
Expedient Access to Type II Kinase Inhibitor Chemotypes by Microwave-Assisted Suzuki Coupling



Abstract
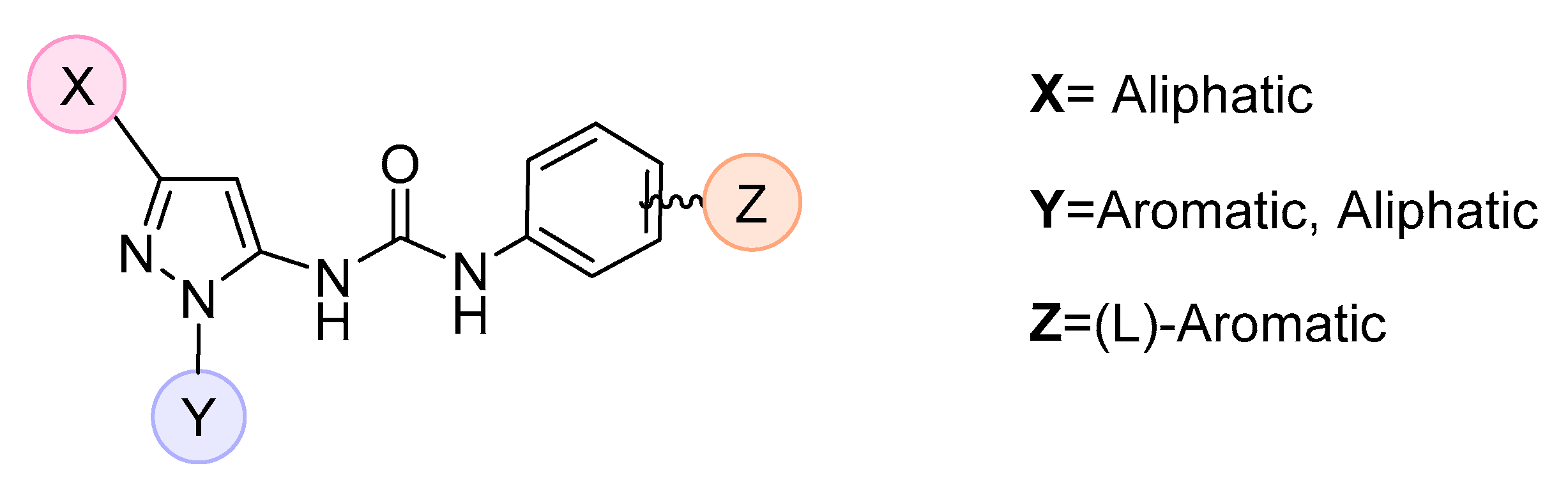
:1. Introduction
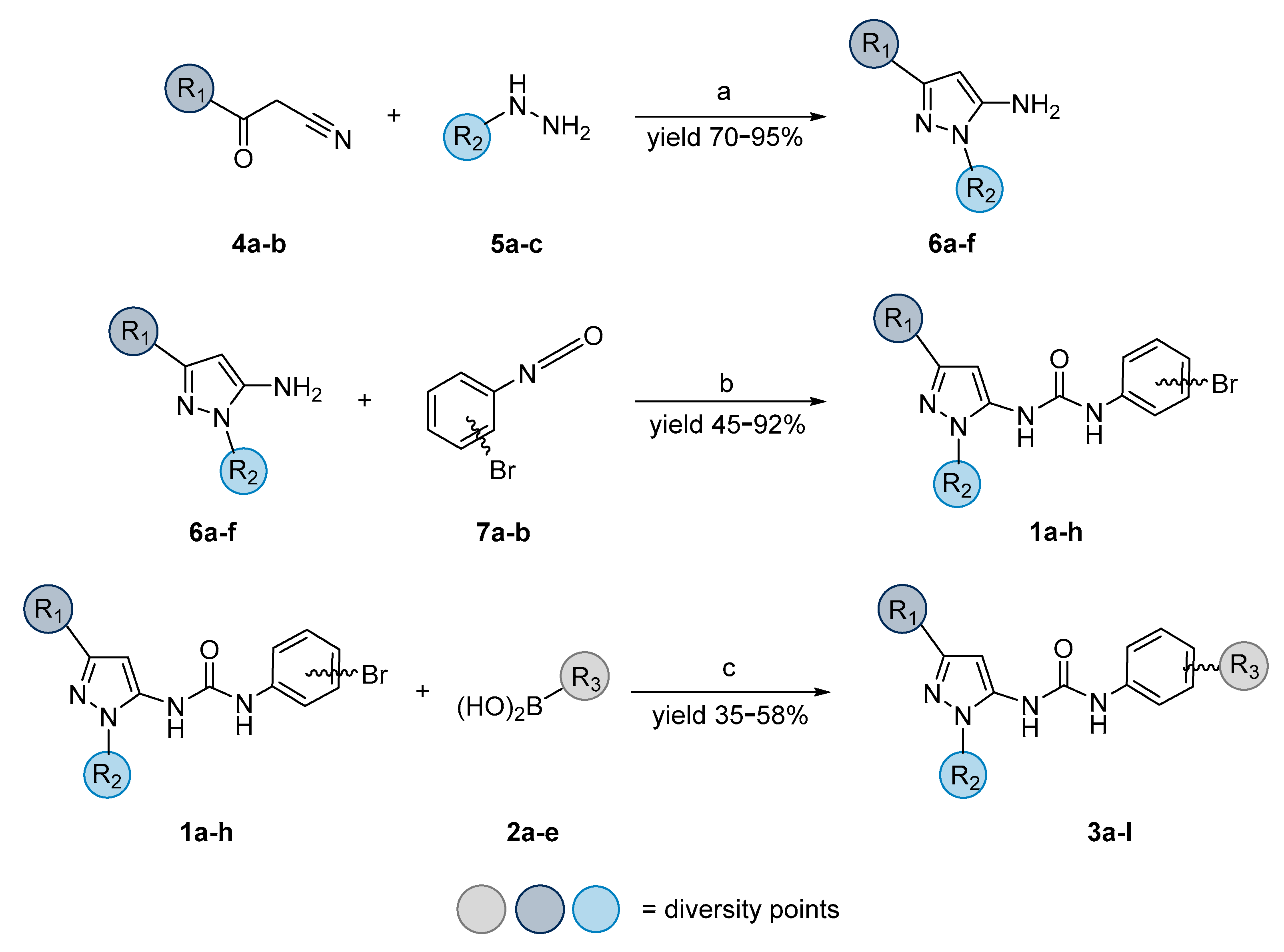
2. Materials and Methods
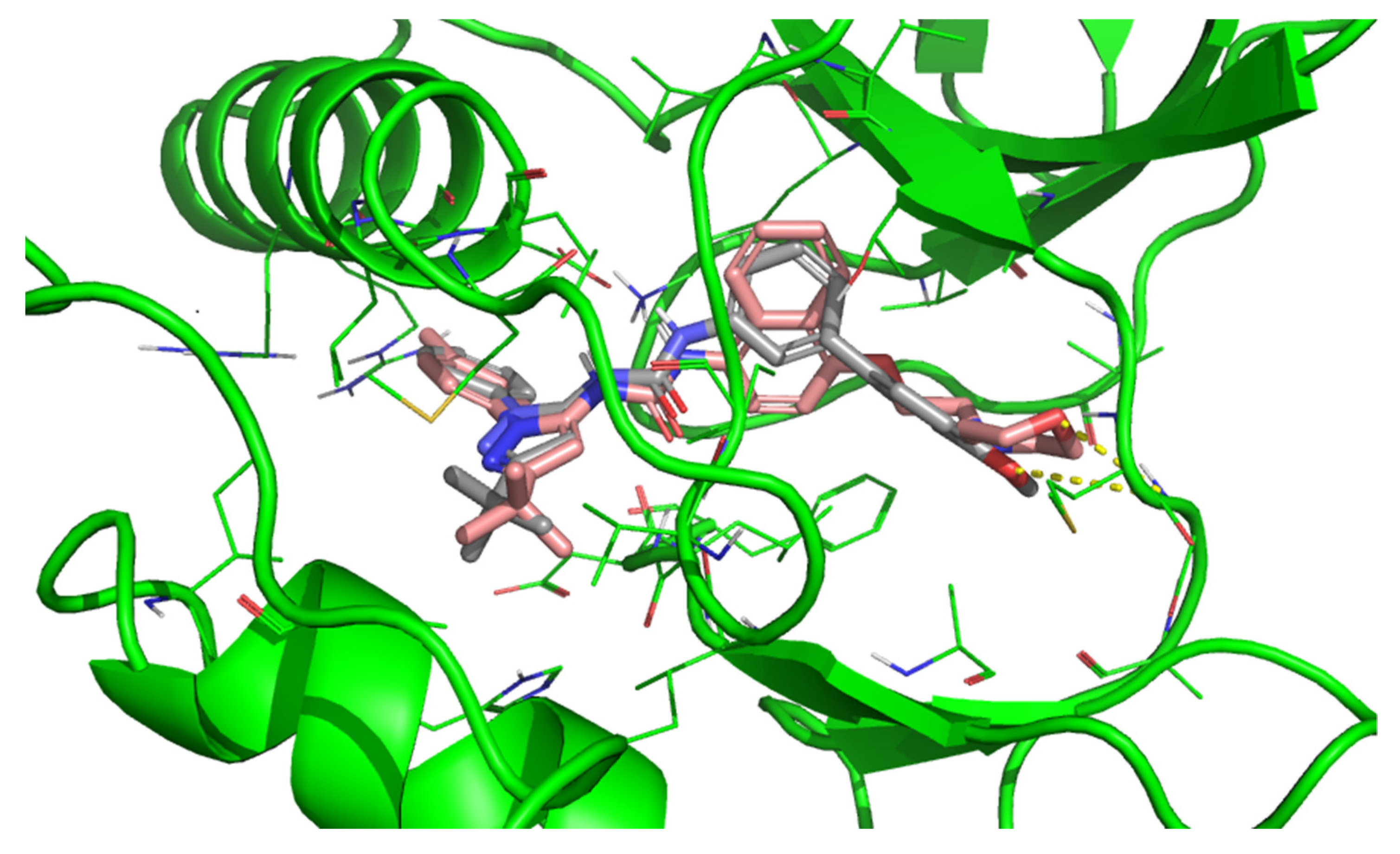
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Garcia, T.G.; Poncet, S.; Derouiche, A.; Shi, L.; Mijakovic, I.; Noirot-Gros, M.-F. Role of Protein Phosphorylation in the Regulation of Cell Cycle and DNA-Related Processes in Bacteria. Front. Microbiol. 2016, 7, 184. [Google Scholar] [CrossRef]
- Niefind, K.; Pütter, M.; Guerra, B.; Issinger, O.G.; Schomburg, D. GTP plus water mimic ATP in the active site of protein kinase CK2. Nat. Genet. 1999, 6, 1100–1103. [Google Scholar] [CrossRef]
- Huse, M.; Kuriyan, J. The Conformational Plasticity of Protein Kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Liao, J.J.-L. Molecular Recognition of Protein Kinase Binding Pockets for Design of Potent and Selective Kinase Inhibitors. J. Med. Chem. 2007, 50, 409–424. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Mol, C.D.; Fabbro, R.; Hosfield, D.J. Structural insights into the conformational selectivity of STI-571 and related kinase inhibitors. Curr. Opin. Drug Discov. Dev. 2004, 7, 639–648. [Google Scholar]
- Vijayan, R.S.K.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, J.R.L.; Levy, R.M. Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Treiber, D.K.; Shah, N.P. Ins and Outs of Kinase DFG Motifs. Chem. Biol. 2013, 20, 745–746. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wu, H.; Wang, L.; Liu, Y.; Knapp, S.; Liu, Q.; Gray, N.S. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS Chem. Biol. 2014, 9, 1230–1241. [Google Scholar] [CrossRef]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Conformation. J. Med. Chem. 2009, 53, 2681–2694. [Google Scholar] [CrossRef]
- Chahrour, O.; Cairns, D.; Omran, Z. Small molecule kinase inhibitors as anti-cancer therapeutics. Mini-Rev. Med. Chem. 2012, 12, 399–411. [Google Scholar] [CrossRef]
- Saturno, G.; Lopes, F.; Niculescu-Duvaz, I.; Zambon, A.; Davies, L.; Johnson, L.; Preece, N.; Lee, R.; Viros, A.; Holovanchuk, D.; et al. The paradox-breaking panRAF plus SRC family kinase inhibitor, CCT3833, is effective in mutant KRAS-driven cancers. Ann. Oncol. 2021, 32, 269–278. [Google Scholar] [CrossRef]
- Girotti, M.R.; Lopes, F.; Preece, N.; Niculescu-Duvaz, D.; Zambon, A.; Davies, L.; Whittaker, S.; Saturno, G.; Viros, A.; Pedersen, M.; et al. Paradox-Breaking RAF Inhibitors that Also Target SRC Are Effective in Drug-Resistant BRAF Mutant Melanoma. Cancer Cell 2015, 27, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Genet. 2002, 9, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Niculescu-Duvaz, D.; Gaulon, C.; Dijkstra, H.P.; Niculescu-Duvaz, I.; Zambon, A.; Ménard, D.; Suijkerbuijk, B.M.J.M.; Nourry, A.; Davies, L.; Manne, H.; et al. Pyridoimidazolones as Novel Potent Inhibitors of v-Raf Murine Sarcoma Viral Oncogene Homologue B1 (BRAF). J. Med. Chem. 2009, 52, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Suijkerbuijk, B.M.J.M.; Niculescu-Duvaz, I.; Gaulon, C.; Dijkstra, H.P.; Niculescu-Duvaz, D.; Ménard, D.; Zambon, A.; Nourry, A.; Davies, L.; Manne, H.A.; et al. Development of Novel, Highly Potent Inhibitors of V-RAF Murine Sarcoma Viral Oncogene Homologue B1 (BRAF): Increasing Cellular Potency through Optimization of a Distal Heteroaromatic Group. J. Med. Chem. 2010, 53, 2741–2756. [Google Scholar] [CrossRef] [PubMed]
- Zambon, A.; Ménard, D.; Suijkerbuijk, B.M.J.M.; Niculescu-Duvaz, I.; Whittaker, S.; Niculescu-Duvaz, D.; Nourry, A.; Davies, L.; Manne, H.A.; Lopes, F.; et al. Novel Hinge Binder Improves Activity and Pharmacokinetic Properties of BRAF Inhibitors. J. Med. Chem. 2010, 53, 5639–5655. [Google Scholar] [CrossRef]
- Whittaker, S.; Ménard, D.; Kirk, R.; Ogilvie, L.; Hedley, D.; Zambon, A.; Lopes, F.; Preece, N.; Manne, H.; Rana, S.; et al. A novel, selective, and efficacious nanomolar pyridopyrazinone inhibitor of V600EBRAF. Cancer Res. 2010, 70, 8036–8044. [Google Scholar] [CrossRef] [Green Version]
- Zambon, A.; Niculescu-Duvaz, D.; Niculescu-Duvaz, I.; Marais, R.; Springer, C.J. BRAF as a therapeutic target: A patent review (2006–2012). Expert Opin. Ther. Pat. 2013, 23, 155–164. [Google Scholar] [CrossRef]
- Gennäs, G.B.A.; Mologni, L.; Ahmed, S.; Rajaratnam, M.; Marin, O.; Lindholm, N.; Viltadi, M.; Gambacorti-Passerini, C.; Scapozza, L.; Yli-Kauhaluoma, J. Design, Synthesis, and Biological Activity of Urea Derivatives as Anaplastic Lymphoma Kinase Inhibitors. ChemMedChem 2011, 6, 1680–1692. [Google Scholar] [CrossRef]
- Suzuki, A. Organoborates in new synthetic reactions. Accounts Chem. Res. 2002, 15, 178–184. [Google Scholar] [CrossRef]
- Miyaura, N.; Yanagi, T.; Suzuki, A. The Palladium-Catalyzed Cross-Coupling Reaction of Phenylboronic Acid with Haloarenes in the Presence of Bases. Synth. Commun. 2006, 11, 513–519. [Google Scholar] [CrossRef]
- Al-Masoudi, N.A.; Essa, A.H.; Alwaaly, A.A.; Saeed, B.A.; Langer, P. Synthesis and conformational analysis of new arylated-diphenylurea derivatives related to sorafenib drug via Suzuki-Miyaura cross-coupling reaction. J. Mol. Struct. 2017, 1146, 522–529. [Google Scholar] [CrossRef]
- Brunner, K.; Maric, S.; Reshma, R.S.; Almqvist, H.; Seashore-Ludlow, B.; Gustavsson, A.-L.; Poyraz, O.; Yogeeswari, P.; Lundbäck, T.; Vallin, M.; et al. Inhibitors of the Cysteine Synthase CysM with Antibacterial Potency against Dormant Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 6848–6859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, G.B.; Richert, R.; Stiegman, A.E. On the existence of and mechanism for microwave-specific reaction rate enhancement. Chem. Sci. 2015, 6, 2144–2152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, C.H.-T.; Lu, Y.; Nagashima, T. A Highly Efficient Microwave-Assisted Suzuki Coupling Reaction of Aryl Perfluorooctylsulfonates with Boronic Acids. Org. Lett. 2004, 6, 1473–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, J.; Breitfelder, S.; Cirillo, P.; Gilmore, T.; Graham, A.G.; Hickey, E.; Klaus, B.; Madwed, J.; Moriak, M.; Moss, N.; et al. Pyrazole Urea-Based Inhibitors of p38 MAP Kinase: From Lead Compound to Clinical Candidate. J. Med. Chem. 2002, 45, 2994–3008. [Google Scholar] [CrossRef]
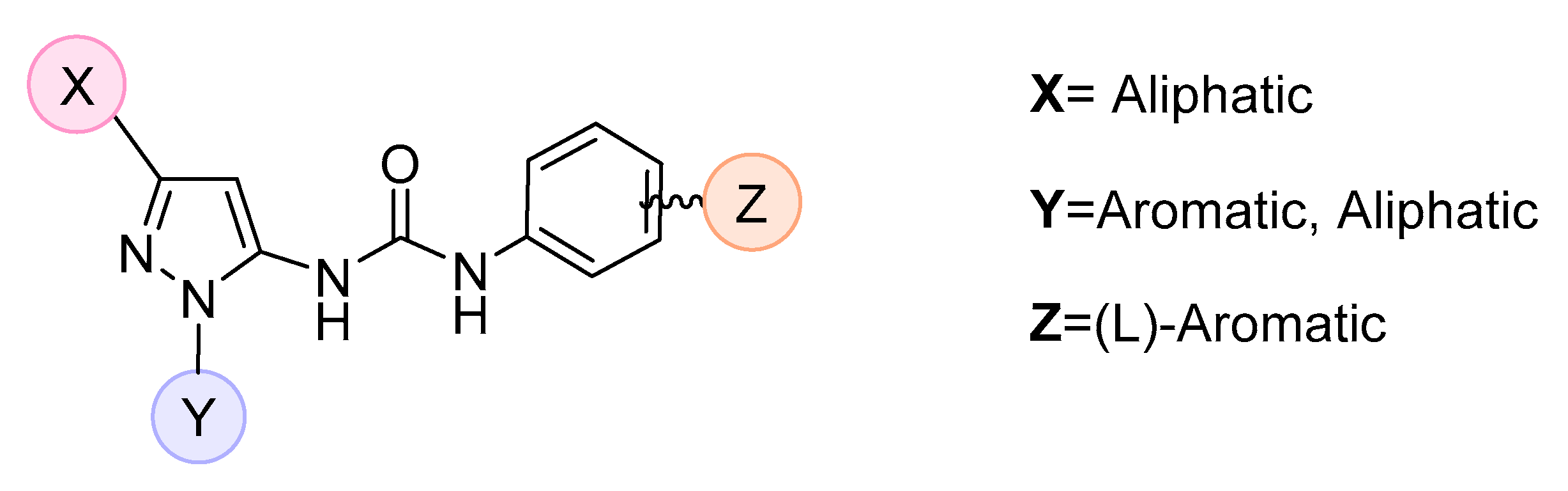
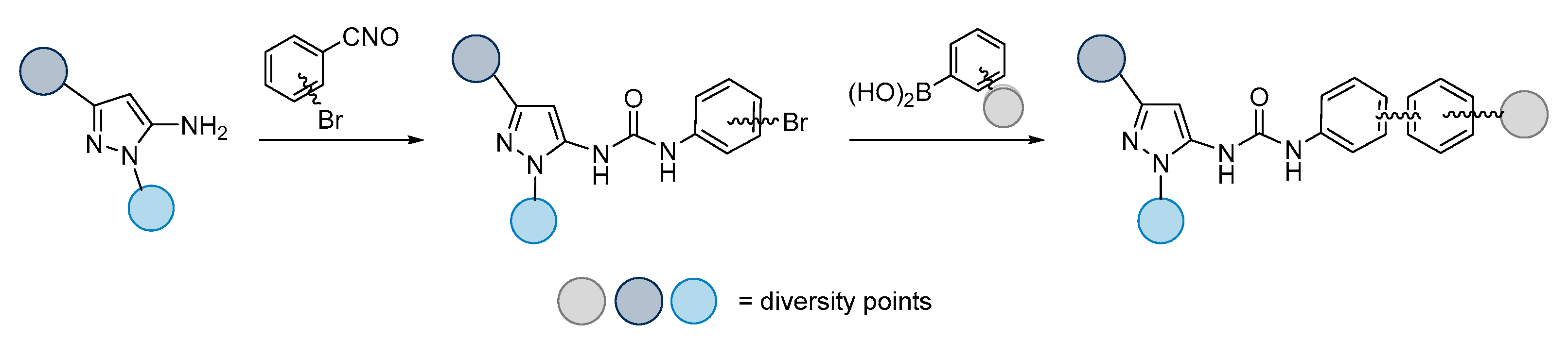
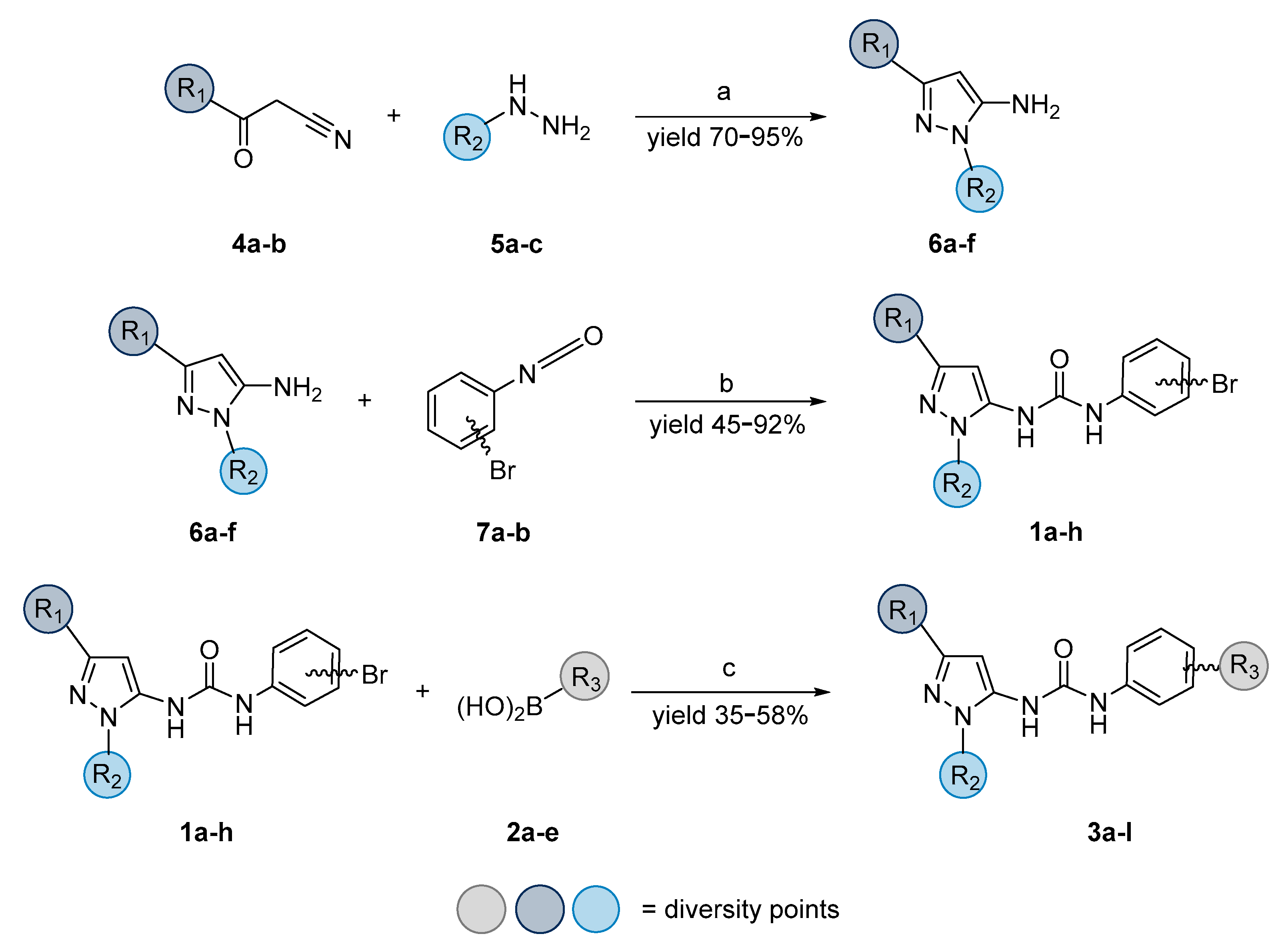
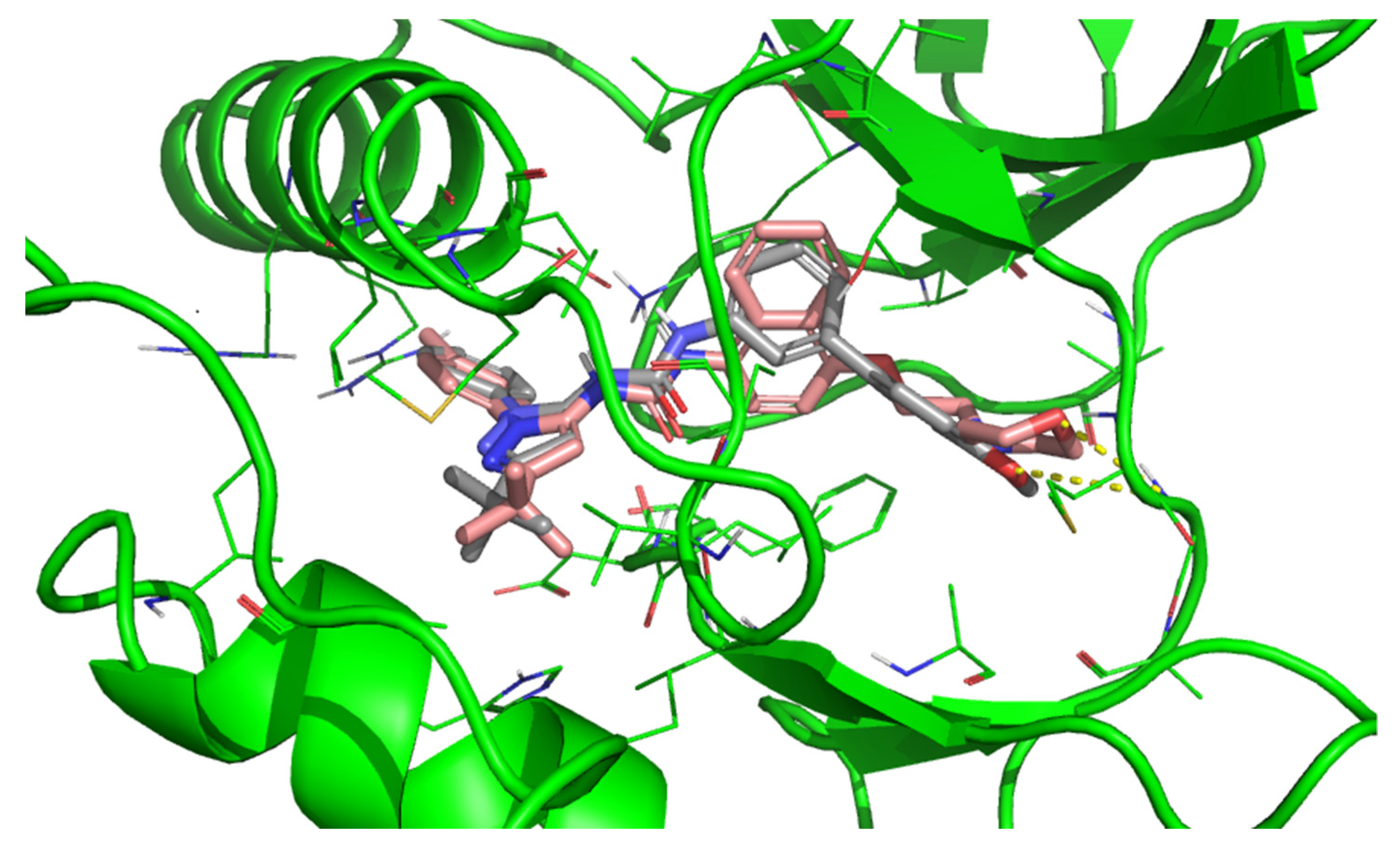

{kind=link}
{kind=link}
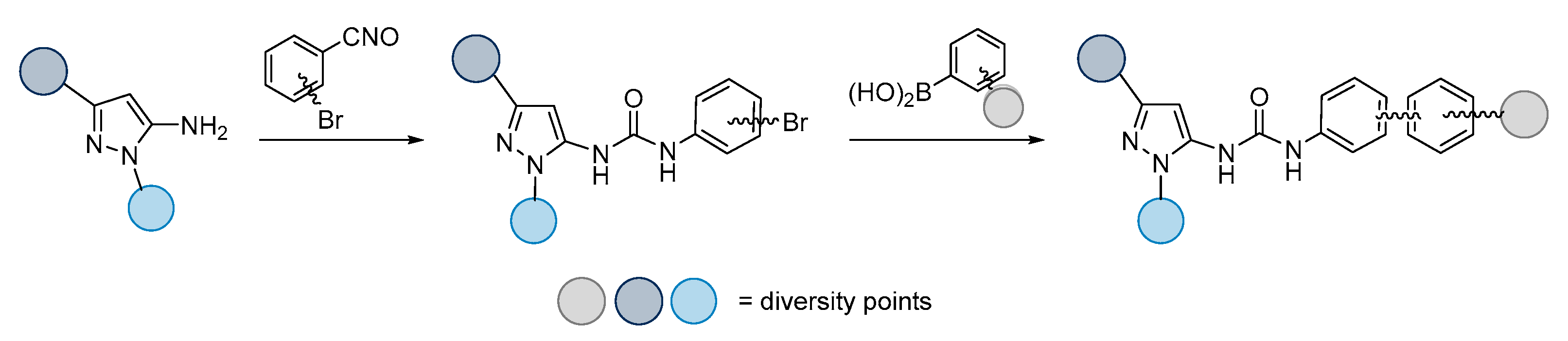{kind=link}
{kind=link}
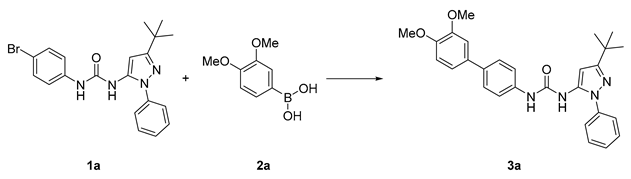 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Conditions | Time (h) | T (°C) | Solvent | Catalyst | Base | Yield |
| 1 | T1 | 18 | 101 | 1,4-dioxane | Pd(PPh3)4 | K2CO3 | N.R. |
| 2 | T2 | 18 | 101 | 1,4-dioxane | Pd(PPh3)4 | Na2CO3 | N.R. |
| 3 | T3 | 18 | 101 | 1,4-dioxane | Pd(dppf)Cl2 | K2CO3 | N.R. |
| 4 | T4 | 18 | 100 | 1:1 DMF/H2O | Pd(PPh3)4 | K2CO3 | N.R. |
| 5 | MW | 1 | 100 | MeCN | Pd(PPh3)4 | Na2CO3 | N.R. |
| 6 | MW | 1.25 | 100 | 1:2 H2O/1,4-dioxane | Pd(PPh3)4 | Na2CO3 | 30% a |
| 7 | MW | 1.25 | 100 | 1:2 H2O/1,4-dioxane | Pd(PPh3)4 | K2CO3 | N.R. |
| 8 | MW | 1.25 | 100 | 1:2 H2O/1,4-dioxane | Pd(dppf)Cl2 | K2CO3 | N.R. |
| 9 | MW5 | 1.25 | 100 | 1:2 H2O/1,4-dioxane | Pd(dppf)Cl2 | Na2CO3 | 50% a |
| 10 | MW6 | 1.25 | 70 | 1:2 H2O/1,4-dioxane | Pd(dppf)Cl2 | Na2CO3 | 15% b |
| 11 | MW7 | 0.5 | 100 | 1:2 H2O/1,4-dioxane | Pd(dppf)Cl2 | Na2CO3 | ~99% b |
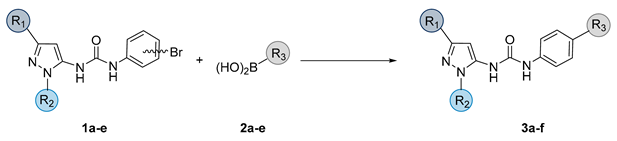 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Yield |
| 3a |  |  | 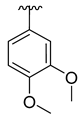 | 35% |
| 3b | 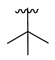 | 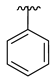 |  | 43% |
| 3c | 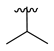 | 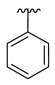 | 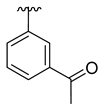 | 36% |
| 3d | 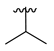 | 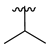 | 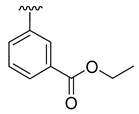 | 52% |
| 3e | 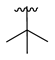 | -Me | 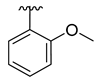 | 48% |
| 3f | 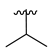 | -Me | 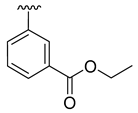 | 58% |
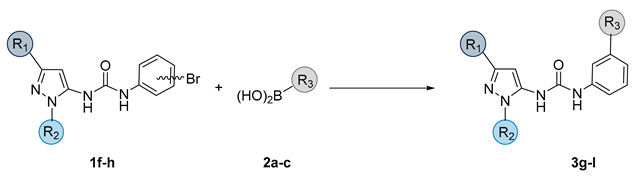 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Yield |
| 3g | 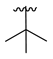 |  | 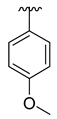 | 36% |
| 3h | 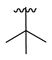 | 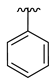 | 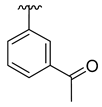 | 48% |
| 3i | 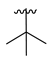 | 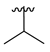 | 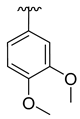 | 36% |
| 3l |  | -Me | 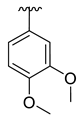 | 41% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Destro, L.; Van Melsen, R.; Gobbi, A.; Terzi, A.; Genitoni, M.; Zambon, A. Expedient Access to Type II Kinase Inhibitor Chemotypes by Microwave-Assisted Suzuki Coupling. Appl. Biosci. 2022, 1, 64-72. https://doi.org/10.3390/applbiosci1010004
Destro L, Van Melsen R, Gobbi A, Terzi A, Genitoni M, Zambon A. Expedient Access to Type II Kinase Inhibitor Chemotypes by Microwave-Assisted Suzuki Coupling. Applied Biosciences. 2022; 1(1):64-72. https://doi.org/10.3390/applbiosci1010004
Chicago/Turabian StyleDestro, Lorenza, Ross Van Melsen, Alex Gobbi, Andrea Terzi, Matteo Genitoni, and Alfonso Zambon. 2022. "Expedient Access to Type II Kinase Inhibitor Chemotypes by Microwave-Assisted Suzuki Coupling" Applied Biosciences 1, no. 1: 64-72. https://doi.org/10.3390/applbiosci1010004
APA StyleDestro, L., Van Melsen, R., Gobbi, A., Terzi, A., Genitoni, M., & Zambon, A. (2022). Expedient Access to Type II Kinase Inhibitor Chemotypes by Microwave-Assisted Suzuki Coupling. Applied Biosciences, 1(1), 64-72. https://doi.org/10.3390/applbiosci1010004