1. Introduction
The vascular system encompasses a monolayer of cells known as the endothelium. These cells release mediators that control several physiological functions, such as vascular homeostasis by maintaining the balance among vasodilation and vasoconstriction, platelet adhesion, and atheromatous plaque formation and progression, among others. Endothelial dysfunction (ED) is recognized as a predictor for the onset of cardiovascular diseases, as it not only accompanies their progression but also involves a reduction in the release of vasodilators and an increase in vasoconstrictor factors by endothelial cells. Nitric oxide (NO), endothelium-derived hyperpolarizing factor (EDHF), and prostacyclin are components acting as vasodilators. Endothelin-1 (ET-1), angiotensin II, thromboxane A2, and prostaglandin H2 [
1,
2,
3] are key contributors acting primarily as vasoconstrictors. Furthermore, ED manifests with aging, even in individuals who are healthy [
4,
5]. With aging, blood vessel walls undergo thickening and increased stiffness, leading to a decline in various processes such as the regulation of vascular tone and the loss of endothelium-dependent factors responsible for inducing vasodilation, including nitric oxide (NO) [
3,
6].
Several studies have indicated that vascular relaxation in the aorta of renal hypertensive rats is impaired due to various factors, one of which is the elevated generation of superoxide anion (O
2•−) [
7,
8,
9]. At high concentrations, superoxide anions react with NO, therefore its bioavailability could be significantly reduced when O
2•− is present. This results in the formation of peroxynitrite, a potent oxidant interacting with DNA, lipids, and proteins triggering cell signaling and or increasing oxidative injury, inducing necrosis or apoptosis [
10].
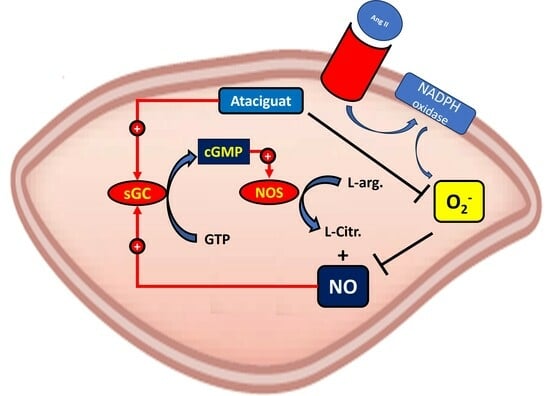
These reactions initiate cellular responses that span from subtle adjustments in cell signaling to the activation of the soluble guanylate cyclase (sGC) enzyme. They interact with the heme group, leading to a modification in the conformation of sGC, rendering it active. The sGC then catalyzes the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP), serving as an intracellular second messenger. This activates the protein kinase C (PKC), inducing several biological effects. The sGC activity is mediated by the sGC activator or sGC stimulator; stimulation is dependent on the sGC heme portion oxidative status, and the ferrous heme (Fe
2+) oxidative state is essential for its activation. Nitric oxide serves as a stimulator for sGC, displaying low affinity for ferric heme (Fe
3+). Consequently, the direct activation of soluble guanylate cyclase (sGC) is regarded as a promising strategy for addressing various cardiovascular disorders linked to endothelial dysfunction [
11].
Ataciguat (HMR 1766) binds to sGC when the iron is in the ferrous heme (Fe
2+), ferric heme (Fe
3+) state or even without this grouping. This characteristic is very important in a biological environment with low redox potential, in which nitric oxide cannot modulate its biological effects by sGC stimulation [
11,
12]. This compound is suggested for managing cardiovascular diseases linked to oxidative stress and pulmonary hypertension [
13]. Since its initial publication in 2004, there has been only a limited number of studies exploring the effects of HMR-1766. Notably, in a rat model with congestive heart failure, prolonged treatment with ataciguat resulted in enhanced vascular function, increased NO sensitivity, and diminished platelet activation [
13]. Moreover, within endothelial cells, ataciguat triggers the synthesis of nitric oxide through a mechanism reliant on nitric oxide synthase activation. This, in turn, leads to a potentiation of vascular relaxation in isolated aortic rings induced by ataciguat [
14].
In this context, the objective of this study is to examine whether the sGC activator ataciguat can reverse the endothelial dysfunction caused by angiotensin II through the inactivation of superoxide anion.
2. Materials and Methods
2.1. Experimental Animals
Male Wistar rats weighing 180–200 g were employed in the study. The rats were kept in a light–dark cycle with unrestricted access to both standard rat chow and water. Renovascular hypertension was induced in the rats utilizing the two-kidney one-clip (2K-1C) model, as outlined in previous descriptions [
7,
15]. This model involves the constriction of only one renal artery to decrease chronic renal perfusion and activate the endogenous renin-angiotensin-aldosterone system. Following a midline laparotomy, anesthetized animals received a silver clip with an internal diameter of 0.20 mm placed around the left renal artery using tribromoethanol (2.5 mg/kg, i.p.). Normotensive two-kidney rats (2K) underwent laparotomy without additional procedures. Post-surgery, the animals were placed in a heated enclosure at approximately 28 °C for optimal anesthesia recovery. In non-anesthetized animals, systolic blood pressure (SBP) was measured on a weekly basis using an indirect tail-cuff method (MLT125R pulse transducer/pressure cuff connected to the PowerLab 4/S analog-to-digital converter; AD Instruments Pty Ltd., Bella Vista, NSW, Australia). To enhance the blood pressure signal, animals were situated in a heated enclosure at approximately 30 °C. Rats were classified as hypertensive if their SBP exceeded 160 mmHg six weeks after surgery. All protocols adhered to the guidelines of the Animal Care and Use Committee of the Federal University of São Carlos and received approval from this committee (CEUA n° 1626101216).
2.2. Vascular Reactivity Studies
At the six week post-surgery mark, male Wistar rats, both hypertensive and normotensive, underwent anesthesia with isoflurane. Following euthanasia via decapitation, the thoracic aortas were isolated and 3 mm long aortic rings were placed in 5 mL bath chambers for isolated organ studies. The chambers contained Krebs solution at 37 °C and pH 7.4 and were continuously aerated with 95% O2 and 5% CO2. The aortic rings were placed in an isometric myograph (Mulvany–Halpern model 610 DMT-USA, Marietta, GA, USA) and were recorded using the PowerLab8/SP as a data acquisition system (ADInstruments Pty Ltd., Colorado Springs, CO, USA).
The aortic rings underwent tension adjustment to 1.5 g, which was recalibrated every 15 min during a 60 min equilibration period before introducing the designated drug. The assessment of endothelial integrity was based on the extent of relaxation induced by 1 μM acetylcholine following the contraction of the aortic ring caused by phenylephrine (0.1 μM). A ring was discarded if acetylcholine-induced relaxation was below 60% in 2K-1C or 80% in 2K rat aortas. Aortic rings from normotensive (2K) and hypertensive (2K-1C) rats were exposed to ataciguat (at a concentration of 0.1 mM) or a control solution (PBS) for a duration of 30 min. Subsequently, the aortic artery rings underwent three washes to eliminate the incubated drug, followed by pre-contraction with phenylephrine (0.1 μM). Following pre-contraction with phenylephrine (0.1 μM), an endothelium-dependent relaxation curve was generated using concentration-effect curves for acetylcholine. Each experiment utilized aortic rings obtained from separate animals.
2.3. Cell Culture
▪ Immortalized human umbilical endothelial cells (HUVEC) were cultured in DMEM (Inlab) supplemented with 10% fetal calf serum, antibiotics, and antimycotics. Cultures were sustained in a 5% CO2 atmosphere at 37 ± 2 °C. Upon reaching 80 to 90% confluence, the cells were trypsinized, centrifuged at 1200 rpm for 5 min, and then seeded in 96-well plates (TPP).
2.4. Nitric Oxide (NO) Measurements
▪ HUVEC were seeded in 96-well plates at a concentration of 5 × 10⁴ cells per well. The plates were then incubated for 24 h in a humidified incubator at 37 °C with 5% CO
2. Following the 24 h incubation, the treatment was removed, and the plates were gently washed with phosphate buffer saline (PBS). To detect intracellular NO, the cells were subjected to incubation with the selective fluorescent probe 4,5-diaminofluorescein (DAF-2T—10 μM) for 30 min. This probe reacts with dinitrogen trioxide (N
2O
3), an oxidation product of NO, producing the fluorescent compound DAF-2T [
16].
▪ This technique enables the quantification of the intracellular concentration of NO, considering its short half-life. The measurements were carried out using the SpectraMax Gemini XS fluorometer (Molecular Devices, San Jose, CA, USA) with an excitation wavelength of 435 nm and an emission wavelength of 538 nm. Angiotensin II incubation was implemented to mimic endothelial dysfunction, while A23187 was utilized to induce an elevation in intracellular calcium, thereby activating NOS.
2.5. Measurement of General Reactive Oxygen Species (ROS) Production
HUVEC were seeded in 96-well plates at a concentration of 5 × 10⁴ cells per well and incubated for 24 h in a humidified incubator with 5% CO2 at 37 °C. After the initial 24 h, the treatment was removed, and the plates were gently washed with phosphate buffer saline (PBS). Subsequently, the cells were incubated for 30 min with ataciguat at concentrations of 0.1, 1, or 10 µM, followed by a 30 min incubation with angiotensin II (Ang II) at 0.1 μM. The detection of intracellular superoxide radical (O2•−) was performed using dihydroethidium (DHE) at a concentration of 50 μM, with an incubation period of 20 min. Fluorescence intensity measurements were conducted using a fluorescence microplate reader (SpectraMax Gemini XS, Molecular Devices) with excitation at 510 nm and emission at 595 nm wavelengths. Angiotensin II incubation was carried out to induce ROS formation, mimicking endothelial dysfunction, and to examine whether ataciguat decreased intracellular ROS concentration through a mechanism independent of nitric oxide formation, as detailed below. Additionally, tempol was employed as a positive control for an agent that reduces intracellular ROS concentration.
ROS detection was also carried out in the presence of hydroxocobalamin (Hcb), a nitric oxide scavenger, to assess whether the reduction of superoxide anions is not attributed to a reaction with NO resulting in peroxynitrite formation. Additionally, this experiment was conducted in the presence of an sGC inhibitor, 1H- [1,2,4] oxadiazolo [4,3-a] quinoxalin-1-one (ODQ), to confirm whether the effect of ataciguat on ROS concentration is linked to intracellular cGMP accumulation. Cells were treated with ataciguat at 10 µM, ODQ, and Hcb for 30 min, followed by treatment with angiotensin II at 0.1 μM for another 30 min. The intracellular detection of superoxide radicals (O2•−) was accomplished using dihydroethidium (DHE) at 50 μM, with an incubation period of 20 min. The fluorescence quantification was conducted as previously described for DHE.
2.6. Measurement of Superoxide Anion Production
In this experiment, the lucigenin probe (5 µM), specifically designed for detecting superoxide anions, was employed. HUVEC were seeded in 96-well plates at a concentration of 5 × 10⁴ cells per well and were maintained in a humidified incubator with 5% CO2 at 37 °C for 24 h. The cells were then treated with ataciguat at concentrations of 0.1, 1, or 10 µM and lucigenin for 30 min, followed by treatment with Ang II at 0.1 μM for an additional 30 min. The increase in fluorescence intensity was monitored using a fluorescence microplate reader (SpectraMax Gemini XS, Molecular Devices) at an excitation wavelength pair of 510 nm and 595 nm. Angiotensin II incubation was conducted to induce O2•− formation, mimic endothelial dysfunction, and determine whether ataciguat reduced the intracellular O2•− concentration. Tempol was utilized as a positive control for an agent that reduces intracellular O2•− concentration.
2.7. Statistical Analysis
Statistical analysis of the results was conducted using GraphPad Prism version 3.0. The one-way ANOVA was employed, with post hoc testing using Newman–Keuls, to assess statistical significance. Data from each set of experiments are presented as mean ± S.E.M., in which ‘n’ indicates the number of animals utilized. Significant values were considered at p < 0.05.
3. Results
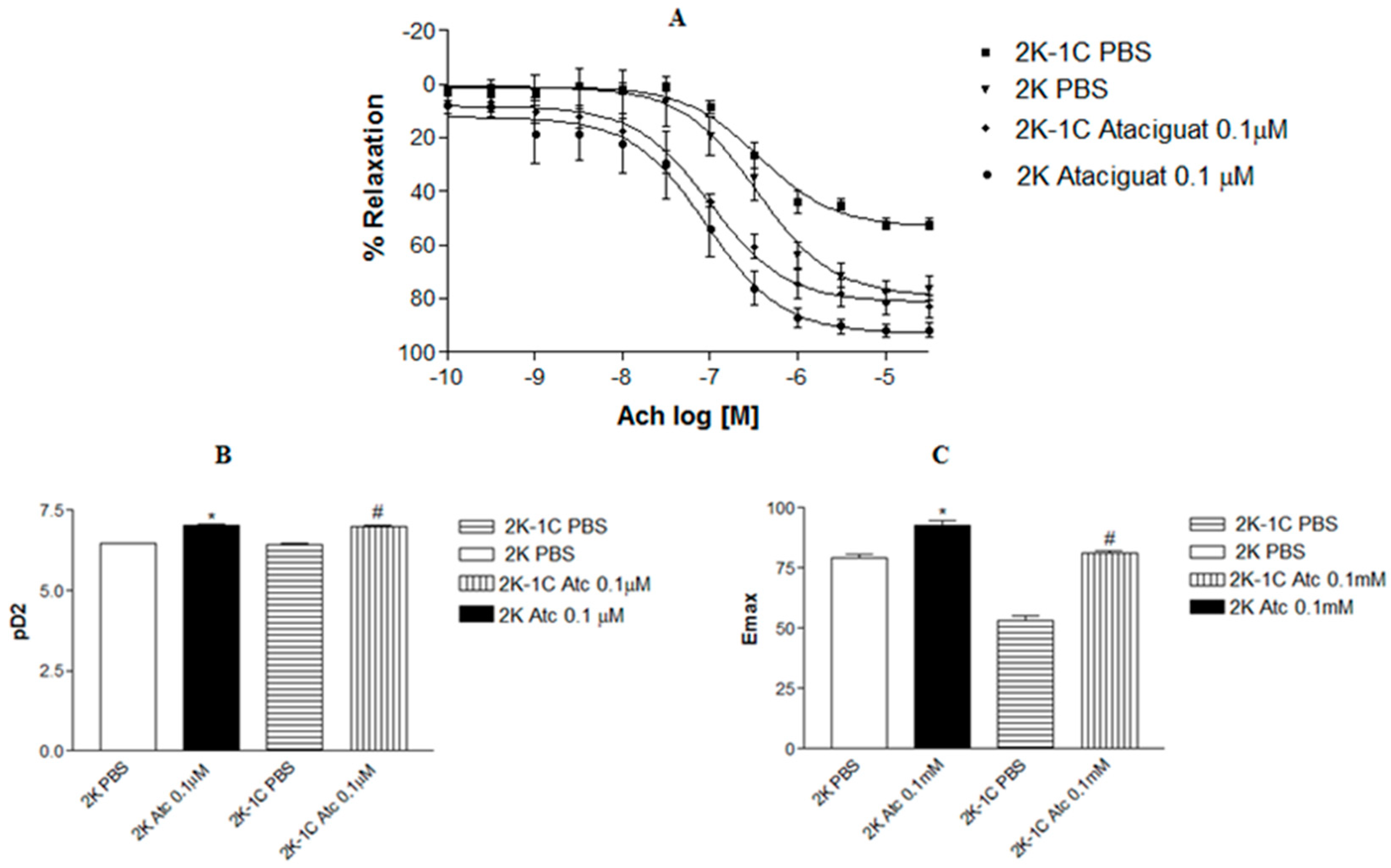
Incubating aortic rings with 0.1 µM ataciguat enhanced the endothelium-dependent relaxation induced by acetylcholine in 2K-1C (pD2: 6.99 ± 0.08,
n = 6) compared to 2K-1C aortic rings treated with PBS (pD2: 6.43 ± 0.07,
n = 6,
p < 0.05), as depicted in
Figure 1A.
A similar outcome was observed in aortic rings from normotensive rats. Treating aortic rings with ataciguat at 0.1 µM in 2K (pD2: 7.04 ± 0.13, n = 6), the rats improved endothelium-dependent relaxation induced by acetylcholine compared to 2K rings treated with PBS (pD2: 6.59 ± 0.07, n = 6). Notably, aortic rings from hypertensive rats treated with ataciguat exhibited superior results compared to normotensive rats without ataciguat treatment.
Figure 1B illustrates the difference in potency (pD2) of acetylcholine-induced relaxation in aortas in the absence or presence of ataciguat treatment. Moreover, the maximum relaxant effect (Emax) was enhanced by ataciguat treatment in the 2K-1C aorta (Emax: 81.0 ± 1.0;
n = 6) and 2K aorta (Emax: 92.98 ± 1.83;
n = 6) compared to the control (Emax 2K-1C: 52.14 ± 2.16,
n = 6; and Emax 2K: 76.07 ± 4.35,
n = 6,
p < 0.05), as depicted in
Figure 1C.
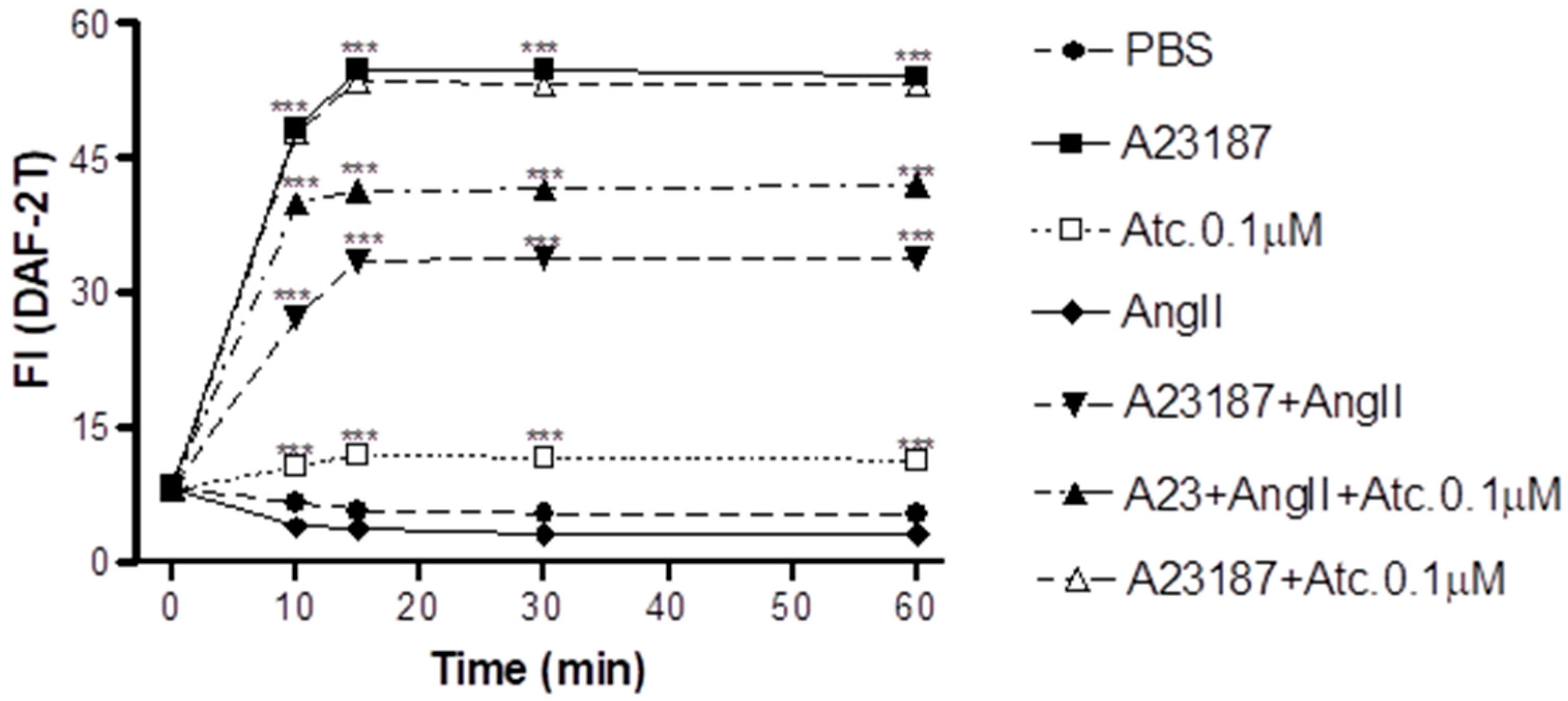
In HUVEC cells, we conducted NO quantification through fluorescence intensity measurement (FI) to assess whether the enhancement in aortic ring vasodilation in hypertensive rats treated with ataciguat is triggered by NO production.
As can be seen in
Figure 2, our findings show that ataciguat induces NO production in endothelial cells (ataciguat 60 min: 11.34 ± 0.34 FI:
n = 5) compared to the control (PBS 60 min: 5.29 ± 0.36 FI;
n = 5,
p < 0.001). After treatment, the NO production stabilized after 15 min for all the protocols, including ataciguat (15 min: 11.94 ± 0.33 FI;
n = 5; 30 min: 11.64 ± 0.56 FI,
n = 5 and 60 min: 11.34 ± 0,34 FI,
n = 6). Ataciguat led to an increase in NO concentration (ataciguat 0.1 µM 60: 11.34 ± 0.34 FI,
n = 5), compared to the control (PBS 60: 5.29 ± 0.36 FI,
n = 5,
p < 0.001). As a positive control, we utilized A23187 to induce the NO production (A23187 10 min: 48.31 ± 0.61,
n = 5; 15 min: 54.75 ± 0.54,
n = 5; 30 min: 54.75 ± 0.55,
n = 5 and 60 min: 54.08 ± 0.52,
n = 5,
p < 0.001. To induce the NO degradation by superoxide, we carried out experiments in the presence of angiotensin II (Ang. II), and we observed that Ang II decreased the NO production induced by A23187 (Ang. II + A23187: 10 min: 27.27 ± 0.67,
n = 5; 15 min: 33.54 ± 0.65,
n = 5; 30 min: 33.80 ± 0.52,
n = 5 and 60 min: 33.93 ± 0.68,
n = 5), compared to A23178 (
p < 0.001). Ataciguat prevented some of the NO degradation induced by angiotensin II (A23187 + Ang. II + ataciguat: 10 min: 39.99 ± 0.52; 15 min: 41.32 ± 0.45; 30 min: 41.51 ± 0.43 and 60 min: 41.98 ± 0.44), compared to A23187 + angiotensin II (
p < 0.001). No additive production of NO was observed for A23187 in the presence of ataciguat (A23187 + ataciguat: 10 min: 47.90 ± 0.66; 15 min: 53.56 ± 0.44; 30 min: 53.23 ± 0.46 and 60 min 53.12 ± 0.44,
p < 0.001).
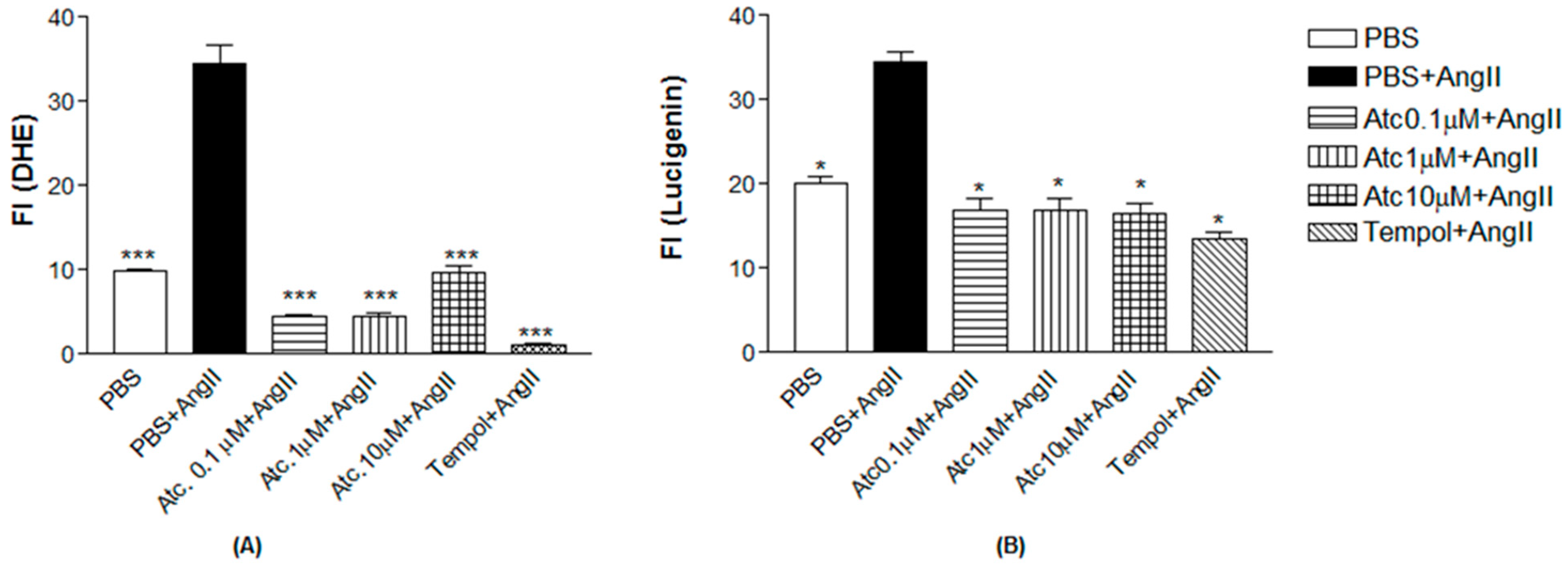
We quantified ROS through fluorescence intensity measurement (FI) using a non-selective ROS probe (DHE) and a selective superoxide probe (lucigenin) to assess whether the enhancement in aortic ring relaxation in hypertensive rats treated with ataciguat is attributed to a reduction in reactive oxygen species.
As a positive control to induce superoxide production, we used angiotensin II.
Figure 3 shows Ang II-induced ROS and superoxide production in endothelial cells (DHE: 34.51 ± 2.20; lucigenin: 34.47 ± 1.22), compared to the control (DHE: 9.97 ± 0.19; lucigenin: 20.10 ± 0.70,
n = 5,
p < 0.05). Ataciguat treatment reduced ROS and superoxide formation, induced by angiotensin II. This effect was observed for all the ataciguat concentrations, including 0.1 uM (DHE: 4.53 ± 0.20; lucigenin: 16.97 ± 1.24,
n = 5,
p < 0.05), 1 uM (DHE: 4.59 ± 0.24; lucigenin: 16.97 ± 1.24,
n = 5,
p < 0.05) and 10 uM (DHE: 9.60 ± 0.93; lucigenin: 16.58 ± 1.03,
n = 5,
p < 0.05). We used tempol as an antioxidant positive control. The treatment using tempol decreased ROS and superoxide formation, induced by angiotensin II (DHE: 1.07 ± 0.25; lucigenin: 13.57 ± 0.68), compared to angiotensin II (DHE: 34.51 ± 2.20); lucigenin: 34.47 ± 1.22,
n = 5,
p < 0.05).
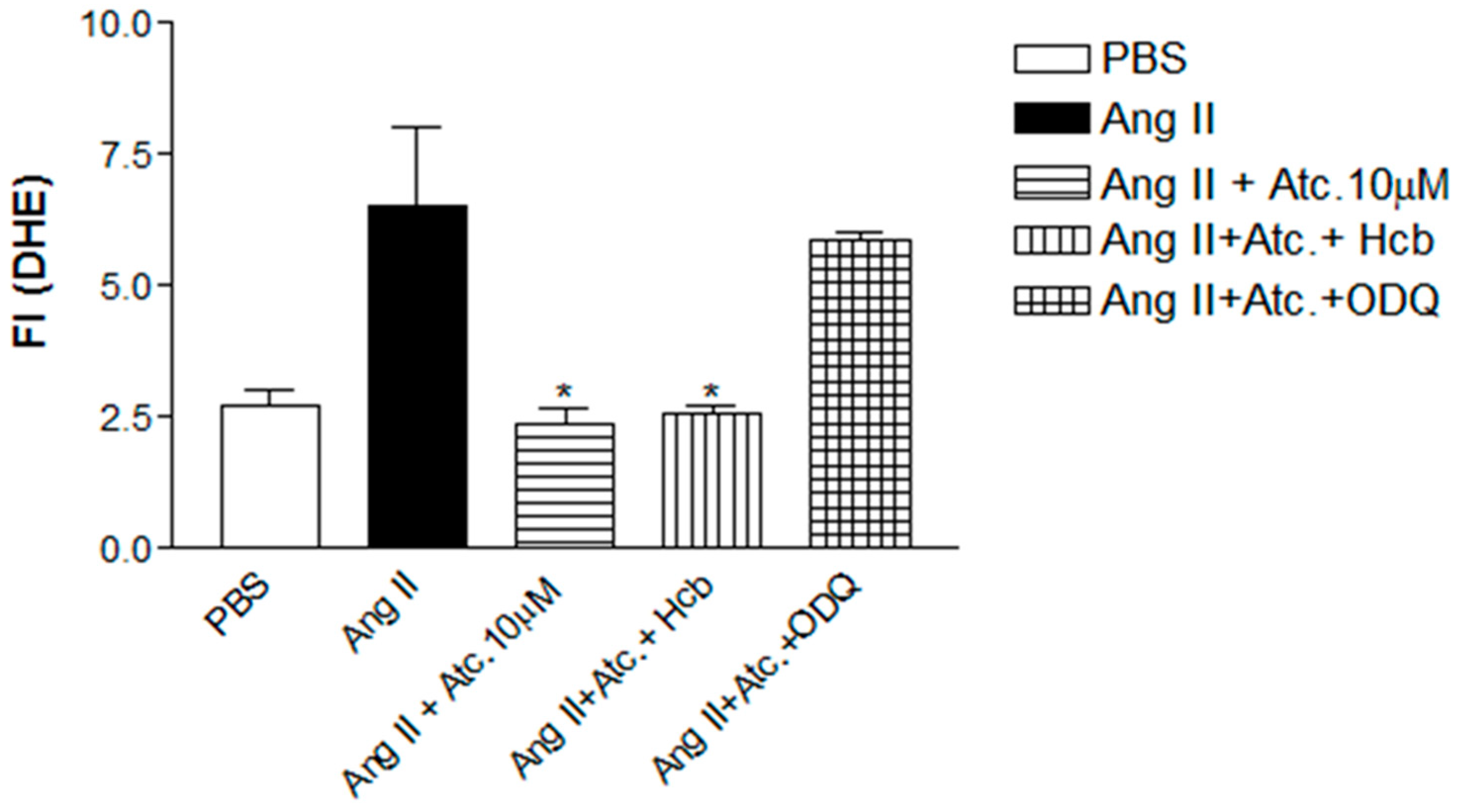
To investigate whether the ataciguat-induced reduction in ROS and superoxide is attributed to sGC activation and/or NO formation, experiments were conducted in the presence of hydroxocobalamin (NO scavenger) or ODQ (sGC inhibitor), as can be seen at
Figure 4. In the presence of hydroxocobalamin, no alteration in the ataciguat effect was observed (Ang. II + ataciguat + Hcb: 2.56 ± 0.16,
n = 5,
p < 0.05), suggesting that the decrease in ROS induced by ataciguat is not dependent on NO production. However, sGC inhibition (ODQ) nullified the ataciguat effect (Ang. II + ataciguat + ODQ: 5.86 ± 0.14,
n = 5,
p < 0.05), indicating the involvement of sGC in the ataciguat effect (
Figure 4).
4. Discussion
The primary discovery of this study was that ataciguat enhances endothelial function in vessels, both with and without endothelial dysfunction, through the inactivation of superoxide anions.
To mitigate the direct vasodilation induced by ataciguat, we incubated aortic rings with a concentration that had been previously confirmed not to induce vasodilation [
14]. Despite the low concentration, ataciguat was still effective in enhancing endothelium-dependent relaxation in isolated aortic rings, both with and without endothelial dysfunction. In rats with cardiac heart failure, chronic treatment with ataciguat demonstrated improvements in both endothelium-dependent (induced by acetylcholine) and non-endothelium-dependent (induced by NO donor) relaxation in aortic rings [
13]. These effects were linked to a decreased concentration of ROS in aortic rings, as measured after chronic ataciguat treatment [
13]. A high concentration of superoxide anions was detected in the aortic rings of rats with renovascular hypertension (2K-1C) [
7], contributing to endothelial dysfunction [
15,
17]. Therefore, the observed improvement in endothelium-dependent relaxation in this study following in vitro treatment with ataciguat can be attributed to a reduction in superoxide anions in aortic rings.
Increased ROS formation results in decreased nitric oxide (NO) bioavailability and is regulated by various endogenous neuro-human systems [
18]. For activation, NO requires the iron heme group of sGC to be in the ferrous state (Fe
2+). However, in conditions of endothelial dysfunction, the sGC iron is in its oxidized state (Fe
3+) or may even be absent [
19]. Consequently, heightened oxidative stress reduces the expression and impairs the NO-induced activation of heme-containing sGC, diminishing the efficacy of vasodilatory therapy with NO donors or eNOS-stimulating compounds.
To assess whether the enhancement in endothelial function induced by ataciguat resulted from a reduction in ROS formation, an experiment with human umbilical vein endothelial cells (HUVEC) was conducted using the dihydroethidium (DHE) probe. An increase in fluorescence intensity was observed when angiotensin II was added, indicating an elevation in ROS production. These findings align with previous publications that have demonstrated increased ROS production in cells stimulated with Ang II [
17]. Treatment with various concentrations of ataciguat (0.1, 1, and 10 µM) decreased the intensity of DHE fluorescence in Ang II-stimulated cells, demonstrating results similar to the treatment with tempol, an SOD mimetic. This result indicates that ataciguat reduces ROS concentration in HUVECs.
To determine whether the reactive oxygen species formed by Ang II stimulation in HUVECs is the superoxide anion (O
2•−), the lucigenin probe was employed [
20]. Our findings demonstrated that after treating HUVECs with angiotensin II, there was an increase in lucigenin fluorescence, suggesting that the ROS being formed is indeed a superoxide anion (O
2•−). Using lucigenin yielded similar results to those obtained with the DHE probe, indicating that ataciguat induces a decrease in superoxide anion levels.
Oxidative stress is characterized by an increase in ROS coupled with a decrease in antioxidant capacity. ROS, particularly the superoxide radical (O
2•−), reacts with nitric oxide (NO), forming peroxynitrite (ONOO
−), a highly oxidant species capable of causing protein nitration and inducing lipid peroxidation [
21]. To assess whether the reduction in ROS in endothelial cells induced by ataciguat does not occur due to a reaction with NO, an experiment was conducted using hydroxocobalamin (Hcb), an NO scavenger. It was observed that in the presence of Hcb, there was a decrease in ROS production similar to ataciguat treatment, indicating that the reduction in ROS induced by ataciguat is not dependent on NO production.
To explore whether the decrease in ROS induced by ataciguat is dependent on sGC stimulation, we utilized an sGC inhibitor (ODQ). ODQ nullified the effect of ataciguat, indicating that the ataciguat effect is contingent on sGC stimulation. The mechanism of sGC inhibition by ODQ involves the oxidation of the sGC heme [
22]. Given that ataciguat activates the sGC when the iron is in the ferrous heme (Fe
2+) state, ferric heme (Fe
3+) state, or even without this grouping [
11], our findings indicate that the reduction of superoxide anion induced by ataciguat is dependent on sGC ferrous heme (Fe
2+). Furthermore, this effect is not attributed to sGC nitric oxide stimulation, as the NO scavenger did not nullify this effect.
Furthermore, we conducted experiments in HUVECs by stimulating (NO) production (A23187) and superoxide anion generation (angiotensin II). In these experiments, we observed that ataciguat can prevent degradation of NO induced by angiotensin II and induce NO production by endothelial cells. These findings align with ROS and superoxide detection, suggesting that ataciguat can mitigate the effects of ROS. These results are consistent with the literature, which demonstrates that ataciguat is a potent stimulus for cGMP production in cells exposed to oxidative stress [
23]. Thus, these results provide additional insights into the biological effects induced by ataciguat, which may enhance various physiological functions dependent on endothelial health, including blood pressure control, the formation and progression of atheromatous plaques, and platelet aggregation, among others.
It is essential to emphasize that the measurement of endothelial function is accessible for clinical use through methods such as flow-mediated dilatation, serving as a crucial tool to guide clinical interventions [
24].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



