
Distinct Phenotypic and microRNA Expression in X-Linked Charcot–Marie–Tooth Correlated with a Novel Mutation in the GJB1 Gene




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Clinical Features and Family Pedigree
2.1.1. Family Pedigree
2.1.2. Patient 1: Clinical Features and Medical Examination
2.1.3. Patient 2: Clinical Features and Medical Examination
2.2. Genetic Analyses
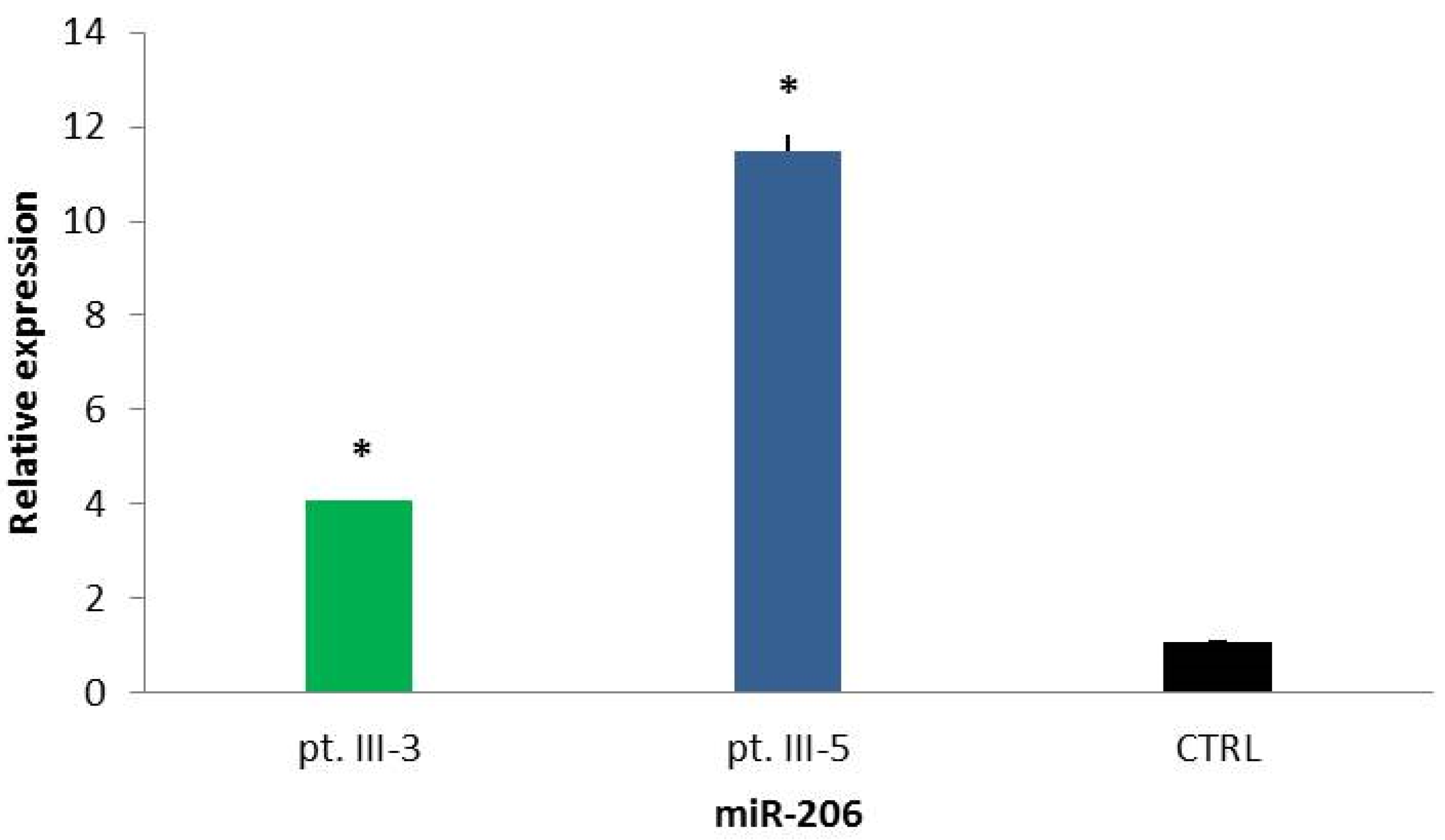
2.3. Circulating miR-206
2.4. Quality of Life in the Two Brothers
3. Discussion and Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pareyson, D.; Marchesi, C. Diagnosis, natural history, and management of Charcot–Marie–Tooth disease. Lancet Neurol. 2009, 8, 654–667. [Google Scholar] [CrossRef]
- Harding, A.E.; Thomas, P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980, 103, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, T. Hereditary neuropathies: An update. Rev. Neurol. 2016, 172, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Morena, J.; Gupta, A.; Hoyle, J.C. Charcot-Marie-Tooth: From Molecules to Therapy. Int. J. Mol. Sci. 2019, 20, 3419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milley, G.M.; Varga, E.T.; Grosz, Z.; Nemes, C.; Arányi, Z.; Boczán, J.; Diószeghy, P.; Molnár, M.J.; Gál, A. Genotypic and phenotypic spectrum of the most common causative genes of Charcot-Marie-Tooth disease in Hungarian patients. Neuromuscul. Disord. 2018, 28, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Saporta, A.S.; Sottile, S.L.; Miller, L.J.; Feely, S.M.; Siskind, C.E.; Shy, M.E. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 2011, 69, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.J. Charcot-Marie-Tooth Disease and Other Hereditary Neuropathies. Contin. Lifelong Learn. Neurol. 2020, 26, 1224–1256. [Google Scholar] [CrossRef]
- Deymeer, F.; Matur, Z.; Poyraz, M.; Battaloglu, E.; Oflazer-Serdaroglu, P.; Parman, Y. Nerve conduction studies in Charcot-Marie-Tooth disease in a cohort from Turkey. Muscle Nerve 2011, 43, 657–664. [Google Scholar] [CrossRef]
- Berciano, J.; García, A.; Gallardo, E.; Peeters, K.; Pelayo-Negro, A.L.; Álvarez-Paradelo, S.; Gazulla, J.; Martínez-Tames, M.; Infante, J.; Jordanova, A. Intermediate Charcot–Marie–Tooth disease: An electrophysiological reappraisal and systematic review. J. Neurol. 2017, 264, 1655–1677. [Google Scholar] [CrossRef]
- Bortolozzi, M. What’s the Function of Connexin 32 in the Peripheral Nervous System? Front. Mol. Neurosci. 2018, 11, 227. [Google Scholar] [CrossRef]
- Cisterna, B.A.; Arroyo, P.; Puebla, C. Role of Connexin-Based Gap Junction Channels in Communication of Myelin Sheath in Schwann Cells. Front. Cell. Neurosci. 2019, 13, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.S.; Koval, M. Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease. Int. J. Mol. Sci. 2018, 19, 1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskaran, M.; Mohan, M. MicroRNAs. Veter- Pathol. 2013, 51, 759–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderón, J.F.; Retamal, M.A. Regulation of Connexins Expression Levels by MicroRNAs, an Update. Front. Physiol. 2016, 7, 558. [Google Scholar] [CrossRef] [Green Version]
- Mitchelson, K.R.; Qin, W.Y. Roles of the canonical myomiRs miR-1, -133 and -206 in cell development and disease. World J. Biol. Chem. 2015, 6, 162–208. [Google Scholar] [CrossRef]
- Horak, M.; Novak, J.; Bienertova-Vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef]
- Pegoraro, V.; Missaglia, S.; Marozzo, R.; Tavian, D.; Angelini, C. MiRNAs as biomarkers of phenotype in neutral lipid storage disease with myopathy. Muscle Nerve 2019, 61, 253–257. [Google Scholar] [CrossRef]
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-generation sequencing in Charcot-Marie-Tooth disease: Opportunities and challenges. Nat. Rev. Neurol. 2019, 15, 644–656. [Google Scholar] [CrossRef]
- Guyton, G.P. Current concepts review: Orthopaedic aspects of Charcot-Marie-Tooth disease. Foot Ankle Int. 2006, 27, 1003–1010. [Google Scholar] [CrossRef]
- Pareyson, D.; Marchesi, C. Natural history and treatment of peripheral inherited neuropathies. Adv. Exp. Med. Biol. 2009, 652, 207–224. [Google Scholar] [CrossRef]
- McCorquodale, D.; Pucillo, E.M.; Johnson, N.E. Management of Charcot-Marie-Tooth disease: Improving long-term care with a multidisciplinary approach. J. Multidiscip. Health 2016, 9, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Reilly, M.M.; Shy, M.E. Diagnosis and new treatments in genetic neuropathies. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Corrado, B.; Ciardi, G.; Bargigli, C. Rehabilitation Management of the Charcot-Marie-Tooth Syndrome: A Systematic Review of the Literature. Medicine 2016, 95, e3278. [Google Scholar] [CrossRef] [PubMed]
- Padua, L.; for the Italian CMT QoL Study Group; Aprile, I.; Cavallaro, T.; Commodari, I.; La Torre, G.; Pareyson, D.; Quattrone, A.; Rizzuto, N.; Vita, G.; et al. Variables influencing quality of life and disability in Charcot Marie Tooth (CMT) patients: Italian multicentre study. Neurol. Sci. 2006, 27, 417–423. [Google Scholar] [CrossRef]
- Kleopa, K.A.; Abrams, C.K.; Scherer, S.S. How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease? Brain Res. 2012, 1487, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Fort, A.; Spray, D.C. Characteristics of gap junction channels in Schwann cells from wild-type and connexin-null mice. Ann. N. Y. Acad. Sci. 1999, 883, 533–537. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Legnini, I.; Martone, J.; Cazzella, V.; D’Amico, A.; Bertini, E.; Bozzoni, I. miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol. Med. 2011, 3, 258–265. [Google Scholar] [CrossRef]
- Koutsoulidou, A.; Kyriakides, T.C.; Papadimas, G.K.; Christou, Y.; Kararizou, E.; Papanicolaou, E.Z.; Phylactou, L.A. Elevated Muscle-Specific miRNAs in Serum of Myotonic Dystrophy Patients Relate to Muscle Disease Progress. PLoS ONE 2015, 10, e0125341. [Google Scholar] [CrossRef]
- Casola, I.; Scicchitano, B.M.; Lepore, E.; Mandillo, S.; Golini, E.; Nicoletti, C.; Barberi, L.; Dobrowolny, G.; Musarò, A. Circulating myomiRs in Muscle Denervation: From Surgical to ALS Pathological Condition. Cells 2021, 10, 2043. [Google Scholar] [CrossRef]
- Macchione, F.; Salviati, L.; Bordugo, A.; Vincenzi, M.; Camilot, M.; Teofoli, F.; Pancheri, E.; Zordan, R.; Bertolin, C.; Rossi, S.; et al. Multiple acyl-COA dehydrogenase deficiency in elderly carriers. J. Neurol. 2020, 267, 1414–1419. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pegoraro, V.; Sabbatini, D.; Salviati, L.; Angelini, C. Distinct Phenotypic and microRNA Expression in X-Linked Charcot–Marie–Tooth Correlated with a Novel Mutation in the GJB1 Gene. Muscles 2022, 1, 66-74. https://doi.org/10.3390/muscles1010007
Pegoraro V, Sabbatini D, Salviati L, Angelini C. Distinct Phenotypic and microRNA Expression in X-Linked Charcot–Marie–Tooth Correlated with a Novel Mutation in the GJB1 Gene. Muscles. 2022; 1(1):66-74. https://doi.org/10.3390/muscles1010007
Chicago/Turabian StylePegoraro, Valentina, Daniele Sabbatini, Leonardo Salviati, and Corrado Angelini. 2022. "Distinct Phenotypic and microRNA Expression in X-Linked Charcot–Marie–Tooth Correlated with a Novel Mutation in the GJB1 Gene" Muscles 1, no. 1: 66-74. https://doi.org/10.3390/muscles1010007
APA StylePegoraro, V., Sabbatini, D., Salviati, L., & Angelini, C. (2022). Distinct Phenotypic and microRNA Expression in X-Linked Charcot–Marie–Tooth Correlated with a Novel Mutation in the GJB1 Gene. Muscles, 1(1), 66-74. https://doi.org/10.3390/muscles1010007