
Agrigenomic Diversity Unleashed: Current Single Nucleotide Polymorphism Genotyping Methods for the Agricultural Sciences


Abstract
1. Introduction
2. PCR-Based SNP Genotyping
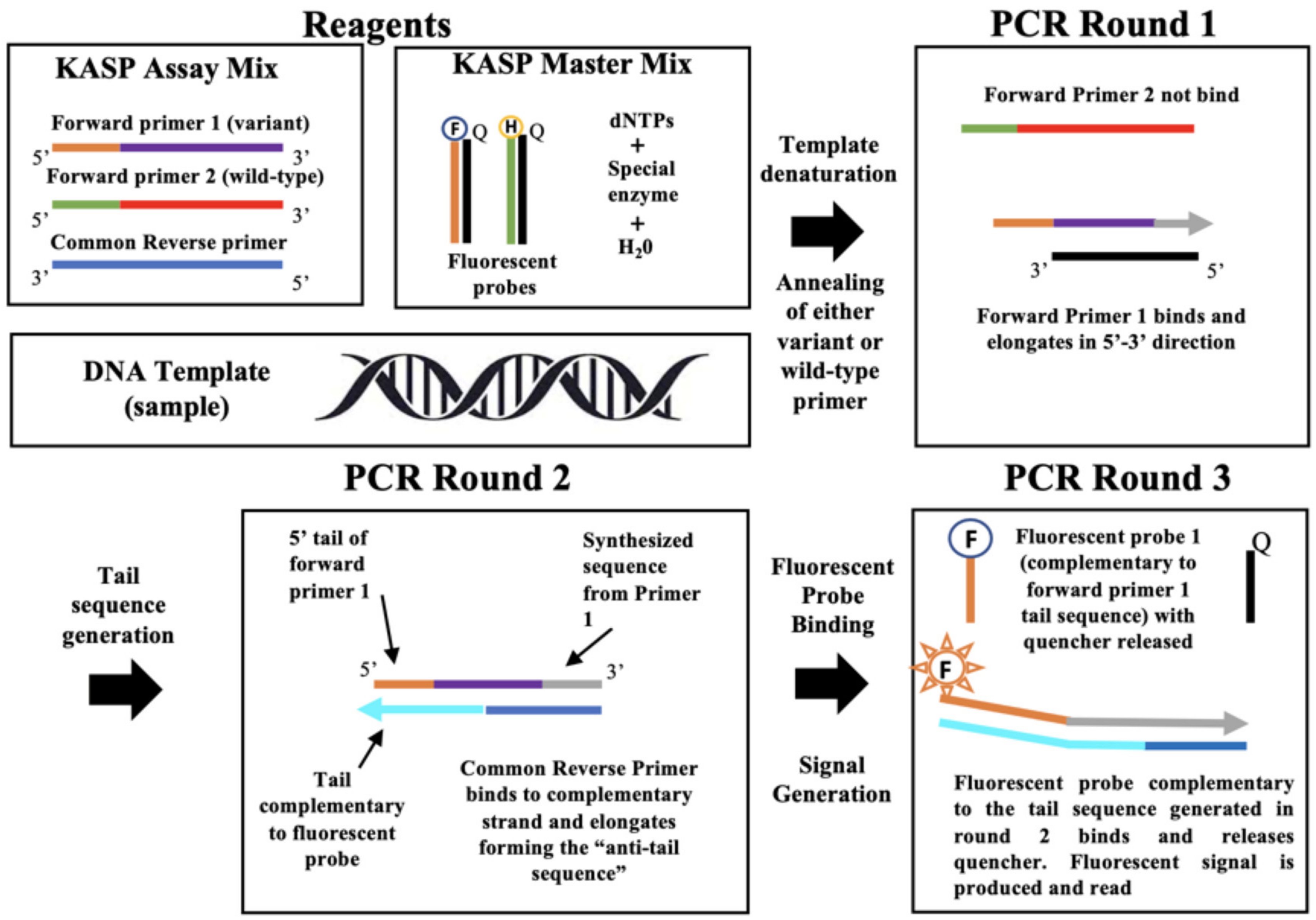
2.1. KASP Method
2.2. PACE Method
2.3. TaqMan Method
2.4. Open Array Method
2.5. rhAMP SNP Genotyping
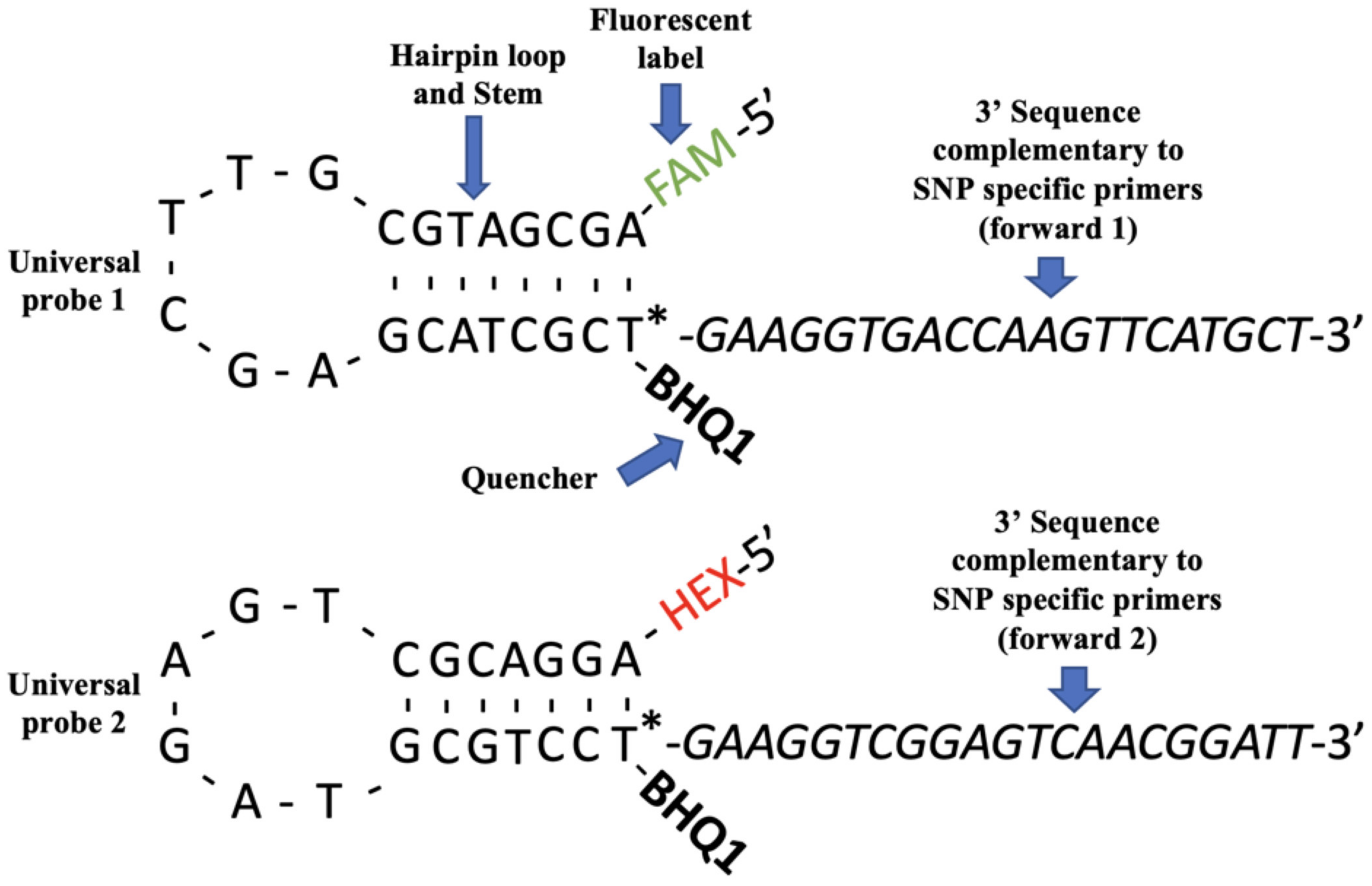
2.6. Amplifluor-Based SNP Genotyping
2.7. Variable Fragment Length Allele Specific PCR (VFLASP)
2.8. High Resolution Melting SNP Genotyping
2.9. MassArray SNP Genotyping System
2.10. PCR-Based Genotyping Conclusions
3. Microarray SNP Genotyping
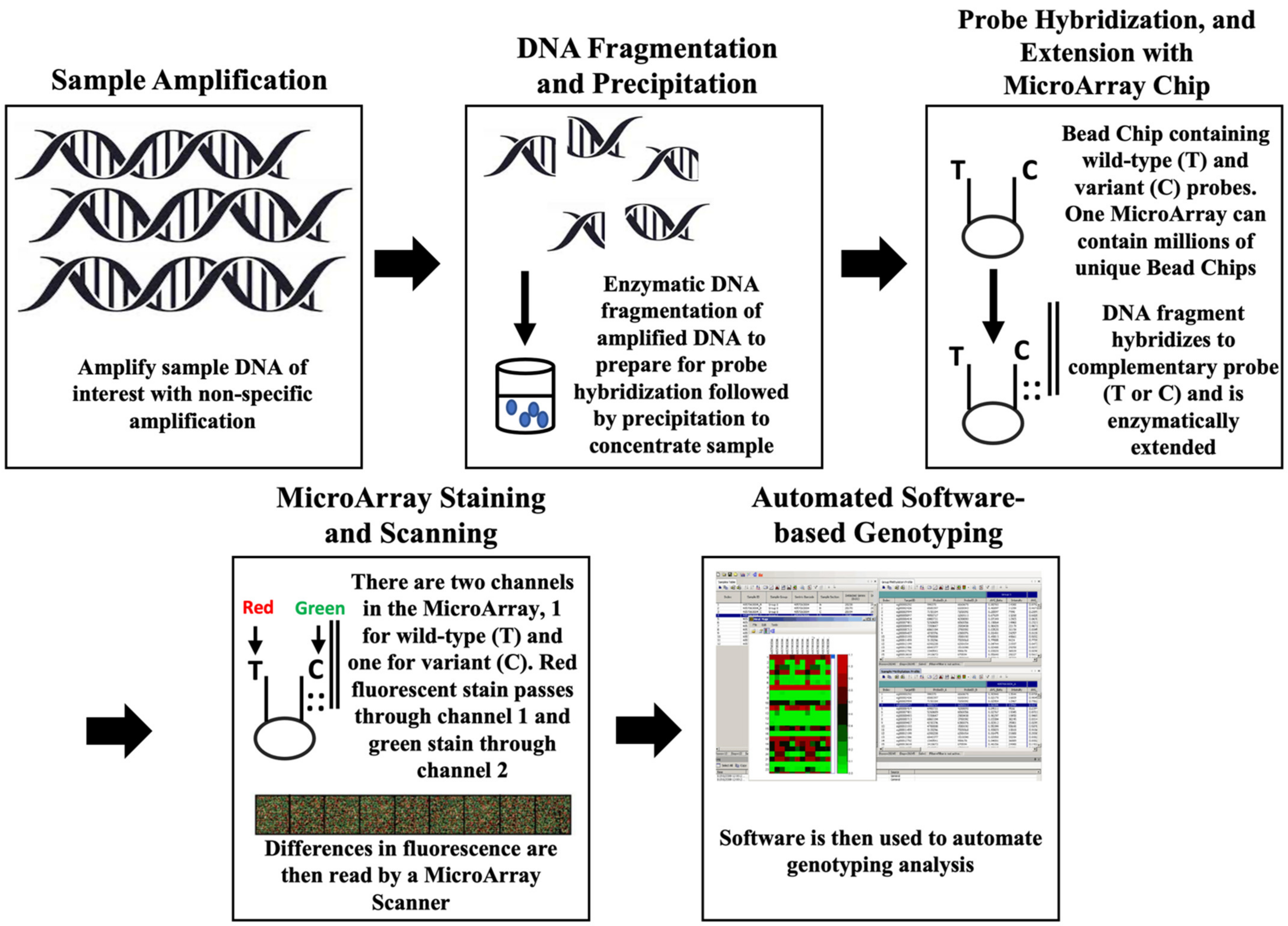
3.1. SNP Arrays
3.2. Microarray-Based Genotyping Conclusions
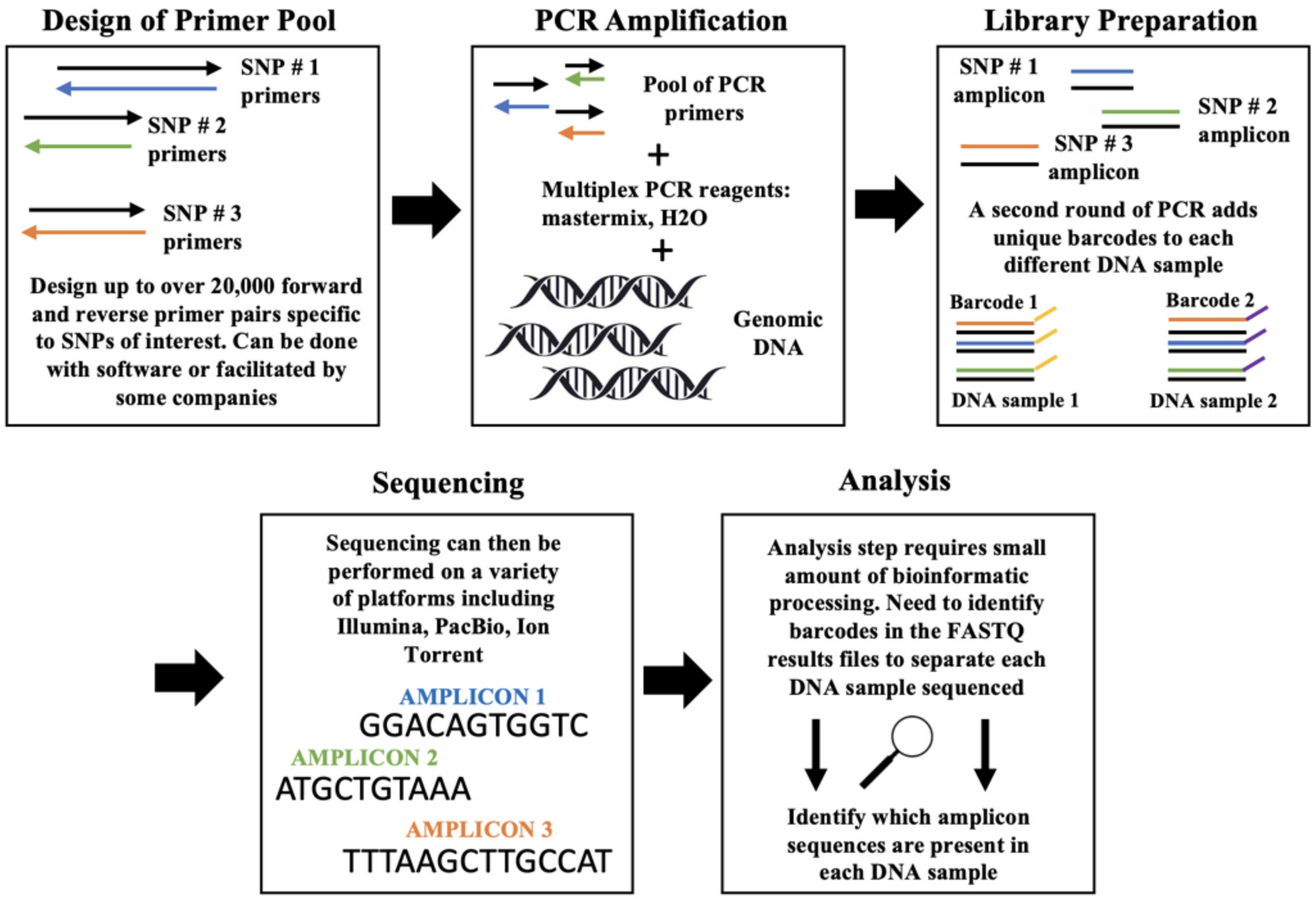
4. Next-Generation Sequencing of SNP Genotyping
4.1. Target SNP-Seq
4.2. Genotyping by Target Sequencing (GBTS)
4.3. AmpliSeq
4.4. MTA-Seq
4.5. Next-Generation Sequencing-Based Genotyping Conclusions
4.6. Potential for MinION in SNP Genotyping to Improve Sequencing-Based Methods
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alvarez-Fernandez, A.; Bernal, M.J.; Fradejas, I.; Martin Ramírez, A.; Md Yusuf, N.A.; Lanza, M.; Hisam, S.; Pérez de Ayala, A.; Rubio, J.M. KASP: A Genotyping Method to Rapid Identification of Resistance in Plasmodium falciparum. Malar. J. 2021, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Sharma, V.; Bhat, A.; Singh, H.; Sharma, I.; Verma, S.; Bhat, G.R.; Sharma, B.; Bakshi, D.; Kumar, R.; et al. MassARRAY Analysis of Twelve Cancer Related SNPs in Esophageal Squamous Cell Carcinoma in J&K, India. BMC Cancer 2020, 20, 497. [Google Scholar] [CrossRef]
- Darrier, B.; Russell, J.; Milner, S.G.; Hedley, P.E.; Shaw, P.D.; Macaulay, M.; Ramsay, L.D.; Halpin, C.; Mascher, M.; Fleury, D.L.; et al. A Comparison of Mainstream Genotyping Platforms for the Evaluation and Use of Barley Genetic Resources. Front. Plant Sci. 2019, 10, 544. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, H.; Tao, J.; Ren, Y.; Xu, C.; Wu, K.; Zou, C.; Zhang, J.; Xu, Y. Development of Multiple SNP Marker Panels Affordable to Breeders through Genotyping by Target Sequencing (GBTS) in Maize. Mol. Breed. 2019, 39, 37. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, Q.; Huang, F.; Zheng, H.; Sang, Z.; Xu, Y.; Zhang, C.; Wu, K.; Tao, J.; Prasanna, B.M.; et al. Development of High-Resolution Multiple-SNP Arrays for Genetic Analyses and Molecular Breeding through Genotyping by Target Sequencing and Liquid Chip. Plant Commun. 2021, 2, 100230. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, R.; Fanelli, R.; Tancredi, F.; Siclari, A.; Garofalo, L. Matching STR and SNP Genotyping to Discriminate between Wild Boar, Domestic Pigs and Their Recent Hybrids for Forensic Purposes. Sci. Rep. 2020, 10, 3188. [Google Scholar] [CrossRef]
- Jatayev, S.; Kurishbayev, A.; Zotova, L.; Khasanova, G.; Serikbay, D.; Zhubatkanov, A.; Botayeva, M.; Zhumalin, A.; Turbekova, A.; Soole, K.; et al. Advantages of Amplifluor-like SNP Markers over KASP in Plant Genotyping. BMC Plant Biol. 2017, 17, 254. [Google Scholar] [CrossRef]
- Kim, N.; Kwon, J.-S.; Kang, W.-H.; Yeom, S.-I. High-Resolution Melting (HRM) Genotyping. Methods Mol Biol. 2023, 2638, 337–349. [Google Scholar] [CrossRef]
- Sato, M.; Hosoya, S.; Yoshikawa, S.; Ohki, S.; Kobayashi, Y.; Itou, T.; Kikuchi, K. A Highly Flexible and Repeatable Genotyping Method for Aquaculture Studies Based on Target Amplicon Sequencing Using Next-Generation Sequencing Technology. Sci. Rep. 2019, 9, 6904. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.; Zhang, L.; Luo, J.; Zhao, H.; Zhang, J.; Wen, C. A New SNP Genotyping Technology Target SNP-Seq and Its Application in Genetic Analysis of Cucumber Varieties. Sci. Rep. 2020, 10, 5623. [Google Scholar] [CrossRef]
- Susi, H.; Burdon, J.J.; Thrall, P.H.; Nemri, A.; Barrett, L.G. Genetic Analysis Reveals Long-Standing Population Differentiation and High Diversity in the Rust Pathogen Melampsora lini. PLoS Pathog. 2020, 16, e1008731. [Google Scholar] [CrossRef] [PubMed]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.L.; Sonstegard, T.S.; et al. Development and Characterization of a High Density SNP Genotyping Assay for Cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Ng, W.-L.; Yang, J.-X.; Li, W.-M.; Ge, X.-J. High Cryptic Species Diversity Is Revealed by Genome-Wide Polymorphisms in a Wild Relative of Banana, Musa Itinerans, and Implications for Its Conservation in Subtropical China. BMC Plant Biol. 2018, 18, 194. [Google Scholar] [CrossRef] [PubMed]
- Lucas, E.R.; Rockett, K.A.; Lynd, A.; Essandoh, J.; Grisales, N.; Kemei, B.; Njoroge, H.; Hubbart, C.; Rippon, E.J.; Morgan, J.; et al. A High Throughput Multi-Locus Insecticide Resistance Marker Panel for Tracking Resistance Emergence and Spread in Anopheles gambiae. Sci. Rep. 2019, 9, 13335. [Google Scholar] [CrossRef]
- Pujolar, J.M.; Limborg, M.T.; Ehrlich, M.; Jaspers, C. High Throughput SNP Chip as Cost Effective New Monitoring Tool for Assessing Invasion Dynamics in the Comb Jelly Mnemiopsis leidyi. Front. Mar. Sci. 2022, 9, 1019001. [Google Scholar] [CrossRef]
- Ayalew, H.; Tsang, P.W.; Chu, C.; Wang, J.; Liu, S.; Chen, C.; Ma, X.-F. Comparison of TaqMan, KASP and RhAmp SNP Genotyping Platforms in Hexaploid Wheat. PLoS ONE 2019, 14, e0217222. [Google Scholar] [CrossRef]
- Zhang, Z.; Cao, Y.; Wang, Y.; Yu, H.; Wu, H.; Liu, J.; An, D.; Zhu, Y.; Feng, X.; Zhang, B.; et al. Development and Validation of KASP Markers for Resistance to Phytophthora capsici in Capsicum annuum L. Mol. Breed. 2023, 43, 20. [Google Scholar] [CrossRef]
- Pradhan, A.K.; Budhlakoti, N.; Chandra Mishra, D.; Prasad, P.; Bhardwaj, S.C.; Sareen, S.; Sivasamy, M.; Jayaprakash, P.; Geetha, M.; Nisha, R.; et al. Identification of Novel QTLs/Defense Genes in Spring Wheat Germplasm Panel for Seedling and Adult Plant Resistance to Stem Rust and Their Validation Through KASP Marker Assays. Plant Dis. 2023, 107, 1847–1860. [Google Scholar] [CrossRef]
- Verma, N.; Garcha, K.S.; Sharma, A.; Sharma, M.; Bhatia, D.; Khosa, J.S.; Kaur, B.; Chuuneja, P.; Dhatt, A.S. Identification of a Major-Effect Quantitative Trait Loci Associated with Begomovirus Resistance in Cucurbita moschata. Phytopathology 2023, 113, 824–835. [Google Scholar] [CrossRef]
- Ilie, D.E.; Gavojdian, D.; Kusza, S.; Neamț, R.I.; Mizeranschi, A.E.; Mihali, C.V.; Cziszter, L.T. Kompetitive Allele Specific PCR Genotyping of 89 SNPs in Romanian Spotted and Romanian Brown Cattle Breeds and Their Association with Clinical Mastitis. Animals 2023, 13, 1484. [Google Scholar] [CrossRef]
- Von Maydell, D. PCR Allele Competitive Extension (PACE). Methods Mol. Biol. 2023, 2638, 263–271. [Google Scholar] [CrossRef] [PubMed]
- von Maydell, D.; Brandes, J.; Lehnert, H.; Junghanns, W.; Marthe, F. Breeding Synthetic Varieties in Annual Caraway: Observations on the Outcrossing Rate in a Polycross Using a High-Throughput Genotyping System. Euphytica 2021, 217, 1. [Google Scholar] [CrossRef]
- Somyong, S.; Phetchawang, P.; Bihi, A.K.; Sonthirod, C.; Kongkachana, W.; Sangsrakru, D.; Jomchai, N.; Pootakham, W.; Tangphatsornruang, S. A SNP Variation in an Expansin (EgExp4) Gene Affects Height in Oil Palm. PeerJ 2022, 10, e13046. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.-Q.; Abdullah, K.G.; Wang, Q.K. The TaqMan Method for SNP Genotyping. Methods Mol. Biol. 2009, 578, 293–306. [Google Scholar] [CrossRef]
- Shumate, S.; Haylett, M.; Nelson, B.; Young, N.; Lamour, K.; Walsh, D.; Bradford, B.; Clements, J. Using Targeted Sequencing and TaqMan Approaches to Detect Acaricide (Bifenthrin, Bifenazate, and Etoxazole) Resistance Associated SNPs in Tetranychus urticae Collected from Peppermint Fields and Hop Yards. PLoS ONE 2023, 18, e0283211. [Google Scholar] [CrossRef]
- Li, H.; Wan, C.; Wang, Z.; Tan, J.; Tan, M.; Zeng, Y.; Huang, J.; Huang, Y.; Su, Q.; Kang, Z.; et al. Rapid Diagnosis of Duck Tembusu Virus and Goose Astrovirus with TaqMan-Based Duplex Real-Time PCR. Front. Microbiol. 2023, 14, 1146241. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, Z.; Zhou, Z.; Sun, J.; Yan, S.; Gao, W.; Shao, Y.; Bai, Y.; Wu, Y.; Yan, Z.; et al. A TaqMan Probe-Based Multiplex Real-Time PCR for Simultaneous Detection of Porcine Epidemic Diarrhea Virus Subtypes G1 and G2, and Porcine Rotavirus Groups A and C. Viruses 2022, 14, 1819. [Google Scholar] [CrossRef]
- Ragazzo, M.; Puleri, G.; Errichiello, V.; Manzo, L.; Luzzi, L.; Potenza, S.; Strafella, C.; Peconi, C.; Nicastro, F.; Caputo, V.; et al. Evaluation of OpenArrayTM as a Genotyping Method for Forensic DNA Phenotyping and Human Identification. Genes 2021, 12, 221. [Google Scholar] [CrossRef]
- Broccanello, C.; Gerace, L.; Stevanato, P. QuantStudioTM 12K Flex OpenArray® System as a Tool for High-Throughput Genotyping and Gene Expression Analysis. In Quantitative Real-Time PCR; Humana: New York, NY, USA, 2020; pp. 199–208. [Google Scholar]
- Noce, A.; Pazzola, M.; Dettori, M.L.; Amills, M.; Castelló, A.; Cecchinato, A.; Bittante, G.; Vacca, G.M. Variations at Regulatory Regions of the Milk Protein Genes Are Associated with Milk Traits and Coagulation Properties in the Sarda Sheep. Anim. Genet. 2016, 47, 717–726. [Google Scholar] [CrossRef]
- Chagné, D.; Vanderzande, S.; Kirk, C.; Profitt, N.; Weskett, R.; Gardiner, S.E.; Peace, C.P.; Volz, R.K.; Bassil, N.V. Validation of SNP Markers for Fruit Quality and Disease Resistance Loci in Apple (Malus × domestica Borkh.) Using the OpenArray® Platform. Hortic. Res. 2019, 6, 30. [Google Scholar] [CrossRef]
- Beltz, K.; Tsang, D.; Wang, J.; Rose, S.; Bao, Y.; Wang, Y.; Larkin, K.; Rupp, S.; Schrepfer, D.; Datta, K.; et al. A High-Performing and Cost-Effective SNP Genotyping Method Using RhPCR and Universal Reporters. Adv. Biosci. Biotechnol. 2018, 9, 497–512. [Google Scholar] [CrossRef]
- Esposito, S.; Taranto, F.; Vitale, P.; Ficco, D.B.M.; Colecchia, S.A.; Stevanato, P.; De Vita, P. Unlocking the Molecular Basis of Wheat Straw Composition and Morphological Traits through Multi-Locus GWAS. BMC Plant Biol. 2022, 22, 519. [Google Scholar] [CrossRef] [PubMed]
- Giglioti, R.; Gutmanis, G.; Katiki, L.M.; Okino, C.H.; de Sena Oliveira, M.C.; Vercesi Filho, A.E. New High-Sensitive RhAmp Method for A1 Allele Detection in A2 Milk Samples. Food Chem. 2020, 313, 126167. [Google Scholar] [CrossRef] [PubMed]
- Mohanrao, M.D.; Senthilvel, S.; Reddy, Y.R.; Kumar, C.A.; Kadirvel, P. Amplifluor-Based SNP Genotyping. Methods Mol. Biol. 2023, 2638, 191–200. [Google Scholar] [CrossRef]
- Khassanova, G.; Khalbayeva, S.; Serikbay, D.; Mazkirat, S.; Bulatova, K.; Utebayev, M.; Shavrukov, Y. SNP Genotyping with Amplifluor-Like Method. Methods Mol. Biol. 2023, 2638, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Shavrukov, Y.; Zhumalin, A.; Serikbay, D.; Botayeva, M.; Otemisova, A.; Absattarova, A.; Sereda, G.; Sereda, S.; Shvidchenko, V.; Turbekova, A.; et al. Expression Level of the DREB2-Type Gene, Identified with Amplifluor SNP Markers, Correlates with Performance, and Tolerance to Dehydration in Bread Wheat Cultivars from Northern Kazakhstan. Front. Plant Sci. 2016, 7, 1736. [Google Scholar] [CrossRef] [PubMed]
- Tóth, T.; Csaba, Á.; Bokor, A.; Ács, N. Variable Fragment Length Allele-Specific Polymerase Chain Reaction (VFLASP), a Method for Simple and Reliable Genotyping. Mol. Cell. Probes 2023, 69, 101910. [Google Scholar] [CrossRef]
- Słomka, M.; Sobalska-Kwapis, M.; Wachulec, M.; Bartosz, G.; Strapagiel, D. High Resolution Melting (HRM) for High-Throughput Genotyping—Limitations and Caveats in Practical Case Studies. Int. J. Mol. Sci. 2017, 18, 2316. [Google Scholar] [CrossRef]
- Chou, L.; Huang, S.-J.; Hsieh, C.; Lu, M.-T.; Song, C.-W.; Hsu, F.-C. A High Resolution Melting Analysis-Based Genotyping Toolkit for the Peach (Prunus persica) Chilling Requirement. Int. J. Mol. Sci. 2020, 21, 1543. [Google Scholar] [CrossRef]
- Chatzidimopoulos, M.; Ganopoulos, I.; Moraitou-Daponta, E.; Lioliopoulou, F.; Ntantali, O.; Panagiotaki, P.; Vellios, E.K. High-Resolution Melting (HRM) Analysis Reveals Genotypic Differentiation of Venturia inaequalis Populations in Greece. Front. Ecol. Evol. 2019, 7, 489. [Google Scholar] [CrossRef]
- Ellis, J.A.; Ong, B. The MassARRAY® System for Targeted SNP Genotyping. Methods Mol. Biol. 2017, 1492, 77–94. [Google Scholar] [CrossRef] [PubMed]
- da Costa Lima Moraes, A.; Sforça, D.A.; Mancini, M.C.; Vigna, B.B.Z.; de Souza, A.P. Polyploid SNP Genotyping Using the MassARRAY System. Methods Mol. Biol. 2023, 2638, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Gan, H.; Lin, X.; Wang, L.; Yao, Y.; Li, L.; Wang, Y.; Zhang, Z. Genome-Wide Association Screening and MassARRAY for Detection of High-Temperature Resistance-Related SNPs and Genes in a Hybrid Abalone (Haliotis discus hannai ♀ × H. fulgens ♂) Based on Super Genotyping-by-Sequencing. Aquaculture 2023, 573, 739576. [Google Scholar] [CrossRef]
- Ji, X.; Cao, Z.; Hao, Q.; He, M.; Cang, M.; Yu, H.; Ma, Q.; Li, X.; Bao, S.; Wang, J.; et al. Effects of New Mutations in BMPRIB, GDF9, BMP15, LEPR, and B4GALNT2 Genes on Litter Size in Sheep. Vet. Sci. 2023, 10, 258. [Google Scholar] [CrossRef] [PubMed]
- Vervalle, J.A.; Costantini, L.; Lorenzi, S.; Pindo, M.; Mora, R.; Bolognesi, G.; Marini, M.; Lashbrooke, J.G.; Tobutt, K.R.; Vivier, M.A.; et al. A High-Density Integrated Map for Grapevine Based on Three Mapping Populations Genotyped by the Vitis18K SNP Chip. Theor. Appl. Genet. 2022, 135, 4371–4390. [Google Scholar] [CrossRef] [PubMed]
- Balog, K.; Mizeranschi, A.E.; Wanjala, G.; Sipos, B.; Kusza, S.; Bagi, Z. Application Potential of Chicken DNA Chip in Domestic Pigeon Species—Preliminary Results. Saudi J. Biol. Sci. 2023, 30, 103594. [Google Scholar] [CrossRef]
- Singh, B.K.; Venkadesan, S.; Ramkumar, M.K.; Shanmugavadivel, P.S.; Dutta, B.; Prakash, C.; Pal, M.; Solanke, A.U.; Rai, A.; Singh, N.K.; et al. Meta-Analysis of Microarray Data and Their Utility in Dissecting the Mapped QTLs for Heat Acclimation in Rice. Plants 2023, 12, 1697. [Google Scholar] [CrossRef]
- Muhu-Din Ahmed, H.G.; Sajjad, M.; Zeng, Y.; Iqbal, M.; Habibullah Khan, S.; Ullah, A.; Nadeem Akhtar, M. Genome-Wide Association Mapping through 90K SNP Array for Quality and Yield Attributes in Bread Wheat against Water-Deficit Conditions. Agriculture 2020, 10, 392. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, H.; Song, N.; Yu, Z.; Imran, K.; Xie, W.; Qiu, S.; Zhou, F.; Wen, J.; Dai, C.; et al. The Bnapus50K Array: A Quick and Versatile Genotyping Tool for Brassica Napus Genomic Breeding and Research. G3 Genes|Genomes|Genet. 2021, 11, jkab241. [Google Scholar] [CrossRef]
- Gunderson, K.L.; Steemers, F.J.; Lee, G.; Mendoza, L.G.; Chee, M.S. A Genome-Wide Scalable SNP Genotyping Assay Using Microarray Technology. Nat. Genet. 2005, 37, 549–554. [Google Scholar] [CrossRef]
- Perkel, J. SNP Genotyping: Six Technologies That Keyed a Revolution. Nat. Methods 2008, 5, 447–453. [Google Scholar] [CrossRef]
- Gardner, S.N.; Thissen, J.B.; McLoughlin, K.S.; Slezak, T.; Jaing, C.J. Optimizing SNP Microarray Probe Design for High Accuracy Microbial Genotyping. J. Microbiol. Methods 2013, 94, 303–310. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rungroj, N.; Nettuwakul, C.; Sudtachat, N.; Praditsap, O.; Sawasdee, N.; Sritippayawan, S.; Chuawattana, D.; Yenchitsomanus, P. A Whole Genome SNP Genotyping by DNA Microarray and Candidate Gene Association Study for Kidney Stone Disease. BMC Med. Genet. 2014, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Balagué-Dobón, L.; Cáceres, A.; González, J.R. Fully Exploiting SNP Arrays: A Systematic Review on the Tools to Extract Underlying Genomic Structure. Brief. Bioinform. 2022, 23, bbac043. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Yang, X.; Peng, Z.; Xu, L.; Wang, J. Development and Applications of a High Throughput Genotyping Tool for Polyploid Crops: Single Nucleotide Polymorphism (SNP) Array. Front. Plant Sci. 2018, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Banks, T.W.; Cloutier, S. SNP Discovery through Next-Generation Sequencing and Its Applications. Int. J. Plant Genom. 2012, 2012, 831460. [Google Scholar] [CrossRef]
- Sekine, D.; Oku, S.; Nunome, T.; Hirakawa, H.; Tsujimura, M.; Terachi, T.; Toyoda, A.; Shigyo, M.; Sato, S.; Tsukazaki, H. Development of a Genome-Wide Marker Design Workflow for Onions and Its Application in Target Amplicon Sequencing-Based Genotyping. DNA Res. 2022, 29, dsac020. [Google Scholar] [CrossRef]
- Takeshima, R.; Ogiso-Tanaka, E.; Yasui, Y.; Matsui, K. Targeted Amplicon Sequencing + Next-Generation Sequencing–Based Bulked Segregant Analysis Identified Genetic Loci Associated with Preharvest Sprouting Tolerance in Common Buckwheat (Fagopyrum esculentum). BMC Plant Biol. 2021, 21, 18. [Google Scholar] [CrossRef]
- Nagano, S.; Hirao, T.; Takashima, Y.; Matsushita, M.; Mishima, K.; Takahashi, M.; Iki, T.; Ishiguri, F.; Hiraoka, Y. SNP Genotyping with Target Amplicon Sequencing Using a Multiplexed Primer Panel and Its Application to Genomic Prediction in Japanese Cedar, Cryptomeria japonica (L.f.) D.Don. Forests 2020, 11, 898. [Google Scholar] [CrossRef]
- Shirasawa, K.; Kuwata, C.; Watanabe, M.; Fukami, M.; Hirakawa, H.; Isobe, S. Target Amplicon Sequencing for Genotyping Genome-Wide Single Nucleotide Polymorphisms Identified by Whole-Genome Resequencing in Peanut. Plant Genome 2016, 9, 1–8. [Google Scholar] [CrossRef]
- Ishikawa, G.; Sakai, H.; Mizuno, N.; Solovieva, E.; Tanaka, T.; Matsubara, K. Developing Core Marker Sets for Effective Genomic-Assisted Selection in Wheat and Barley Breeding Programs. Breed. Sci. 2022, 72, 22004. [Google Scholar] [CrossRef] [PubMed]
- Gashururu, R.S.; Maingi, N.; Githigia, S.M.; Getange, D.O.; Ntivuguruzwa, J.B.; Habimana, R.; Cecchi, G.; Gashumba, J.; Bargul, J.L.; Masiga, D.K. Trypanosomes Infection, Endosymbionts, and Host Preferences in Tsetse Flies (Glossina Spp.) Collected from Akagera Park Region, Rwanda: A Correlational Xenomonitoring Study. One Health 2023, 16, 100550. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Kim, Y.-C.; Won, S.-Y.; Jeong, M.-J.; Park, K.-J.; Park, H.-C.; Roh, I.-S.; Kang, H.-E.; Sohn, H.-J.; Jeong, B.-H. Identification of a Novel Risk Factor for Chronic Wasting Disease (CWD) in Elk: S100G Single Nucleotide Polymorphism (SNP) of the Prion Protein Gene (PRNP). Vet. Res. 2023, 54, 48. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Ogata, T.; Shimada, T.; Endo, T.; Iketani, H.; Shimizu, T.; Yamamoto, T.; Omura, M. Minimal Marker: An algorithm and computer program for the identification of minimal sets of discriminating DNA markers for efficient variety identification. J. Bioinform. Comput. Biol. 2013, 11, 1250022. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Ren, Y.; Jian, Y.; Guo, Z.; Zhang, Y.; Xie, C.; Fu, J.; Wang, H.; Wang, G.; Xu, Y.; et al. Development of a Maize 55 K SNP Array with Improved Genome Coverage for Molecular Breeding. Mol. Breed. 2017, 37, 20. [Google Scholar] [CrossRef]
- Onda, Y.; Takahagi, K.; Shimizu, M.; Inoue, K.; Mochida, K. Multiplex PCR Targeted Amplicon Sequencing (MTA-Seq): Simple, Flexible, and Versatile SNP Genotyping by Highly Multiplexed PCR Amplicon Sequencing. Front. Plant Sci. 2018, 9, 201. [Google Scholar] [CrossRef]
- Singh, A.; Bhatia, P. Comparative Sequencing Data Analysis of Ion Torrent and MinION Sequencing Platforms Using a Clinical Diagnostic Haematology Panel. Int. J. Lab. Hematol. 2020, 42, 833–841. [Google Scholar] [CrossRef]
- Nygaard, A.B.; Tunsjø, H.S.; Meisal, R.; Charnock, C. A Preliminary Study on the Potential of Nanopore MinION and Illumina MiSeq 16S RRNA Gene Sequencing to Characterize Building-Dust Microbiomes. Sci. Rep. 2020, 10, 3209. [Google Scholar] [CrossRef]
- Whitford, W.; Hawkins, V.; Moodley, K.S.; Grant, M.J.; Lehnert, K.; Snell, R.G.; Jacobsen, J.C. Proof of Concept for Multiplex Amplicon Sequencing for Mutation Identification Using the MinION Nanopore Sequencer. Sci. Rep. 2022, 12, 8572. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.-L.; Zhang, J.-R.; Zhang, X.-M.; Liu, X.; Lin, Y.-F.; Bai, H.; Wang, M.-C.; Cheng, F.; Liu, J.-D.; Li, P.; et al. Forensic Nanopore Sequencing of STRs and SNPs Using Verogen’s ForenSeq DNA Signature Prep Kit and MinION. Int. J. Legal Med. 2021, 135, 1685–1693. [Google Scholar] [CrossRef]
- Cornelis, S.; Gansemans, Y.; Vander Plaetsen, A.-S.; Weymaere, J.; Willems, S.; Deforce, D.; Van Nieuwerburgh, F. Forensic Tri-Allelic SNP Genotyping Using Nanopore Sequencing. Forensic Sci. Int. Genet. 2019, 38, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Tabata, Y.; Matsuo, Y.; Fujii, Y.; Ohta, A.; Hirota, K. Rapid Detection of Single Nucleotide Polymorphisms Using the MinION Nanopore Sequencer: A Feasibility Study for Perioperative Precision Medicine. JA Clin. Rep. 2022, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, S.; Gansemans, Y.; Deleye, L.; Deforce, D.; Van Nieuwerburgh, F. Forensic SNP Genotyping Using Nanopore MinION Sequencing. Sci. Rep. 2017, 7, 41759. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP Characterization a | Number of Targetable SNPs b | Monetary Cost c,* | Time Cost d | Labor Cost e | Experimental Accuracy f | Equipment Needs g | Experimental and Analytical Complexity h | Use Cases i | |
|---|---|---|---|---|---|---|---|---|---|
| KASP Method | High | 1–50 | Low | Low | Low | 39.38–91% | General qPCR | Simple | Plasmodium falciparum, Capsicum annuum, Triticum aestivum, Cucurbita moschata, Bos taurus [1,17,18,19,20] |
| PACE Method | High | 1–50 | Low | Low | Low | 51.6–82% | General qPCR | Simple | Triticum species, Carum carvi [22,23] |
| TaqMan Method | High | 1–50 | Low | Low | Low | 88.26–95% | General qPCR | Simple | Tetranychus urticae, tembusus virus, astrovirus, Sus domesticus [25,26,27] |
| Open Array Method | High | 1–200 | Low | Low | Low | 85.9% | Specialized (open array equipment) | Simple | Ovis aries, Malus × Domestica borkh [30,31] |
| rhAMP method | High | 1–50 | Low | Low | Low | 100% | General qPCR | Simple | Triticum aestivum, Bos taurus, Hordeum vulgare [33,34] |
| Amplifluor-based Method | High | 1–50 | Low | Low | Low | N/A (authors did not discuss rate of genotyping success) | General qPCR | Simple | Triticum species [36,37] |
| VFLASP Method | High | 1–50 | Low | Low | Low | 100% (authors note this method is not suitable for all SNPs, based on assay design) | General PCR and gel electrophoresis | Simple | Homo sapiens [38] |
| High Resolution Melting | High | 1–50 | Low | Low | Low | 55.6–80% | General qPCR | Simple | Triticum species, Prunus persica, Venturia inaequalis [33,40,41] |
| MassArray Genotyping | High | 1–50 | Low | Low | Low | N/A (authors did not discuss rate of genotyping success) | Specialized (mass spectrometer) | Moderate | Triticum species, Haliotis discus, Ovis aries [43,44,45] |
| Microarray-based Genotyping | Low | 100,000+ | High | High development time, low experimental time | Medium | 49–91.5% | Specialized (microarray plate reader and genotyping software) | Complex, but design is outsourced to manufacturers | Vitis viníferaa, Columba livia domestica, Oriza sativa, Triticum aestivum, Brassica napus [46,47,48,49,50] |
| Next-Generation Sequencing-Based Genotyping | Low–Medium | One–several thousand | Medium | Medium–high (depending on sequencing availability) | Medium–high | 58.9–98% | General PCR and access to sequencing services | Experimentally simple, analysis requires some bioinformatics | Allium cepa, Fagopyrum esculentum, Cryptomeria japonica, Arachis hypogaea, Hordeum vulgare, Glossina species, Cervidae species [58,59,60,61,62,63,64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawrie, R.D.; Massey, S.E. Agrigenomic Diversity Unleashed: Current Single Nucleotide Polymorphism Genotyping Methods for the Agricultural Sciences. Appl. Biosci. 2023, 2, 565-585. https://doi.org/10.3390/applbiosci2040036
Lawrie RD, Massey SE. Agrigenomic Diversity Unleashed: Current Single Nucleotide Polymorphism Genotyping Methods for the Agricultural Sciences. Applied Biosciences. 2023; 2(4):565-585. https://doi.org/10.3390/applbiosci2040036
Chicago/Turabian StyleLawrie, Roger D., and Steven E. Massey. 2023. "Agrigenomic Diversity Unleashed: Current Single Nucleotide Polymorphism Genotyping Methods for the Agricultural Sciences" Applied Biosciences 2, no. 4: 565-585. https://doi.org/10.3390/applbiosci2040036
APA StyleLawrie, R. D., & Massey, S. E. (2023). Agrigenomic Diversity Unleashed: Current Single Nucleotide Polymorphism Genotyping Methods for the Agricultural Sciences. Applied Biosciences, 2(4), 565-585. https://doi.org/10.3390/applbiosci2040036