
AssayBLAST: A Bioinformatic Tool for In Silico Analysis of Molecular Multiparameter Assays

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. AssayBLAST Architecture
2.2. BLAST Search Adaptations
- -
- dust = ’no’—Disables the filtering of low complexity regions so as not to miss possible binding sites.
- -
- word_size = 7—Reducing the word size is crucial for detecting short sequences and makes BLAST more sensitive to short, exact matches.
- -
- Gapopen = 10 and gapextend = 6—The gap penalties have been adjusted to prioritize hits without gaps, as primers and probes are strongly affected by them.
- -
- E-value = 1000—An e-value of 1000 ensures that all bindings are found, not just the best ones.
- -
- Reward = 5 and penalty = -4—The high reward value of 5 favors exact matches, which are critical for detecting short oligos. The penalty of −4 discourages mismatches, as they can significantly affect the binding efficiency.
- -
- Strand = ‘plus’—This parameter ensures that only one strand of the genome is searched and enables a second search with the reverse complementary sequences to differentiate binding strands safely.
- -
- max_target_seqs = 50,000—The maximum number of returns is very high to ensure that all potential binding sites within a genome are captured.
2.3. AssayBLAST User Parameters
2.4. DNA Microarray Data
2.5. Binary Data Classification
2.6. qPCR Data
2.7. Statistical Analysis
3. Results
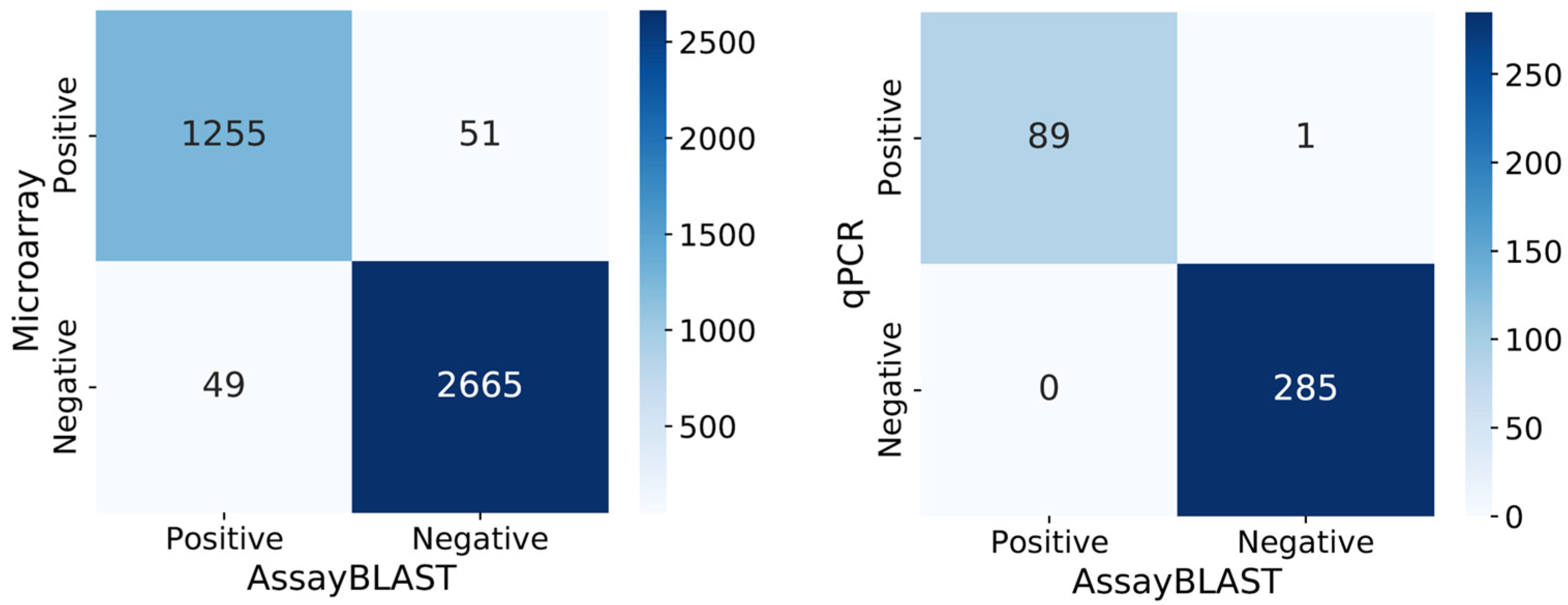
3.1. Analysis of the AssayBLAST and Microarray Results
3.2. Analysis of the Mismatch Count and Microarray Intensity Thresholds
3.3. Analysis of the AssayBLAST and qPCR Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mullis, K.B. The unusual origin of the polymerase chain reaction. Sci. Am. 1990, 262, 56–65. [Google Scholar] [CrossRef]
- Guatelli, J.C.; Whitfield, K.M.; Kwoh, D.Y.; Barringer, K.J.; Richman, D.D.; Gingeras, T.R. Isothermal, in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication. Proc. Natl. Acad. Sci. USA 1990, 87, 1874–1878. [Google Scholar] [CrossRef]
- Southern, E.M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 1975, 98, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Hassibi, A.; Vikalo, H.; Riechmann, J.L.; Hassibi, B. Real-time DNA microarray analysis. Nucleic Acids Res. 2009, 37, e132. [Google Scholar] [CrossRef] [PubMed]
- Panjkovich, A.; Norambuena, T.; Melo, F. dnaMATE: A consensus melting temperature prediction server for short DNA sequences. Nucleic Acids Res. 2005, 33 (Suppl. 2), W570–W572. [Google Scholar] [CrossRef]
- Taylor, S.C.; Nadeau, K.; Abbasi, M.; Lachance, C.; Nguyen, M.; Fenrich, J. The ultimate qPCR experiment: Producing publication quality, reproducible data the first time. Trends Biotechnol. 2019, 37, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.A.; Rosenzweig, A. DNA microarrays: Implications for cardiovascular medicine. Circ. Res. 2002, 91, 559–564. [Google Scholar] [CrossRef]
- Bumgarner, R. Overview of DNA microarrays: Types, applications, and their future. Curr. Protoc. Mol. Biol. 2013, 101, 22.1.1–22.1.11. [Google Scholar] [CrossRef]
- Rodríguez, A.; Rodríguez, M.; Córdoba, J.J.; Andrade, M.J. Design of primers and probes for quantitative real-time PCR methods. In PCR Primer Design; Springer: Berlin/Heidelberg, Germany, 2015; pp. 31–56. [Google Scholar] [CrossRef]
- Vázquez-González, L.; Regueira-Iglesias, A.; Balsa-Castro, C.; Vila-Blanco, N.; Tomás, I.; Carreira, M.J. PrimerEvalPy: A tool for in-silico evaluation of primers for targeting the microbiome. BMC Bioinform. 2024, 25, 189. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chapman, B.; Chang, J. Biopython: Python tools for computational biology. ACM Sigbio Newsl. 2000, 20, 15–19. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Monecke, S.; Jatzwauk, L.; Weber, S.; Slickers, P.; Ehricht, R. DNA microarray-based genotyping of methicillin-resistant Staphylococcus aureus strains from Eastern Saxony. Clin. Microbiol. Infect. 2008, 14, 534–545. [Google Scholar] [CrossRef]
- Monecke, S.; Coombs, G.; Shore, A.C.; Coleman, D.C.; Akpaka, P.; Borg, M.; Chow, H.; Ip, M.; Jatzwauk, L.; Jonas, D.; et al. A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS ONE 2011, 6, e17936. [Google Scholar] [CrossRef]
- Monecke, S.; Gavier-Widen, D.; Mattsson, R.; Rangstrup-Christensen, L.; Lazaris, A.; Coleman, D.C.; Shore, A.C.; Ehricht, R. Detection of mecC-positive Staphylococcus aureus (CC130-MRSA-XI) in diseased European hedgehogs (Erinaceus europaeus) in Sweden. PLoS ONE 2013, 8, e66166. [Google Scholar] [CrossRef]
- Monecke, S.; Roberts, M.C.; Braun, S.D.; Diezel, C.; Müller, E.; Reinicke, M.; Linde, J.; Joshi, P.R.; Paudel, S.; Acharya, M.; et al. Sequence analysis of novel Staphylococcus aureus lineages from wild and captive macaques. Int. J. Mol. Sci. 2022, 23, 11225. [Google Scholar] [CrossRef]
- Collatz, M.; Reinicke, M.; Diezel, C.; Braun, S.D.; Monecke, S.; Reissig, A.; Ehricht, R. ConsensusPrime—A Bioinformatic Pipeline for Efficient Consensus Primer Design—Detection of Various Resistance and Virulence Factors in MRSA—A Case Study. BioMedInformatics 2024, 4, 1249–1261. [Google Scholar] [CrossRef]
- Collatz, M.; Braun, S.D.; Monecke, S.; Ehricht, R. ConsensusPrime—A Bioinformatic Pipeline for Ideal Consensus Primer Design. BioMedInformatics 2022, 2, 637–642. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Gall, J.G.; Pardue, M.L. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc. Natl. Acad. Sci. USA 1969, 63, 378–383. [Google Scholar] [CrossRef]
- Pardue, M.L.; Gall, J.G. Molecular hybridization of radioactive DNA to the DNA of cytological preparations. Proc. Natl. Acad. Sci. USA 1969, 64, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Nurmi, J.; Wikman, T.; Karp, M.; Lövgren, T. High-performance real-time quantitative RT-PCR using lanthanide probes and a dual-temperature hybridization assay. Anal. Chem. 2002, 74, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Mueckstein, U.; Leparc, G.G.; Posekany, A.; Hofacker, I.; Kreil, D.P. Hybridization thermodynamics of NimbleGen microarrays. BMC Bioinform. 2010, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Naqib, A.; Jeon, T.; Kunstman, K.; Wang, W.; Shen, Y.; Sweeney, D.; Hyde, M.; Green, S.J. PCR effects of melting temperature adjustment of individual primers in degenerate primer pools. PeerJ 2019, 7, e6570. [Google Scholar] [CrossRef]
- Alwine, J.C.; Kemp, D.J.; Stark, G.R. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc. Natl. Acad. Sci. USA 1977, 74, 5350–5354. [Google Scholar] [CrossRef]
- Thomas, P.S. Hybridization of denatured RNA and small DNA fragments transferred to nitrocellulose. Proc. Natl. Acad. Sci. USA 1980, 77, 5201–5205. [Google Scholar] [CrossRef]

{kind=link}
| mm count/Threshold: | 1/0.3 | 1/0.5 | 1/0.7 | 2/0.1 | 2/0.2 | 2/0.3 | 2/0.4 | 2/0.5 | 2/0.6 | 2/0.7 | 2/0.8 | 3/0.3 | 3/0.5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy | 97.0% | 97.7% | 96.9% | 96.0% | 96.9% | 97.4% | 97.4% | 97.5% | 97.6% | 96.0% | 96.0% | 95.9% | 95.3% |
| Specificity | 99.9% | 99.7% | 97.6% | 99.1% | 99.0% | 98.8% | 98.4% | 98.2% | 98.1% | 95.6% | 95.6% | 95.4% | 94.3% |
| Precision | 99.8% | 99.4% | 94.5% | 98.2% | 97.9% | 97.5% | 96.8% | 96.2% | 95.9% | 90.5% | 90.5% | 91.3% | 89.2% |
| Sensitivity | 91.2% | 93.5% | 95.4% | 90.4% | 93.0% | 94.6% | 95.2% | 96.1% | 96.5% | 97.0% | 97.0% | 96.8% | 97.2% |
| F1-Score | 95.3% | 96.4% | 95.0% | 94.1% | 95.4% | 96.0% | 96.0% | 96.2% | 96.2% | 93.6% | 93.6% | 94.0% | 93.0% |
| GenBank Accession No. | primer_lukF_11b_forward | primer_lukF_11b_revcomp | probe_lukF_10_forward | probe_lukF_10_revcomp | Interpreted Theoretical Result | Microarray Signal Intensity | Interpreted Microarray Result |
|---|---|---|---|---|---|---|---|
| CP102974 | 0 (pos: 1913864–1913881) | 0 (pos: 1913835–1913860) | positive | 0.82 | positive | ||
| CP102961 | 1 (pos: 784154–784171) | 2 (pos: 784175–784200) | positive | 0.48 | negative | ||
| CP102972-973 | 0 (pos: 796245–796262) | 0 (pos: 796266–796291) | positive | 0.81 | positive | ||
| CP102960 | 0 (pos: 1931674–1931691) | 0 (pos: 1931645–1931670) | positive | 0.81 | positive | ||
| CP102971 | 0 (pos: 254183–254200) | 0 (pos: 254154–254179) | positive | 0.81 | positive | ||
| CP102970 | 0 (pos: 287837–287854) | 0 (pos: 287808–287833) | positive | 0.78 | positive | ||
| CP102959 | 0 (pos: 1940682–1940699) | 0 (pos: 1940653–1940678) | positive | 0.82 | positive | ||
| CP102968-969 | 0 (pos: 1889784–1889801) | 0 (pos: 1889755–1889780) | positive | 0.81 | positive | ||
| CP102958 | 0 (pos: 2344146–2344163) | 0 (pos: 2344167–2344192) | positive | 0.79 | positive | ||
| CP102967 | 0 (pos: 2388703–2388720) | 0 (pos: 2388724–2388749) | positive | 0.8 | positive | ||
| CP102957 | 0 (pos: 2204112–2204129) | 0 (pos: 2204133–2204158) | positive | 0.8 | positive | ||
| CP102956 | 0 (pos: 1942449–1942466) | 0 (pos: 1942420–1942445) | positive | 0.81 | positive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collatz, M.; Braun, S.D.; Reinicke, M.; Müller, E.; Monecke, S.; Ehricht, R. AssayBLAST: A Bioinformatic Tool for In Silico Analysis of Molecular Multiparameter Assays. Appl. Biosci. 2025, 4, 18. https://doi.org/10.3390/applbiosci4020018
Collatz M, Braun SD, Reinicke M, Müller E, Monecke S, Ehricht R. AssayBLAST: A Bioinformatic Tool for In Silico Analysis of Molecular Multiparameter Assays. Applied Biosciences. 2025; 4(2):18. https://doi.org/10.3390/applbiosci4020018
Chicago/Turabian StyleCollatz, Maximilian, Sascha D. Braun, Martin Reinicke, Elke Müller, Stefan Monecke, and Ralf Ehricht. 2025. "AssayBLAST: A Bioinformatic Tool for In Silico Analysis of Molecular Multiparameter Assays" Applied Biosciences 4, no. 2: 18. https://doi.org/10.3390/applbiosci4020018
APA StyleCollatz, M., Braun, S. D., Reinicke, M., Müller, E., Monecke, S., & Ehricht, R. (2025). AssayBLAST: A Bioinformatic Tool for In Silico Analysis of Molecular Multiparameter Assays. Applied Biosciences, 4(2), 18. https://doi.org/10.3390/applbiosci4020018