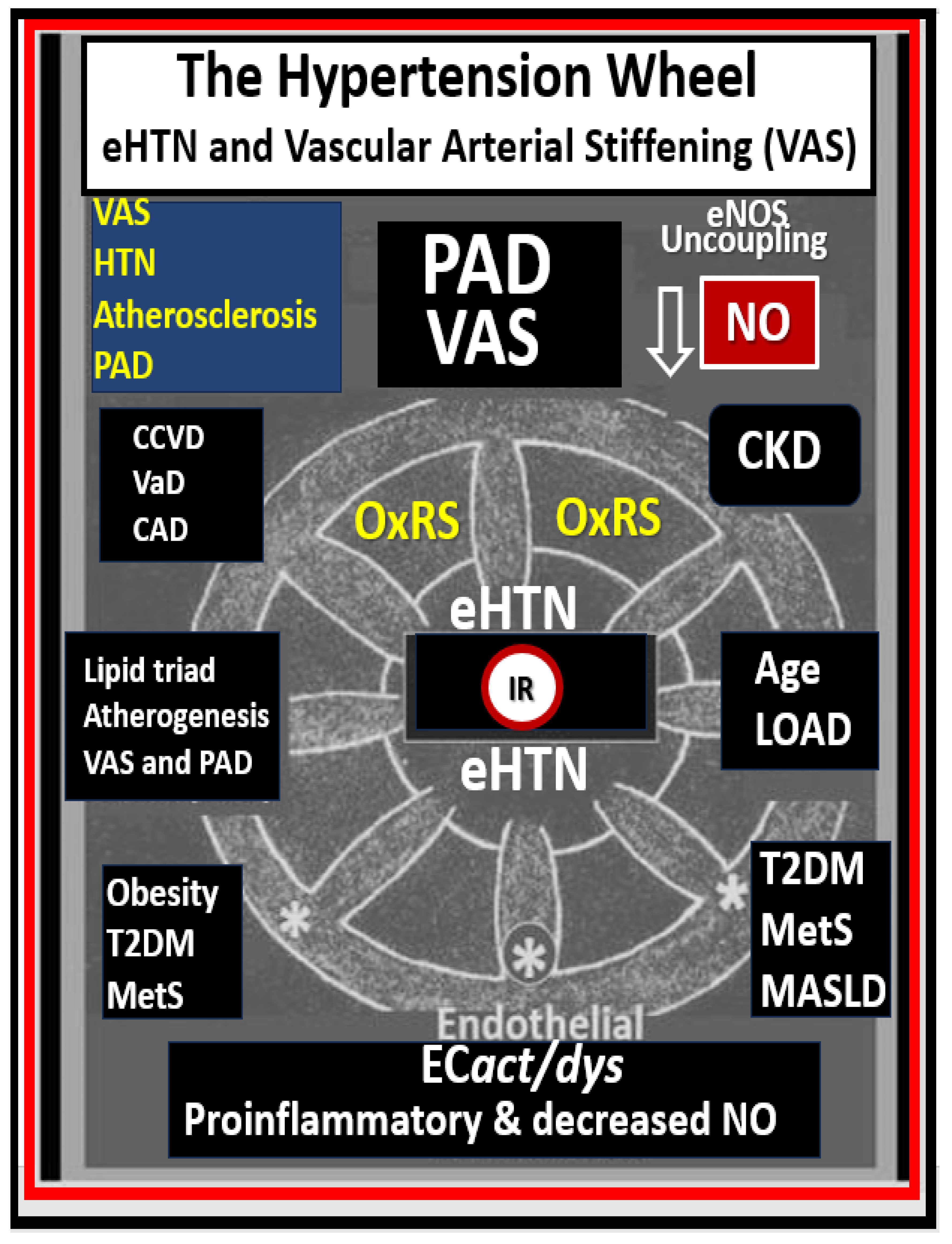
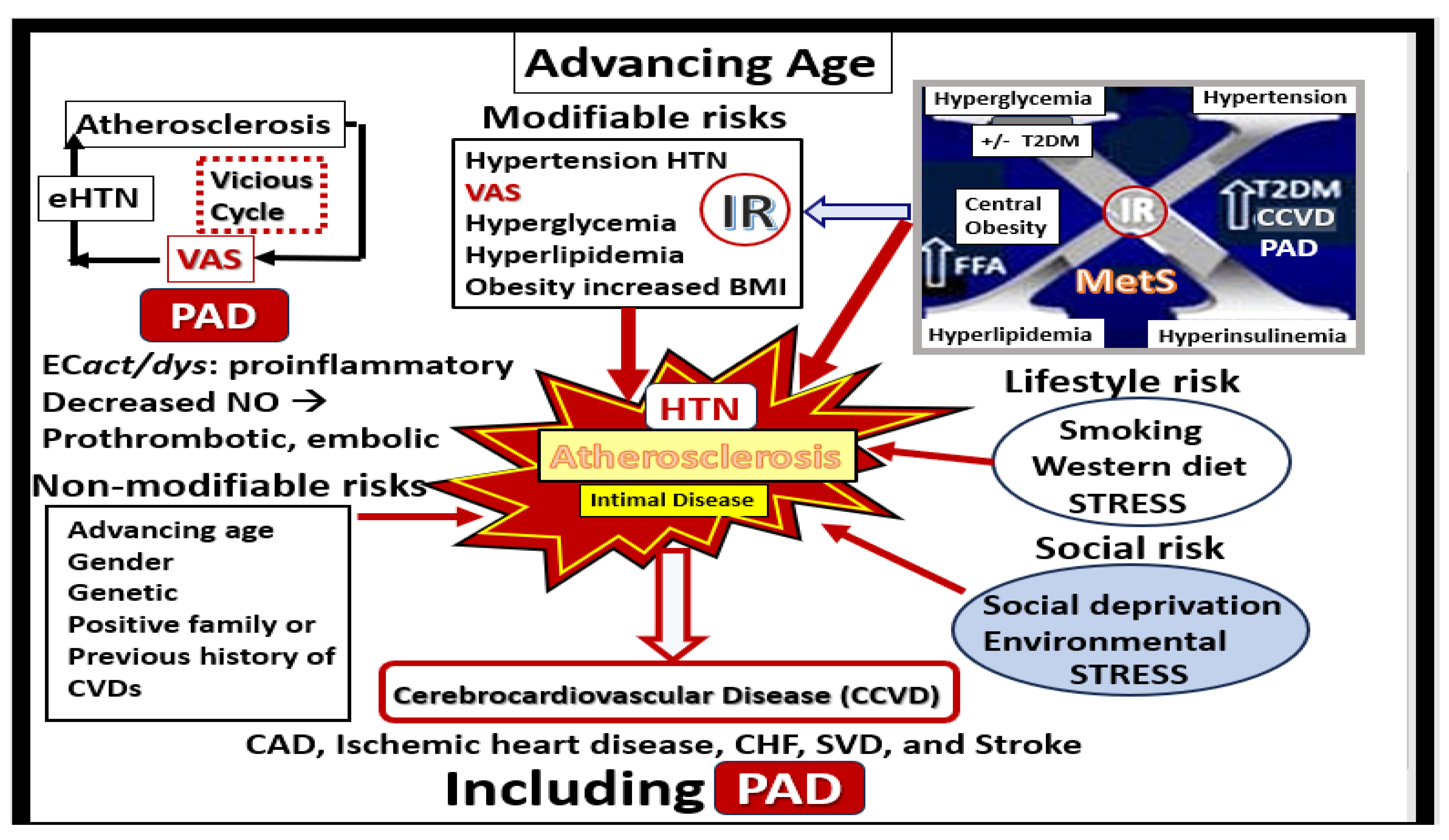

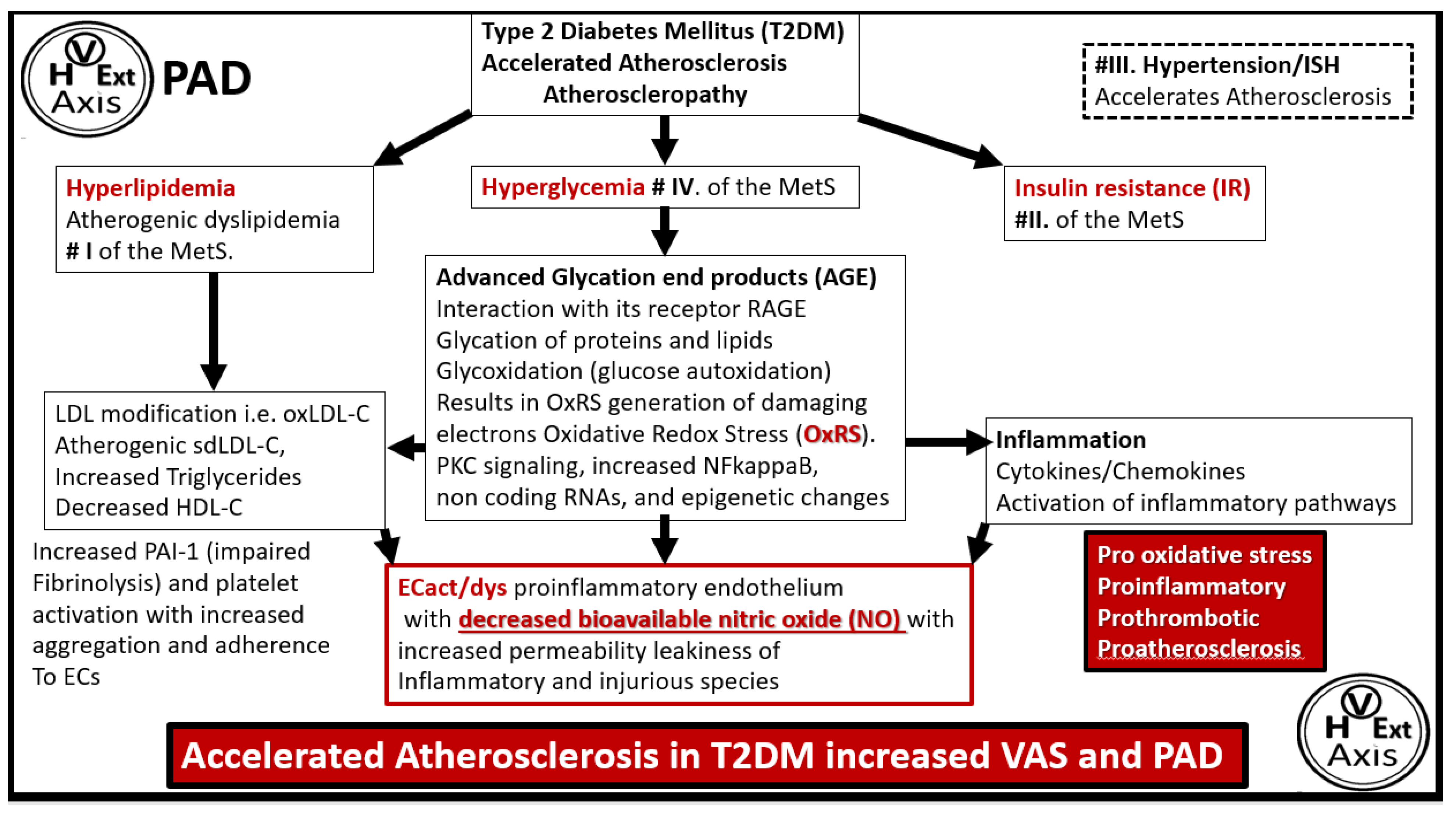
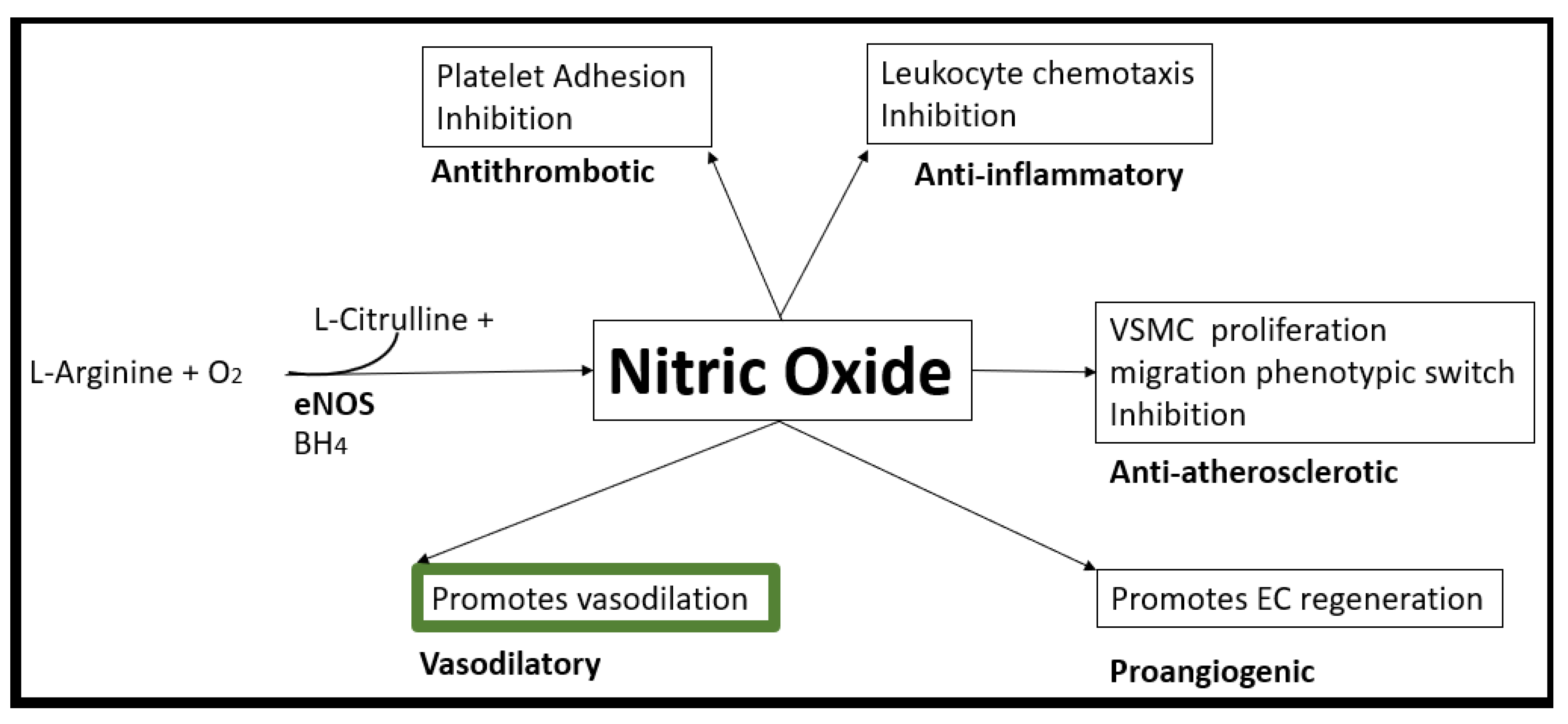
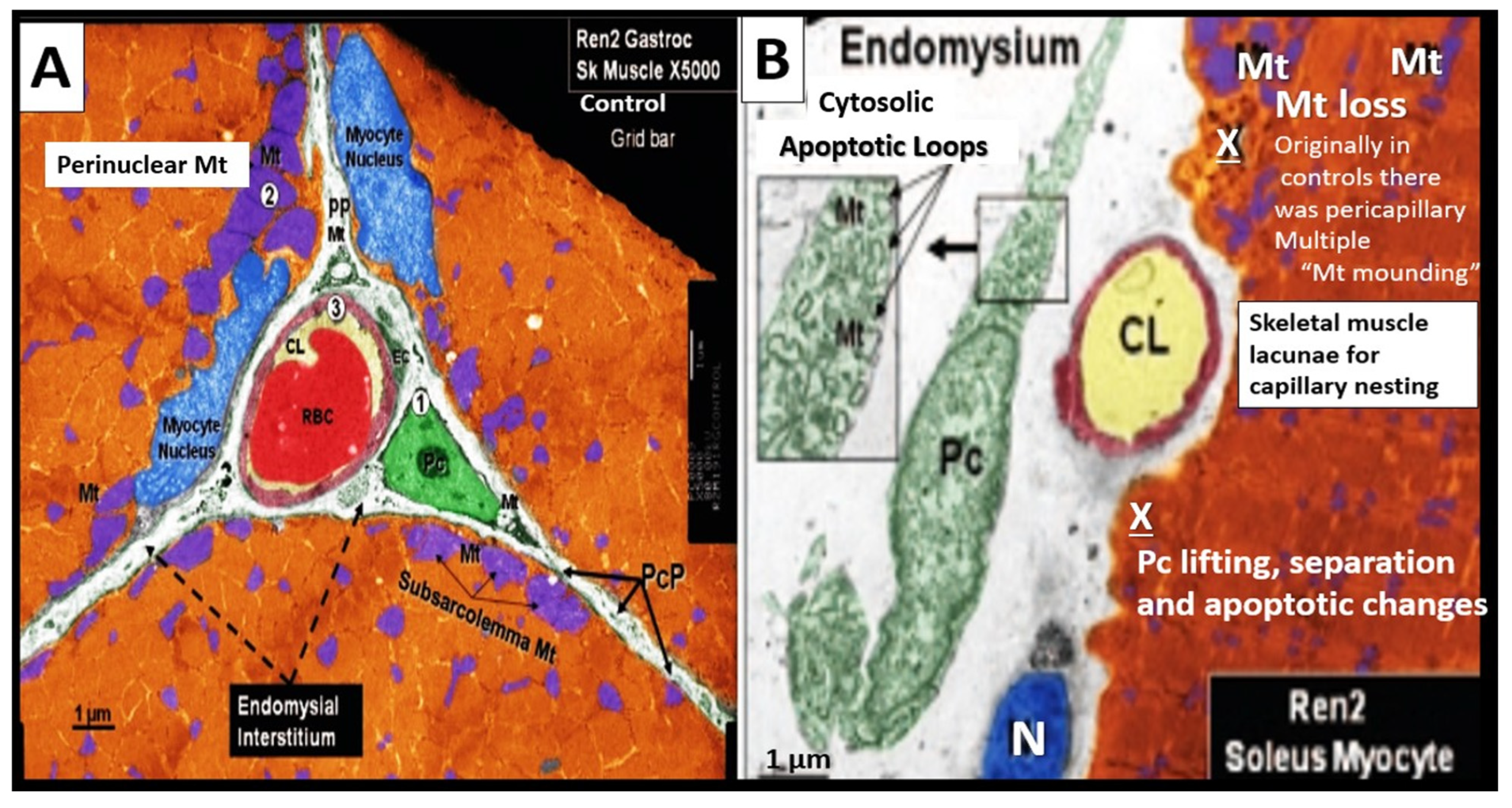
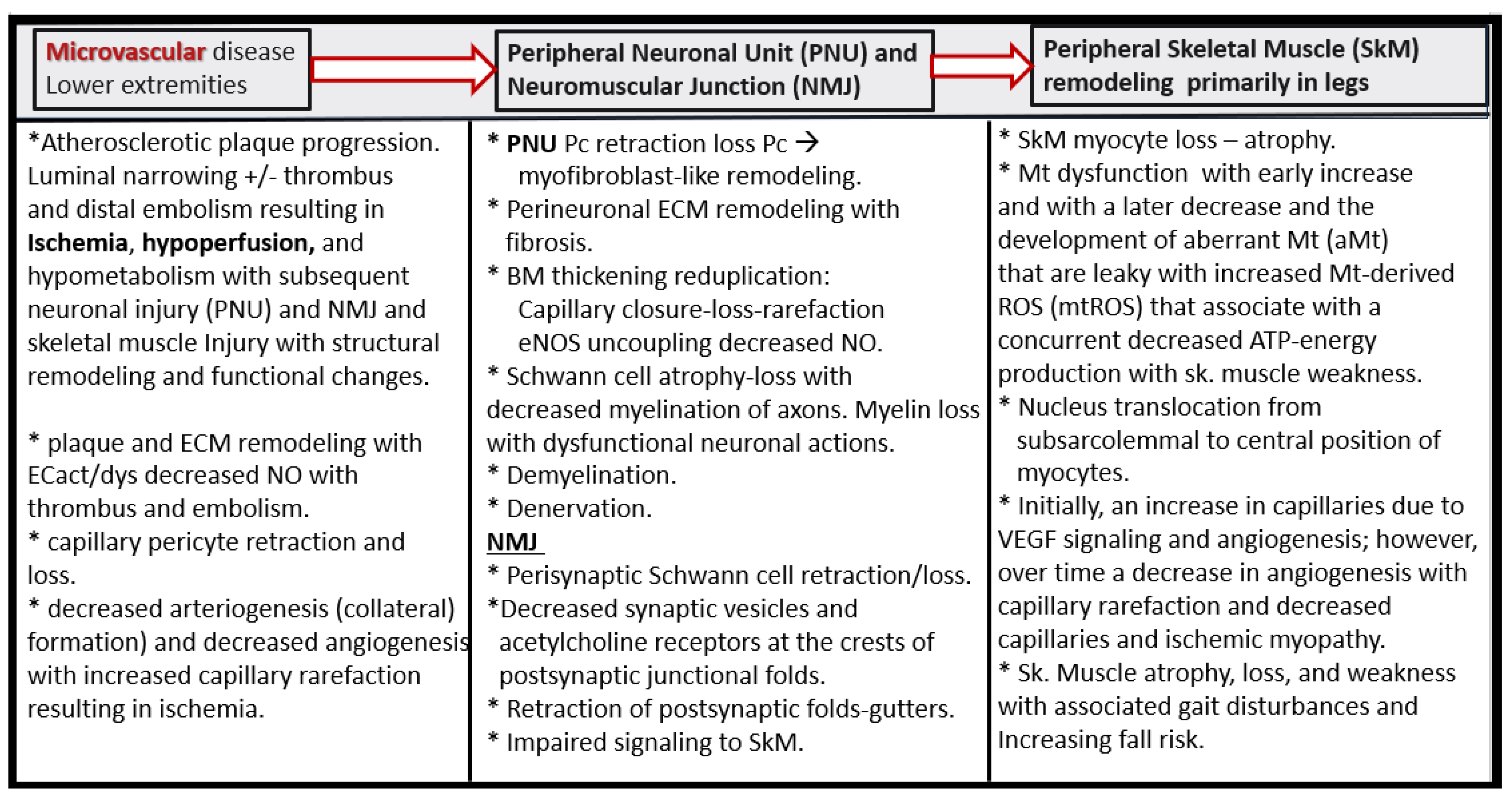
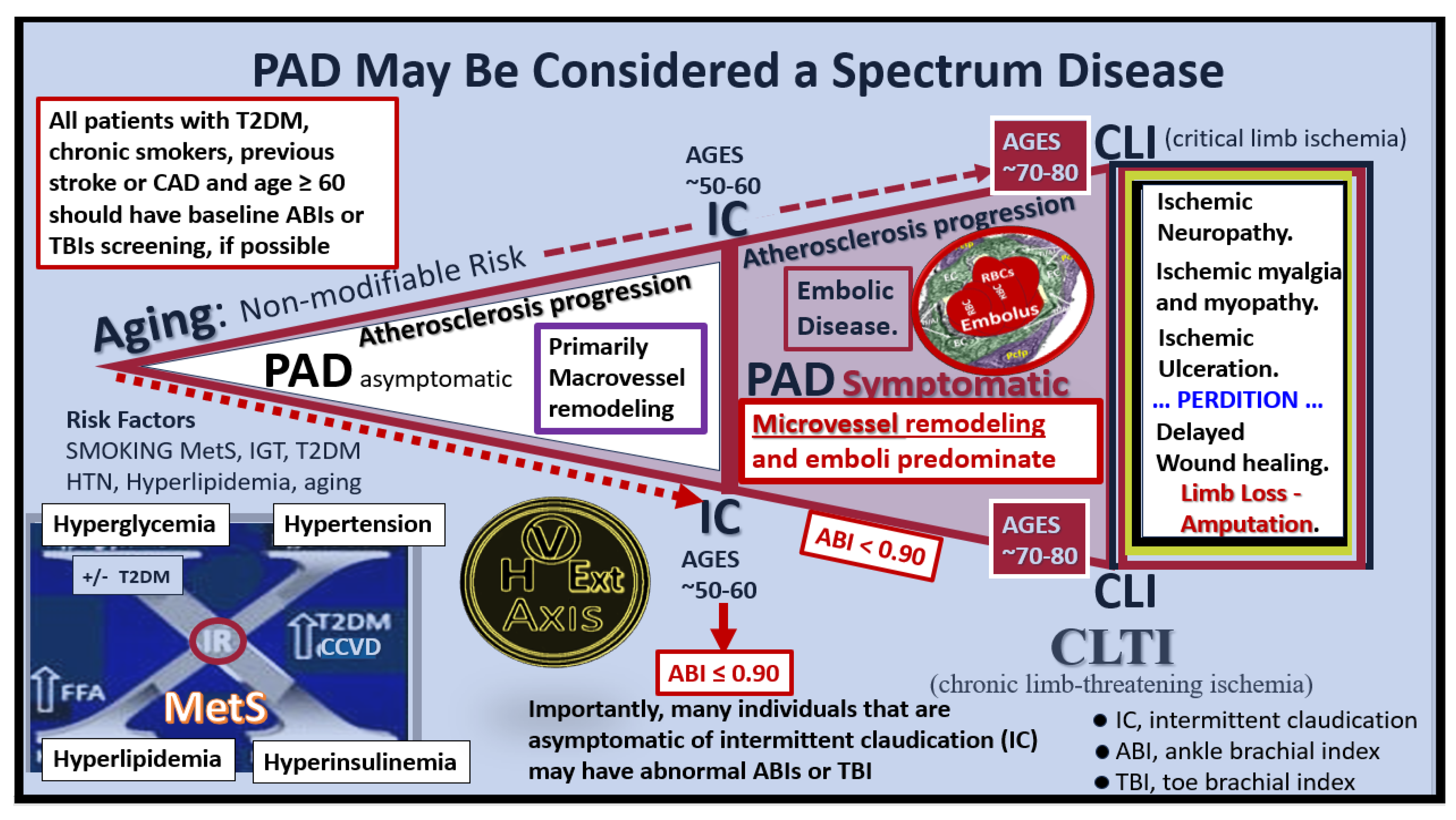
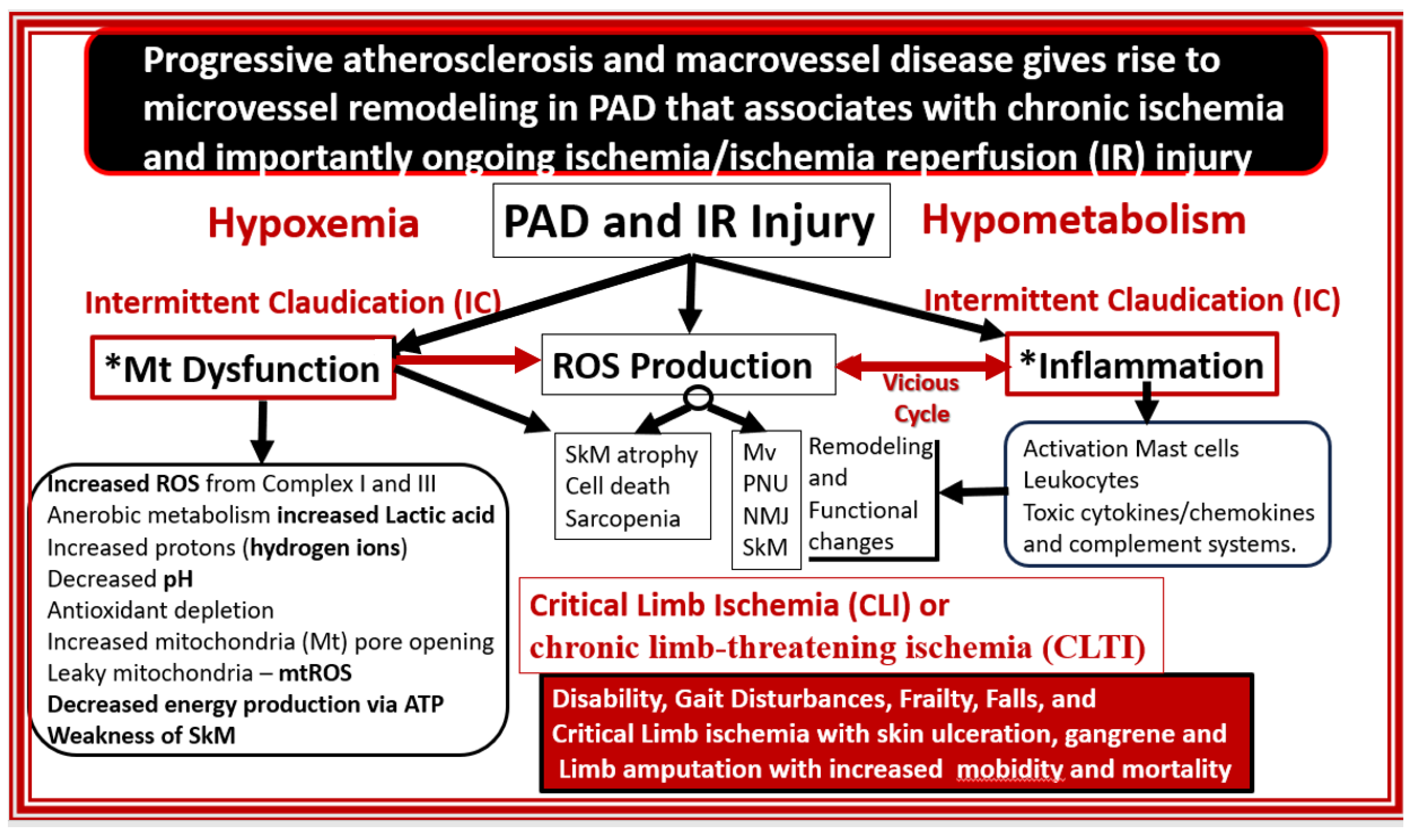
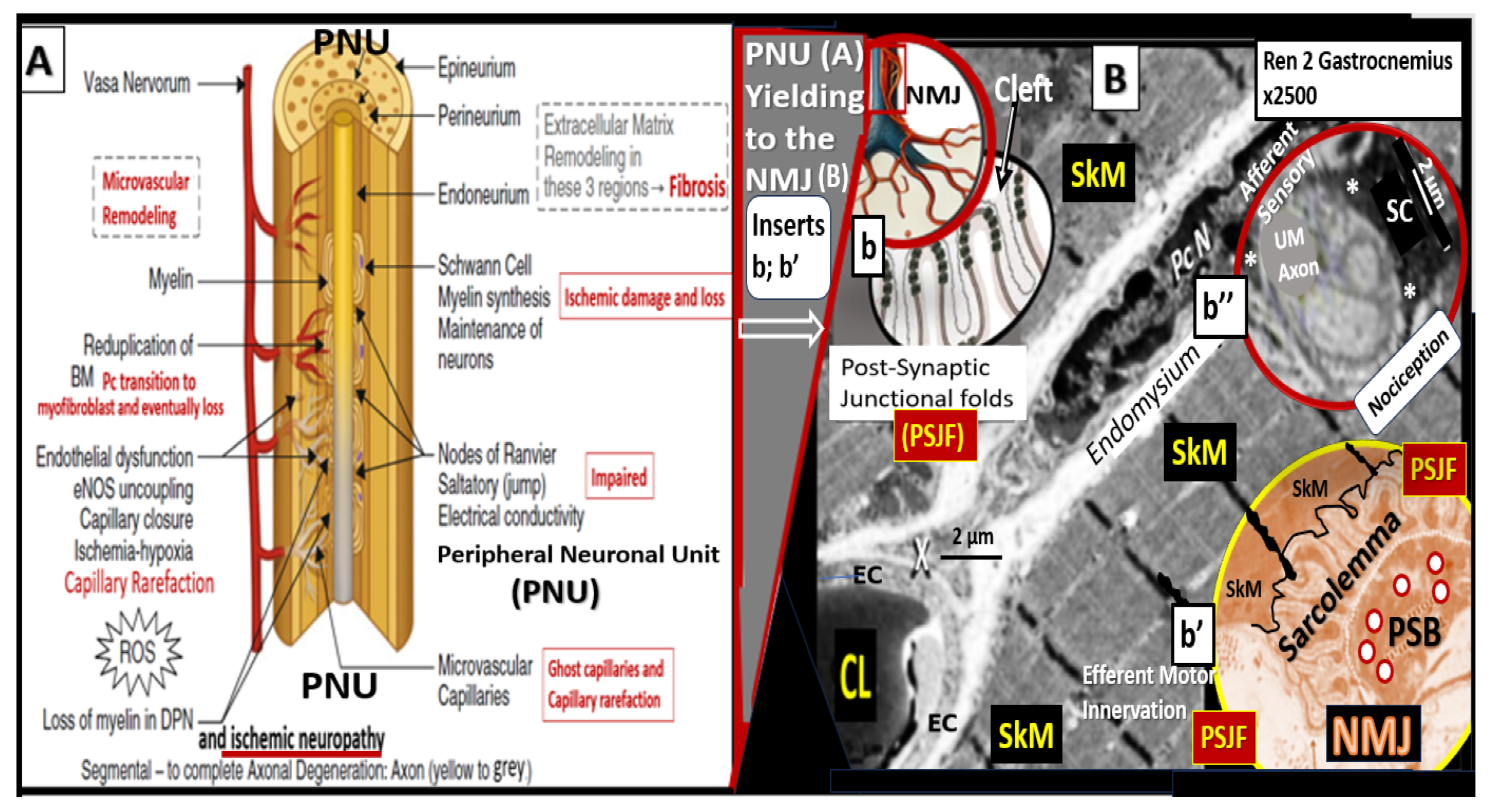
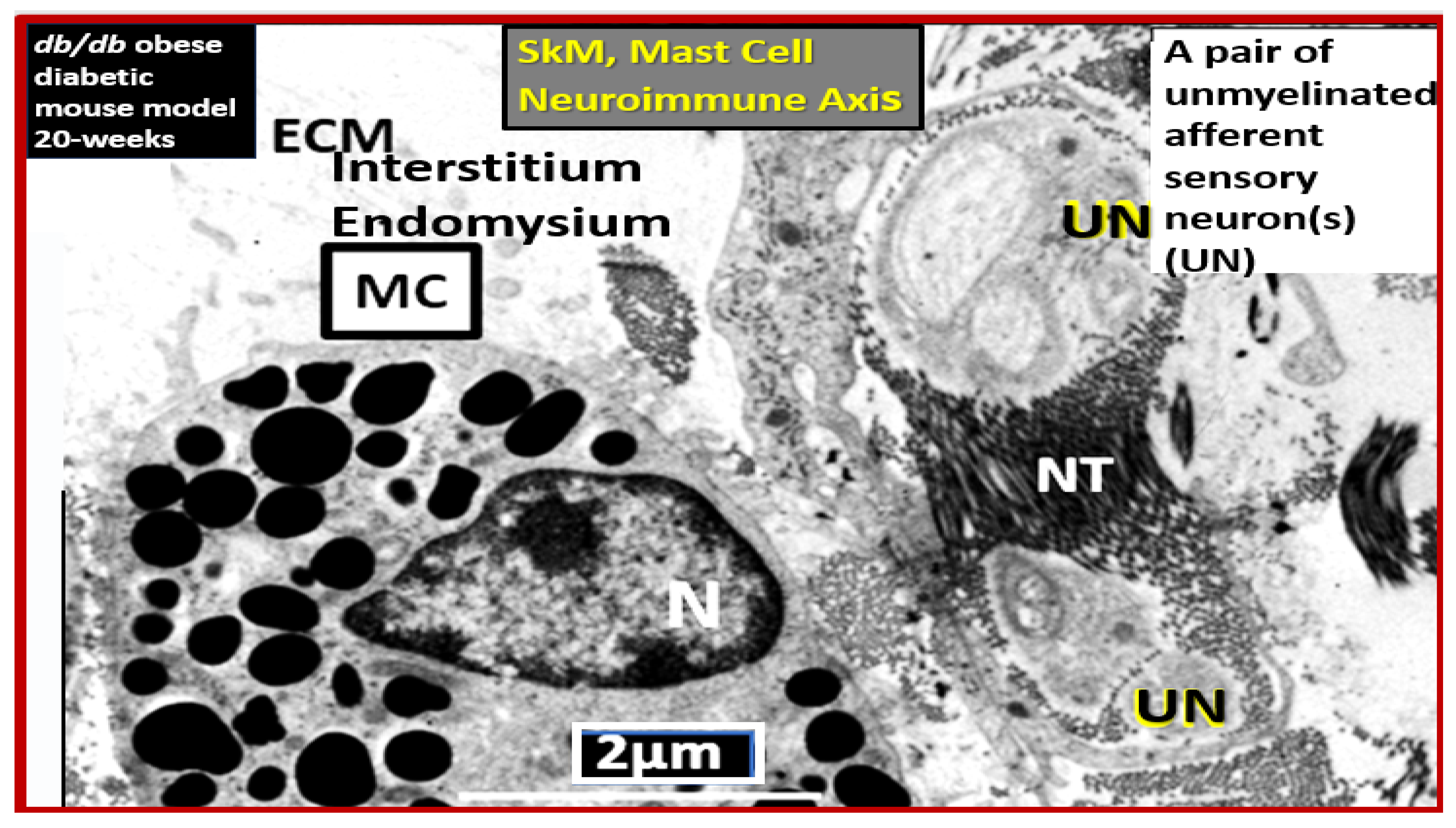
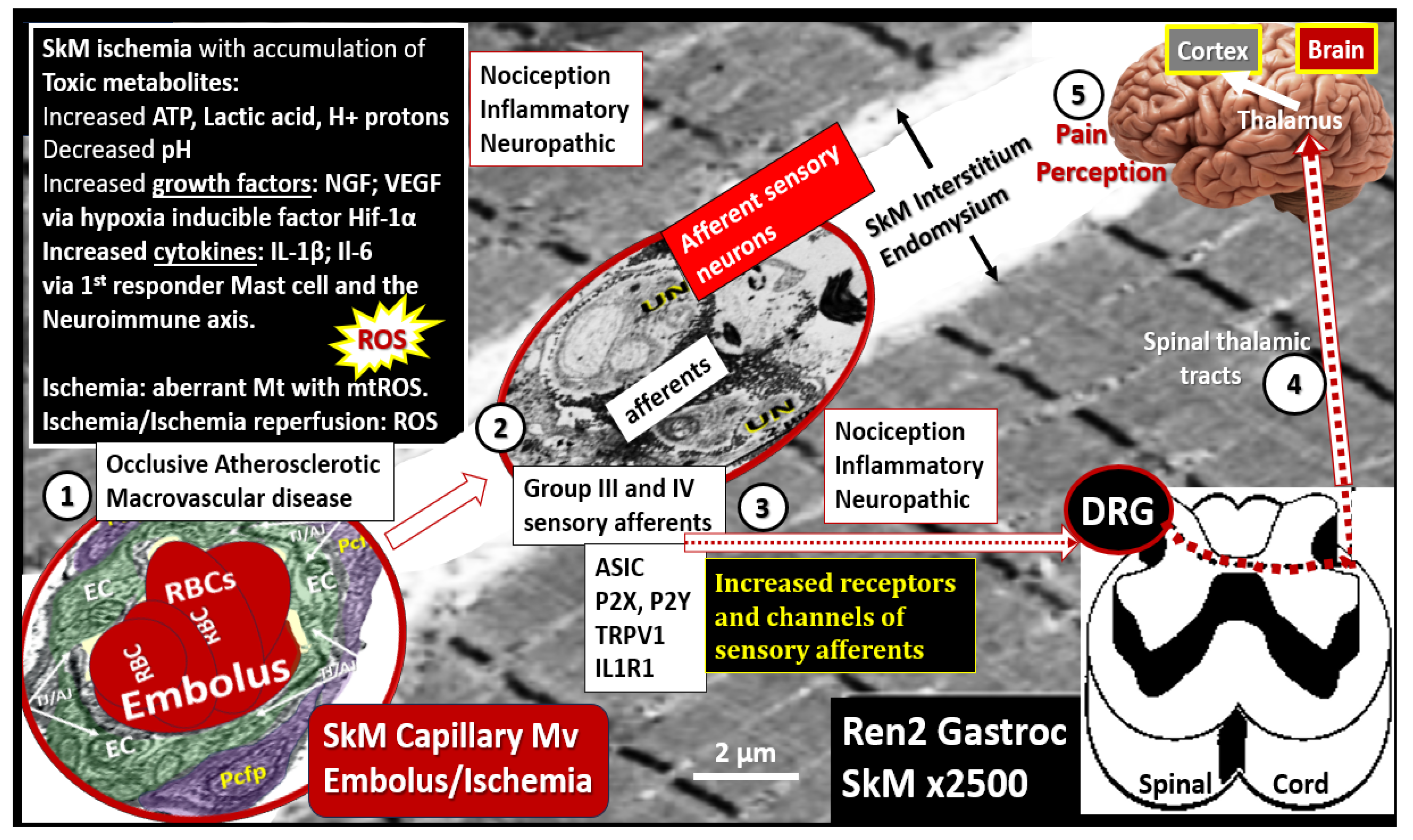
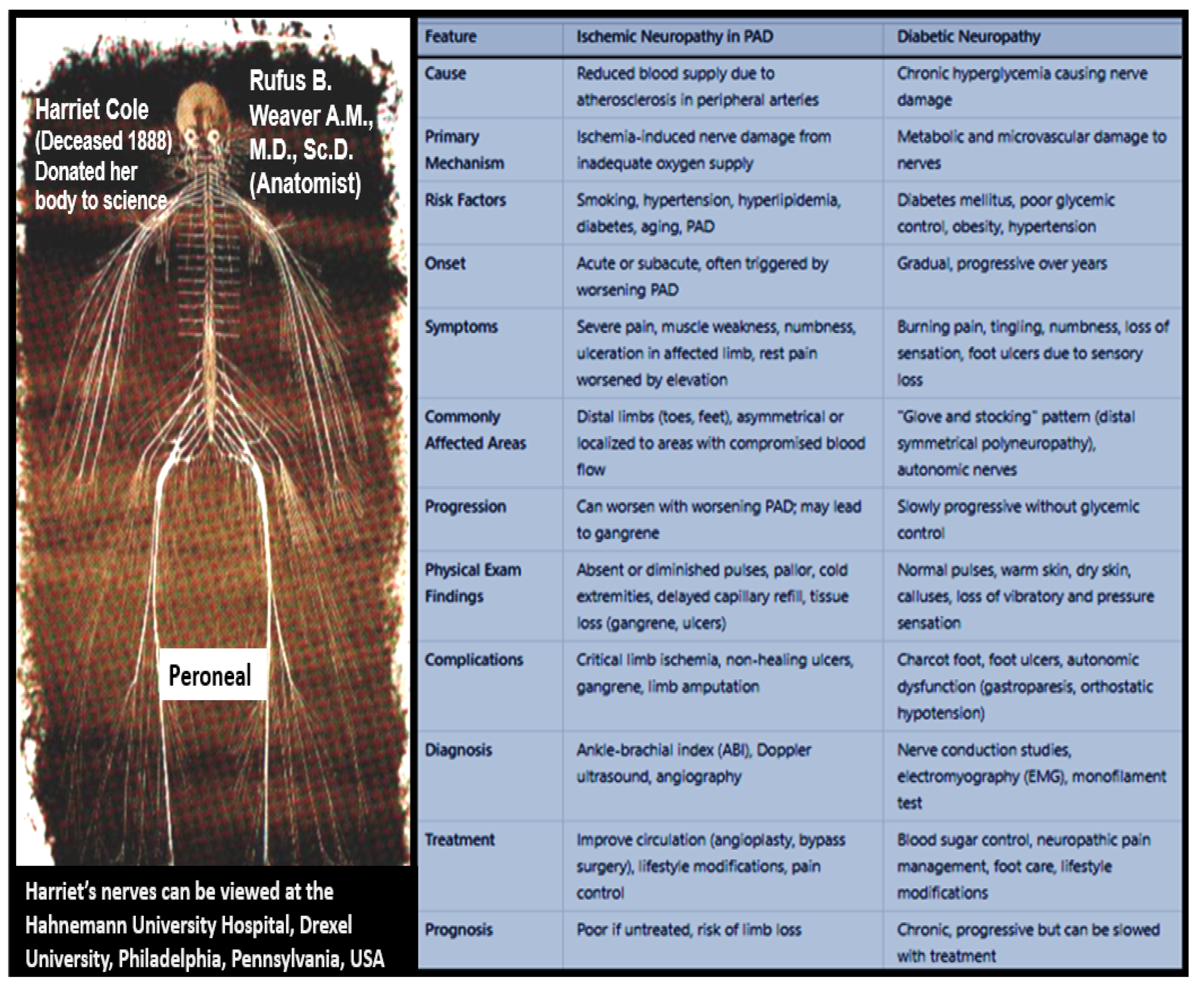

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayden, M.R. Peripheral Artery Disease: Atherosclerosis, Decreased Nitric Oxide, and Vascular Arterial Stiffening. J. Vasc. Dis. 2025, 4, 21. https://doi.org/10.3390/jvd4020021
Hayden MR. Peripheral Artery Disease: Atherosclerosis, Decreased Nitric Oxide, and Vascular Arterial Stiffening. Journal of Vascular Diseases. 2025; 4(2):21. https://doi.org/10.3390/jvd4020021
Chicago/Turabian StyleHayden, Melvin R. 2025. "Peripheral Artery Disease: Atherosclerosis, Decreased Nitric Oxide, and Vascular Arterial Stiffening" Journal of Vascular Diseases 4, no. 2: 21. https://doi.org/10.3390/jvd4020021
APA StyleHayden, M. R. (2025). Peripheral Artery Disease: Atherosclerosis, Decreased Nitric Oxide, and Vascular Arterial Stiffening. Journal of Vascular Diseases, 4(2), 21. https://doi.org/10.3390/jvd4020021