Abstract
In recent years, 3D cell culture systems have emerged as sophisticated in vitro models, providing valuable insights into human physiology and diseases. The transition from traditional 2D to advanced 3D cultures has introduced novel obstacles, complicating the characterization and analysis of these models. While the lactate dehydrogenase (LDH) activity assay has long been a standard readout for viability and cytotoxicity assessments in 2D cultures, its applicability in long-term 3D cultures is hindered by inappropriate normalization and low LDH stability over time. In response to these challenges, we propose an optimization of LDH assays, including a crucial normalization step based on total protein quantification and a storage method using an LDH preservation buffer. We applied it to compare unexposed cerebral organoids with organoids exposed to a toxic dose of valproic acid, and showed efficient normalization of cellular viability as well as enhanced LDH stability within the buffer. Importantly, normalized LDH activity results obtained were independent of organoid dimension and cell density. This refined LDH assay, tailored to address 3D culture constraints, allows for the transposition of this routine test from 2D to 3D cultures.
1. Introduction
In vitro biological research has witnessed a surge in the adoption of 3D cell culture models, ranging from cellular aggregates, microtissues, and spheroids to more complex organoid models [1]. This growing interest can be attributed to the inherent advantages these 3D culture systems offer over traditional 2D cultures, including a better recapitulation of physiological cellular organization, enhanced cell-to-cell and cell-to-matrix communications, as well as improved cellular differentiation and signaling [1,2,3]. Particularly, organoids stand as complex 3D systems capable of recapitulating some aspects of tissue-specific organogenesis, cellular heterogeneity, cytoarchitectures, and functions [3,4,5,6,7]. From this complexity, 3D culture systems hold significant promise across a range of applications, including fundamental research, drug development, personalized medicine, and regenerative medicine [4,8].
As the popularity of 3D cultures continues to rise, various methods and protocols for their generation have emerged [1,2]. These methods encompass diverse culture supports, such as low-adherence culture plates, enabling 3D cell culture in suspension; bioreactors, permitting large-volume dynamic flow conditions that improve 3D system oxygenation [9,10]; microfluidic chips, referred to as organ-on-chip systems, allowing for the precise control of fluid flows and recapitulation of cellular microenvironment, improving cellular viability, maturation, and homogeneity [11,12,13,14,15,16]; as well as bioengineering strategies, including the use of 3D scaffolds and bioprinting [2,6,17,18]. These advancements have provided researchers with greater control over critical parameters, such as fluid flows, nutrients/oxygen supply, and waste management, therefore improving 3D culture conditions.
Nevertheless, the transition from 2D to 3D culture systems presents a unique set of challenges, with one of the foremost being cell viability assessment over time. Historically, lactate dehydrogenase (LDH) activity assays have served for assessing cellular viability, particularly in the context of 2D cell cultures [19,20,21]. LDH, a cytoplasmic enzyme, present in almost all cell types [22,23], is released into the extracellular environment when the cell membrane is damaged [21]. The LDH activity assay, performed on a conditioned culture medium, is based on the enzymatic conversion of lactate to pyruvate by LDH, carried out in the presence of nicotinamide adenine dinucleotide (NAD+). This enzymatic process results in the reduction in NAD+ to NADH, H+, and the concomitant oxidation of lactate to pyruvate [24]. Then, the regeneration of NAD+ by oxidation of NADH, H+, is generally coupled to the production of either a colored or a luminescent molecule, which can be detected using a spectrophotometer or a luminometer. The amount detected is directly proportional to the amount of LDH present in the medium, and thus is correlated to the cell viability in the culture conditions. Described for the first time in 1960 [19], the most common LDH assay relies on a tetrazolium salt, which is reduced into formazan, a red product that can be quantified using spectrometry [25,26,27]. Variants of this historical assay have been described [28,29,30,31], and several commercially available LDH activity detection kits have been developed. An LDH assay provides rapid and robust results to assess cell viability in a non-invasive manner, and enables the monitoring of cell culture over time thanks to regular sampling of a conditioned medium. This assay is also adapted to cytotoxicity evaluation of compounds, and cytoprotective studies of drug candidates [21].
However, implementing the LDH assay to 3D cell cultures faces a range of challenges, necessitating tailored solutions. Among these, the normalization step is of critical importance. In 2D cultures, the LDH activity value is usually normalized either by the number of seeded cells, the LDH activity in cell lysates, or the protein concentration. However, the inherent complexity of 3D cultures makes the precise quantification of cell density impossible while preserving the culture integrity. A potential strategy could be relying on the 3D culture dimension [32]; however, this measurement may not always be proportional to the actual cell number. Moreover, complex 3D culture systems like organoids commonly exhibit high heterogeneity within a single batch, including variations of dimensions, cell densities, cellular contents, and structures. Also, depending on the working hypothesis, the experimental settings may affect the volumes of culture medium, timepoints for medium renewal, or the culture support employed between 3D cultures. The LDH activity in cell lysates could effectively address these limitations. However, it requires lysing the organoid, therefore preventing longitudinal studies, and restricting its use for the normalization of endpoint analyses. These highlight the need for a robust normalization step, based on protein concentration, to allow for the direct comparison of LDH activity results between organoids.
Furthermore, LDH is not stable, and its activity diminishes over time, which requires us to perform LDH quantification on fresh conditioned medium. This limited stability poses a substantial obstacle to longitudinal comparisons, impeding comprehensive analyses of cellular dynamics over extended timeframes. One solution can be the use of a storage buffer to preserve LDH activity at temperatures below −20 °C for several days. Hence, it is imperative to adapt and optimize the LDH assay protocol to effectively address these limitations and ensure accurate and reliable viability assessments in 3D cell cultures.
In response to these needs, this article focuses on the optimization of the LDH activity assay specifically tailored for 3D cell cultures, with a particular emphasis on surmounting the aforementioned limitations. Our approach integrates a normalization method based on the quantification of total protein content in the conditioned culture medium, complemented by the characterization of a dedicated preservation buffer capable of maintaining LDH stability at −20 °C for one month. Using these strategic adaptations, we compared control cerebral organoids with organoids exposed to valproic acid, a known neurotoxin [33,34,35], and demonstrated the effective normalization of cytotoxicity levels between the organoids. By introducing this novel LDH assay methodology, our aim is to provide researchers with a standardized framework, paving the way for an improved LDH assay adapted to 3D culture analyses.
2. Material and Methods
Human-induced pluripotent stem cells culture—Human-induced Pluripotent Stem Cells (hiPSCs) were obtained with the reprogramming of BJ primary foreskin fibroblasts obtained from ATCC (CRL-2522). BJ fibroblasts were reprogrammed using the non-integrative Sendai virus vectors, following the manufacturer’s recommendations (A16517, ThermoFisher Scientific, Waltham, MA, USA). Pluripotency was confirmed with the detection of pluripotency markers using Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR), and the absence of mycoplasma was regularly checked. The culture and maintenance of hiPSCs were conducted as previously reported [36,37]. hiPSCs were maintained in feeder-free conditions on Geltrex-coated cell culture plates (A1569601, Gibco, Grand Island, NY, USA) in mTeSR+ medium (100-0276, STEMCELL Technologies, Madison, WI, USA) supplemented with mTeSR+ 5X supplement and 1% Penicillin/Streptomycin (P/S) (15140122, Gibco, Grand Island, NY, USA) at 37 °C, in a 5% enriched CO2 atmosphere.
Generation of cerebral organoids—A cerebral organoid generation protocol was adapted from the previously described protocols by Xiang and colleagues [38,39] to produce cortical organoids. Briefly, on day 0, hiPSCs were detached with 0.02% EDTA treatment (E8008, Sigma-Aldrich, Irvine, UK) and dissociated in single-cell suspension using Accutase (AT-104, STEMCELL Technologies, San Diego, CA, USA). V-bottom cell-repellent 96-well plates (651970, Greiner Bio-One, Frickenhausen, Germany) were seeded with 20,000 cells/well in neural induction medium composed of Dulbecco’s Modified Eagle Medium:Nutrient Mixture F-12 (DMEM-F12), GlutaMAX supplement (10565018, Gibco, Paisley, UK), 15% (v/v) KnockOut Serum Replacement (KOSR) (10828010, Gibco, Grand Island, NY, USA), 1% Minimum Essential Medium-Non-Essential Amino Acids (MEM-NEAA) (1140035, Gibco, Grand Island, NY, USA), 1% P/S, 100 nM LDN-193189 (72147, STEMCELL Technologies, Vancouver, BC, Canada), 10 μM SB-431542 (72232, STEMCELL Technologies, Vancouver, BC, Canada), 2 μM XAV-939 (X3004-5MG, Sigma-Aldrich, Saint Louis, MO, USA), 100 μM β-Mercaptoethanol (21985023, Gibco, Grand Island, NY, USA), and supplemented with 5% Fetal Bovine Serum (FBS) (10270106, Gibco, Paisley, UK) and 50 µM Y-27632 (72304, STEMCELL Technologies, Vancouver, BC, Canada). At day 2, embryoid bodies (EBs) were harvested and transferred in suspension cell culture 24-well plates (144530, Nunc, Roskilde, Denmark). The neural induction medium was changed every two days until day 10, without FBS supplement from day 2, and without Y-27632 from day 4 onwards. From day 10 to day 18, EBs were cultured in differentiation medium without vitamin A, composed of DMEM-F12:NeuroBasal Medium at 1:1 ratio (NeuroBasal Medium: 21103049, Gibco, Paisley, UK), supplemented with 0.5% (v/v) MEM-NEAA, 1% P/S, 0.5% N2 supplement 100X (17502-048, Gibco, Grand Island, NY, USA), 1% B-27 Supplement minus vitamin A (12587010, Gibco, Grand Island, NY, USA), 1% HEPES solution (H0887, Sigma-Aldrich, Irvine, UK), 0.025% human insulin (19278-5 mL, Sigma-Aldrich, Saint Louis, MO, USA), and 50 µM β-Mercaptoethanol. From day 18, EBs were cultured in differentiation medium with vitamin A, with the same composition as the previous medium, except for the B-27 Supplement with vitamin A (17504044, Gibco, Grand Island, NY, USA), and supplemented with 20 ng/mL BDNF (78005, STEMCELL Technologies, Vancouver, BC, Canada), 200 μM ascorbic acid (A9290225G, Sigma-Aldrich, Saint Louis, MO, USA), and 200 µM cAMP (73886, STEMCELL Technologies, Vancouver, BC, Canada). Organoids were cultured in a 24-well plate in 500 µL of culture medium changed every two days from day 2 to day 28, then in 1 mL of medium changed once per week, under agitation (80 rpm/min) at 37 °C, in a 5% enriched CO2 atmosphere.
Valproic acid exposure on cerebral organoids—Cerebral organoids were exposed to valproic acid (P4543, Sigma-Aldrich, Saint Louis, MO, USA) at two concentrations (1 mM and 10 mM) from day 18 to day 21 of culture.
Cerebral organoid surface area measurements—To assess cerebral organoid growth evolutions over time, brightfield images (5X) of the organoids were acquired at different timepoints during the culture, and organoid surface areas were measured using the ImageJ—Fiji software, version 1.54f [40].
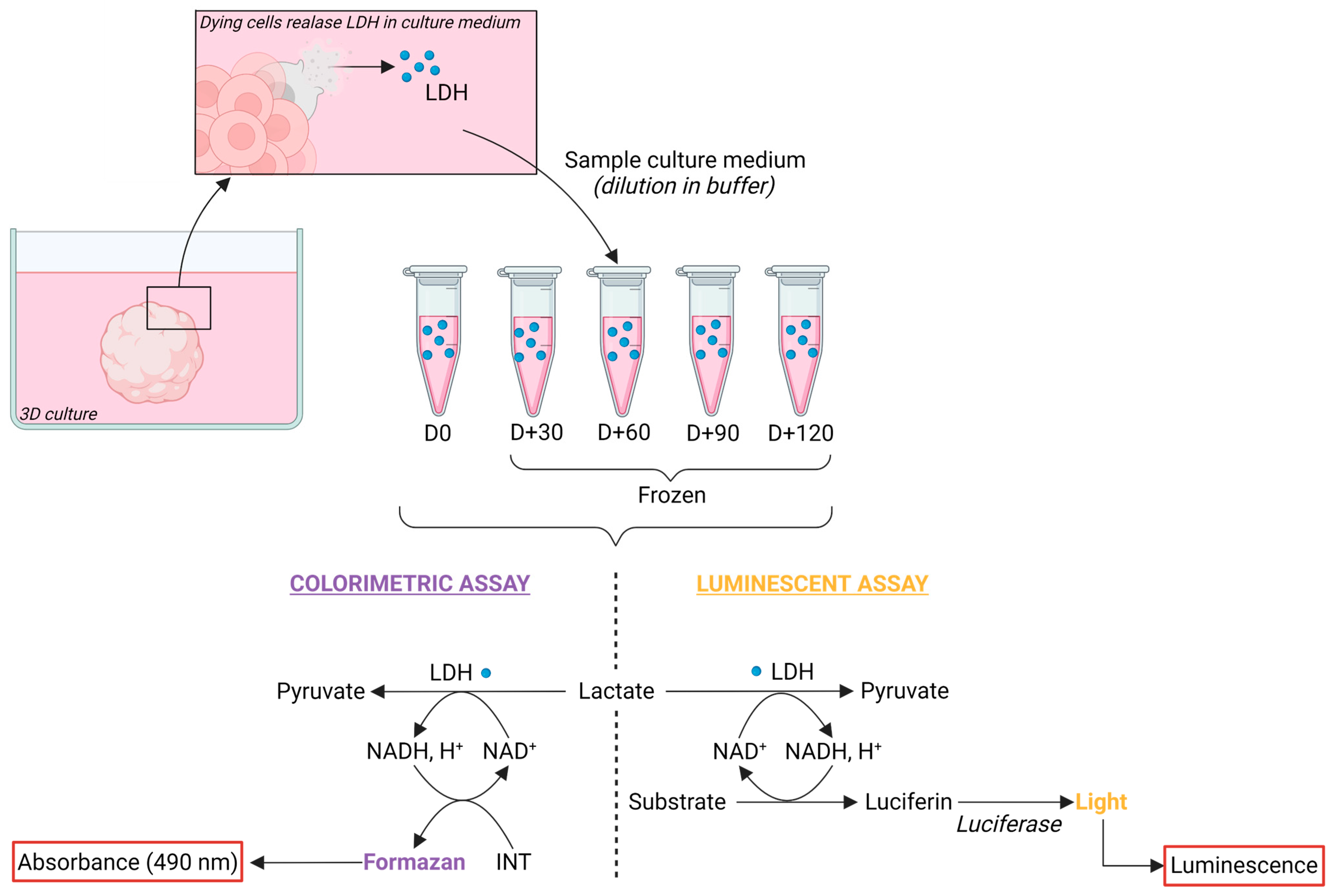
Conditioned culture medium (CM) sampling and processing for lactate dehydrogenase (LDH) activity preservation—To preserve LDH activity, CM was mixed with an LDH-preservation buffer containing Bovine Serum Albumin (BSA) as a stabilizing agent at a 1:1 ratio. To test for LDH stability, aliquots of CM sampled from 2 months of cerebral organoids were frozen, with (CMB) or without preservation buffer, for monthly analysis over a span of 4 months (Figure 1). LDH preservation buffer was composed of 200 mM Tris-HCl buffer solution at pH 7.3 (T2194-1L, Sigma-Aldrich, Saint Louis, MO, USA), 10% Glycerol (15523-1L-R, Sigma-Aldrich, Saint Louis, MO, USA), and 1% Bovine Serum Albumin (BSA) (A2153, Sigma-Aldrich, Saint Louis, MO, USA).
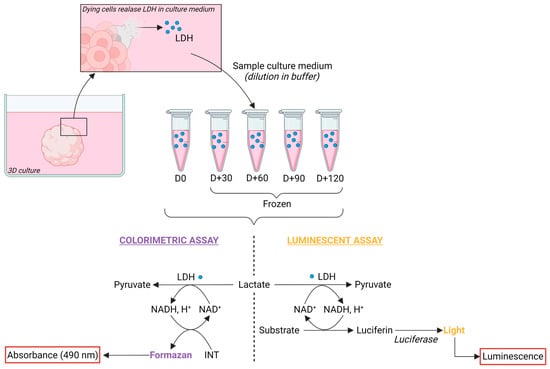
Figure 1.
Preservation and detection of Lactate Dehydrogenase in conditioned medium of cerebral organoids. Principle of LDH detection kits: upon cell death or membrane damage, LDH leaks into the culture medium. Conditioned medium was collected and processed with the addition of an LDH-preservation buffer at a 1:1 ratio prior freezing. Subsequent LDH detection employed either colorimetric or luminescent assays. In brief, LDH catalyzes the conversion of lactate to pyruvate while reducing NAD+ to NADH, H+. NADH, H+ is then oxidized back to NAD+ with the concomitant conversion of a substrate to a product. In the colorimetric assay, iodonitrotetrazolium (INT) is reduced to formazan, yielding a purple color detected at 490 nm. In the luminescent assay, luciferin is produced and then metabolized with the addition of luciferase to produce light. Created with BioRender.com (accessed on 3 November 2023).
Dilution assay of the conditioned medium in LDH-preservation buffer solution (CMB)—To search for an optimal dilution that would prevent saturation of the measured absorbance or luminescence, a range of CMB dilutions in distilled water was tested. The assessed dilutions ranged from 1 (100% CMB) to 1/8 (12.5% CMB; 87.5% water). After optimization, the working dilution of 1/4 (25% CMB; 75% water) was validated and used for the cytotoxic analysis of valproic acid exposures.
LDH activity assays for cytotoxicity evaluation—The LDH activity was measured using two commercial kits, following the manufacturers’ instructions: CyQUANT LDH Cytotoxicity Assay Kit (C20301, ThermoFisher Scientific) and LDH-Glo Cytotoxicity Assay (J2380, Promega) (Figure 1). For cytotoxic analysis of valproic acid exposures, CM was sampled at day 21 before medium renewal. For the positive control of maximum LDH release, a cerebral organoid was lysed using incubation in 1 mL of RIPA Lysis and Extraction Buffer (89900, ThermoFisher Scientific) supplemented with Pierce Protease and Phosphatase Inhibitor Mini Tablets (A32959, ThermoFisher Scientific) for 15 min on ice, followed by gentle mechanical dissociation. Negative control of fresh culture medium containing 10 mM valproic acid was used.
Bicinchoninic assay (BCA) for determination of total protein concentration—Detection and quantitation of total protein were determined using the Pierce BCA Protein Assay Kit (23225, ThermoFisher Scientific), following the manufacturer’s recommendations.
3. Results
3.1. Lactate Dehydrogenase (LDH)-Preservation Buffer Allows for the Conservation of LDH for One Month at −20 °C
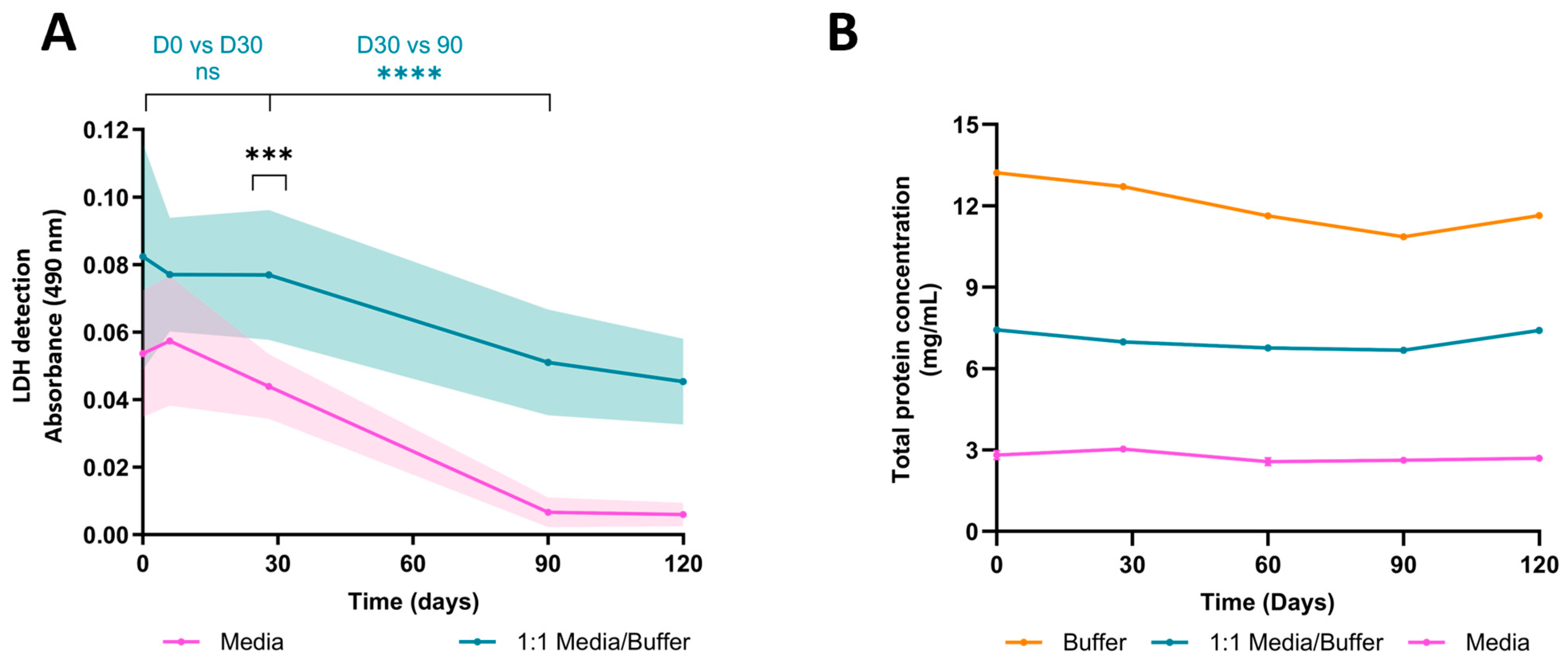
Measuring LDH content in conditioned medium (CM) is a step typically conducted post-exposure to search for potential effects. The prolonged culture period of organoids offers the possibility of longitudinal studies and comparisons across various culture timepoints. However, to do so, meticulous handling and preservation of the CM is required to ensure LDH integrity at the time of analysis. To address this, we prepared an LDH-preservation buffer and used it to dilute the CM at a 1:1 ratio. Our findings reveal that without the buffer, LDH stability starts to diminish within a week at −20 °C to reach a significant difference at one month (absorbance, D30w/o buffer = 0.044 ± 0.008 SEM, D30buffer = 0.077 ± 0.016 SEM), whereas the buffer extended enzyme stability for up to a month (absorbance, D0buffer = 0.077 ± 0.014 SEM, D30buffer = 0.077 ± 0.016 SEM) (Figure 2A). Remarkedly, LDH activity in CM without a buffer dramatically decreased over time until becoming undetectable after three months of storage, whereas LDH activity in CMB steadily diminished and remained detectable after four months. Notably, these overall declines appear to be specific to LDH activity, or at least do not impact all protein types. Indeed, we assessed total protein concentration via a bicinchoninic assay (BCA), and showed that the concentration remained stable throughout the four-month study (Average protein concentration, Cbuffer = 12.01 mg/mL ± 0.38 SEM, CCMB = 7.05 mg/mL ± 0.14 SEM, CCM = 2.74 mg/mL ± 0.07 SEM) (Figure 2B). Furthermore, the preservation buffer markedly increases total protein concentration due to its abundant BSA content, suggesting a necessity for dilutions to prevent saturation in subsequent analyses.
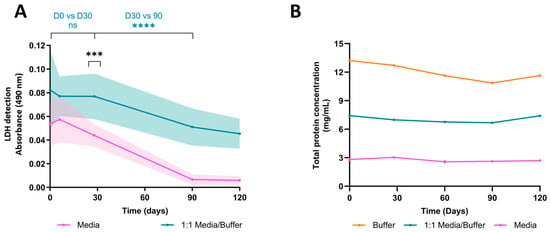
Figure 2.
Stability of Lactate Dehydrogenase and total protein concentration in preservation buffer at −20 °C over time. (A) LDH stability was evaluated over time. Samples of cerebral organoids conditioning the medium, with or without preservation buffer, were frozen and analyzed at day 0, 30, 60, 90, and 120 post-collection. Without a preservation buffer, LDH stability declines after one week of preservation at −20 °C (D30w/o buffer vs. D30buffer, p = 0.0008, nested t-test, n = 3 organoids). Preservation buffer maintains LDH stability for up to one month at −20 °C (D0buffer vs. D30buffer, p = 0.1807; D30buffer vs. D90buffer, p < 0.0001, nested t-test, n = 3 organoids). (B) Total protein concentration was quantified in the conditioned medium, conditioned medium diluted in preservation buffer, and preservation buffer alone. Total protein concentration remained stable over time (D90buffer vs. D120buffer, p = 0.6629; D90media/buffer vs. D120media/buffer, p = 0.3810; mixed-effects analysis followed by Tukey’s post hoc, n = 3). ns: not significant; ****: p < 0.0001; ***: p < 0.001.
3.2. Optimal Dilution of Conditioned Medium Mixed with Buffer (CMB) for Consistent Analyses
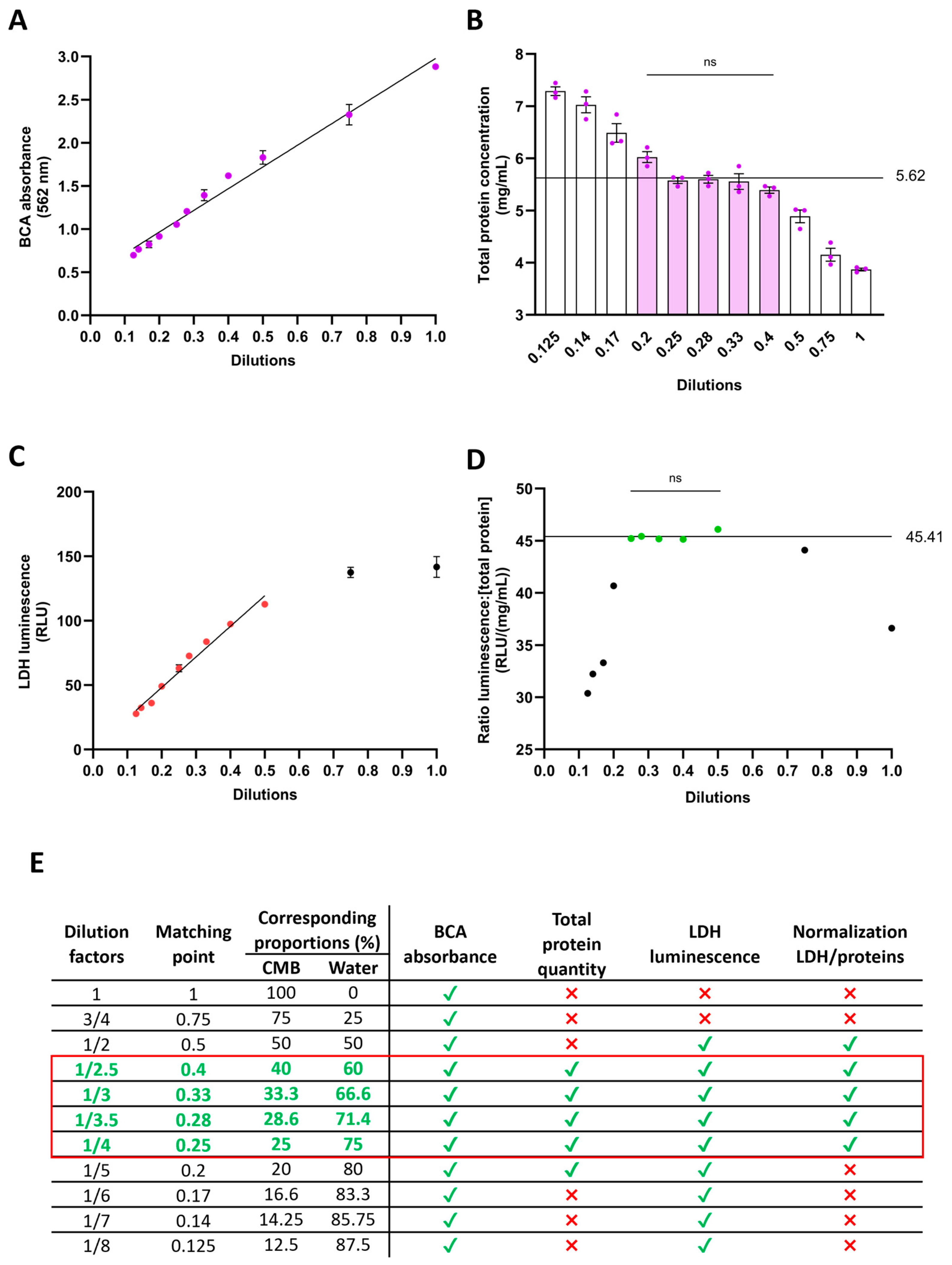
Signals such as absorbance or luminescence can easily saturate in the presence of abundant substrates, posing a challenge to data reliability. To ensure signal linearity and coherence between actual and measured values, we diluted the CMB sampled from 2 months of cerebral organoids with distilled water across various factors ranging from 1 to 1/8 (Figure 3E). We then assessed total protein concentration over this dilution range. Notably, the signal maintained linearity (R2 = 0.9811), suggesting that the preservation buffer had no discernible impact on the proportionality between dilution and absorbance (Figure 3A). However, converting absorbance to protein concentration using a BSA standard curve revealed discrepancies. Among the eleven dilutions tested, only five consistently converged to a concentration of 5.62 mg/mL (Figure 3B), validating their reliability. Regarding LDH quantification via the luminescent assay, signal linearity persisted within dilutions ranging from 1/2 to 1/8 (R2 = 0.9773) (Figure 3C), indicating potential saturation above 1/2 dilution and the kit’s high sensitivity in detecting low LDH concentrations. To normalize LDH measurements, we utilized the total protein concentration of CMB. Normalizing luminescence by protein concentration across the eleven dilutions revealed convergence for five dilutions, yielding a consistent ratio value of 45.41 (Figure 3D), affirming their reliability. Overall, our findings support the consistency of measurements within working dilutions ranging from 1/2.5 to 1/4 (Figure 3E). For our next experiment, we decided to use a dilution factor of 1/4.
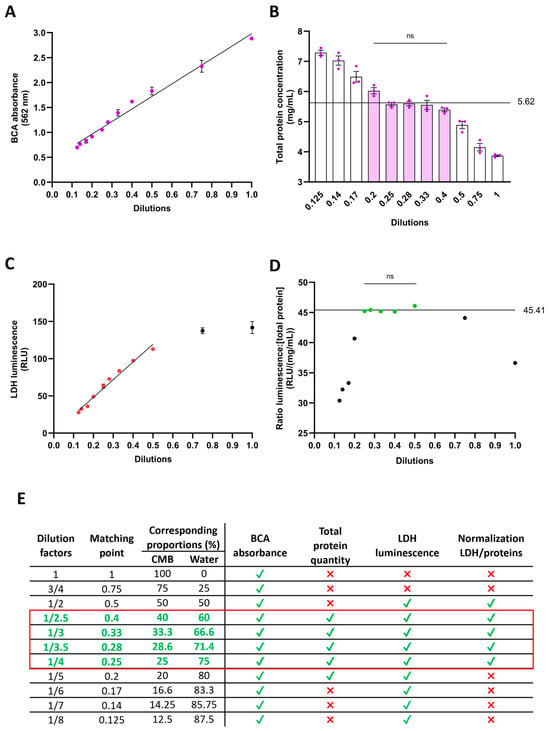
Figure 3.
Dilution optimization of 1:1 conditioned medium in LDH-preservation buffer. The addition of preservation buffer to conditioned medium increases the protein content. To prevent saturation of absorbance or luminescence detection, various dilutions of the 1:1 medium/buffer in water were assessed. (A) BCA reaction to quantify protein content. Linearity was maintained across all dilutions. (B) Total protein concentration detection, taking into account the dilution factor. Dilutions ranging from 1/2.5 to 1/5 yield protein concentration detection comparable to 5.62 mg/mL (filled bars). (C) LDH detection using the luminescent assay. Signal linearity was maintained for dilutions from 1/2 to 1/8 (colored dots). (D) Validation of luminescence to total protein ratio. Ratios within dilutions ranging from 1/2 to 1/4 are not significantly different from 45.41 RLU/(mg/mL) (colored dots). (E) Summary Table of validated dilutions. For each test, green ticks indicate validation of the dilution and red crosses non validation. The red square highlights the dilutions that were validated with all tests. ns: not significant.
3.3. Efficient Normalization of LDH Activity to Evaluate Cerebral Organoid Cytotoxicity
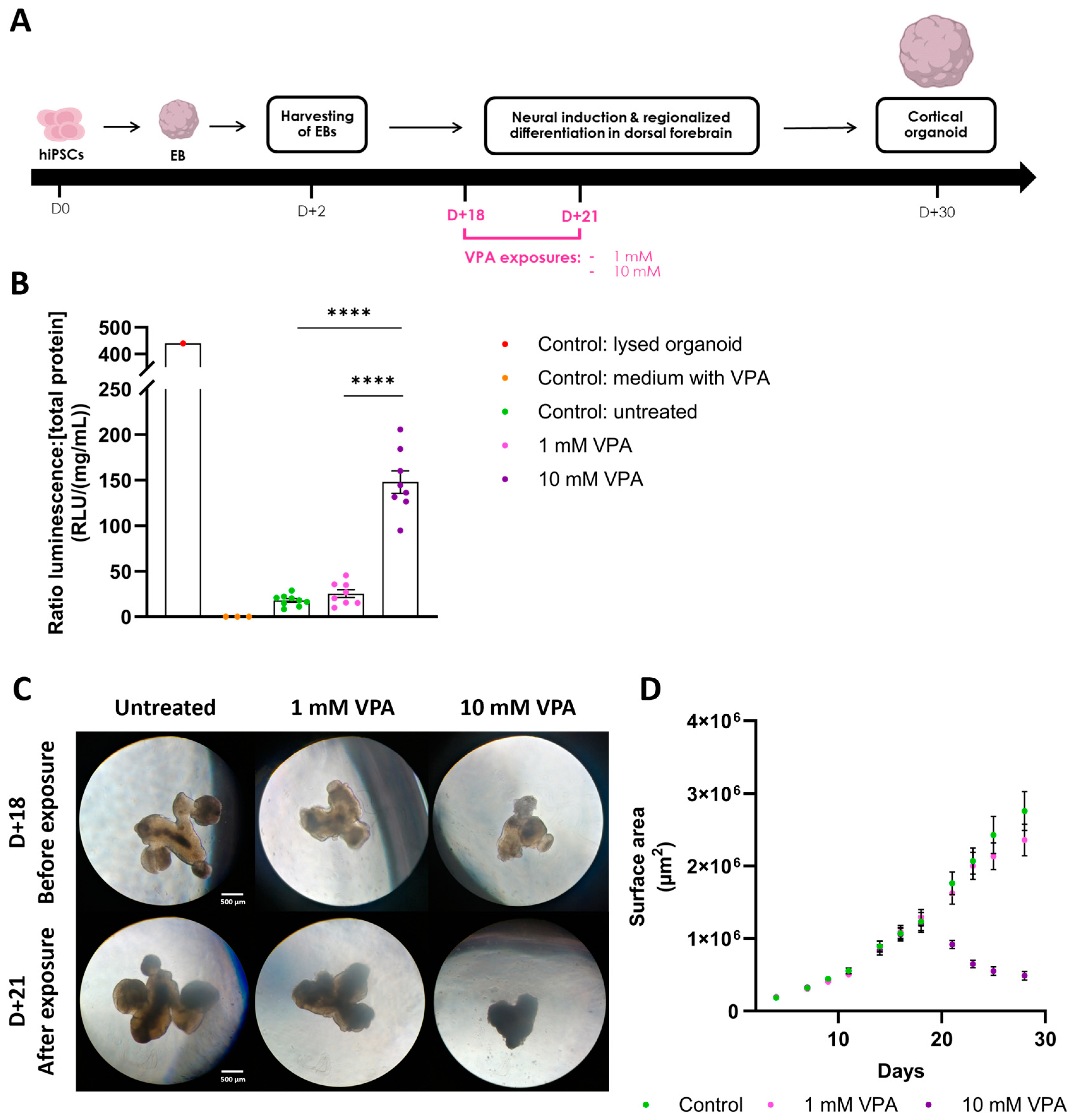
To validate our technique, we applied the methodology following exposure of cerebral organoids to valproic acid (VPA), a known developmental neurotoxin (Figure 4A). Exposure to 1 mM VPA showed no significant impact on LDH release into the CM, evidenced by comparable LDH levels between the treated and control conditions (ratio LDH, LDHuntreated = 17.87 RLU/(mg/mL) ± 1.91 SEM, LDH1 mM VPA = 25.41 RLU/(mg/mL) ± 4.07 SEM) (Figure 4B). Conversely, exposure to 10 mM VPA markedly elevated LDH levels in the CM (ratio LDH, LDH10 mM VPA = 147.89 RLU/(mg/mL) ± 11.53 SEM), indicating, as anticipated, compromised cell membrane integrity. These LDH measures are in accordance with the morphological observations of the organoids, as the 1 mM VPA-exposed organoids are similar to controls, whereas the 10 mM VPA-exposed organoids exhibit a cellular loss associated with a compromised structural integrity, suggesting non-viability (Figure 4C). This is further confirmed by the growth evolutions of the organoids, as the surface of organoids exposed to 10 mM VPA began to decrease immediately upon exposure, indicating cellular loss (Figure 4D). These results confirm the validity of our approach to effectively normalize distinct cytotoxicity levels. Comparisons with all the controls (ratio LDH, LDHlysed = 439.82 RLU/(mg/mL), LDHCtrl VPA = 0 RLU/(mg/mL) ± 0 SEM) underscored the precision of the luminescent detection technique, as well as the potential of our methodology to discriminate between experimental conditions.
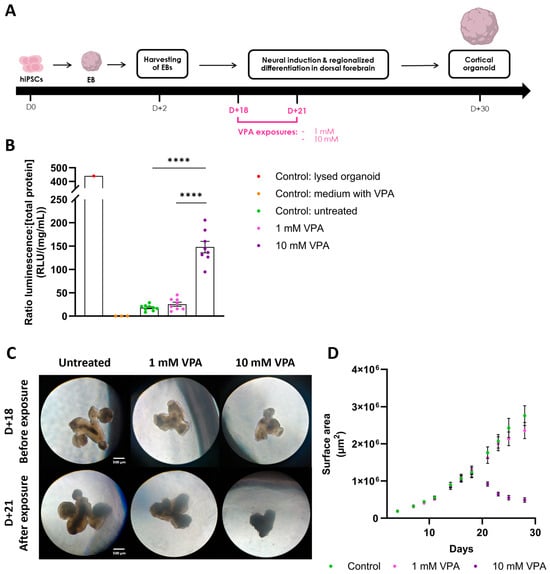
Figure 4.
Valproic acid exposure on cerebral organoids increases LDH release in the medium. (A) Cortical organoid generation timeline. hiPSCs were aggregated into embryoid bodies (EB) before being differentiated into dorsal forebrain organoids. Organoids were exposed to either 1 mM or 10 mM VPA at day 18 for 72 h. Created with BioRender.com. (B) Subsequently, at day 21, LDH activity was detected using the luminescent assay, and was normalized using total protein concentration. Treatment with 10 mM VPA significantly increased LDH levels in the medium compared to untreated organoids (LDHuntreated = 17.87 RLU/(mg/mL) ± 1.91 SEM; LDH10 mM VPA = 147.89 RLU/(mg/mL) ± 11.53 SEM; n = 8–9 organoids per condition; one-Way ANOVA followed by Tukey’s multiple comparison; F(4, 24) = 142.2; p < 0.0001). Control: medium with VPA containing 10 mM VPA. (C) Organoids observed with optical brightfield microscopy (5×) before and after VPA exposure. Control and 1 mM VPA exposed organoids showed no morphological difference. Organoids exposed to 10 mM VPA exhibited cellular loss associated with smaller organoid size and darker color, suggesting cell death. (D) Growth profiles of the cerebral organoids over 30 days. After exposure to 10 mM VPA at day 18, the area of the organoids started to decrease, further confirming cell death induction. In contrast, organoids treated with 1 mM VPA exhibited the same growth profile as the controls, suggesting no detrimental effects of VPA at this lower concentration. ****: p < 0.0001.
4. Discussion
The widespread utilization of lactate dehydrogenase (LDH) activity measurement in 2D in vitro cytotoxicity studies suggests a potential need for applications in 3D cultures. Transitioning LDH assays from 2D to 3D cell models could substantially aid in evaluating the viability of 3D systems over time or in following compound exposures. Traditionally, LDH activity values in 2D cultures are normalized against either the initially seeded cell population, the protein concentration, or as a percentage of total LDH activity in cell lysates. However, in the context of 3D cultures, particularly with organoids displaying complex self-organizations, precise cell counting poses a considerable challenge [41]. Notably, the absence of non-invasive methods for accurate cell counting in 3D cultures presents an obstacle for long-term studies. Similarly, normalization based on total LDH activity in cell lysates requires dissociation and lysis of the organoids, therefore rendering it unsuitable for longitudinal studies. To address this, we developed a methodology to normalize LDH activity values using the total protein concentration in the culture medium, presuming a proportional relationship with cell number.
Moreover, the prolonged culture duration of organoids, enabling longitudinal studies, necessitates the collection of conditioned medium (CM) at various timepoints during the culture. Consequently, it becomes imperative to develop a method for CM long-term storage without compromising LDH enzyme integrity or activity. Indeed, proteins diluted in solution are more prone to inactivation and activity loss due to low-level binding [42]. To stabilize LDH by binding, we decided to use Bovine Serum Albumin (BSA) as a carrier. BSA not only has the capability to bind proteins and maintain their activity, but is also known to protect proteins from heat [43], aggregation [44], and reactive oxygen species [45], all of which can impact enzyme functionality by altering their conformation. Additionally, it has been shown that LDH isoform B exhibits favorable stability when diluted with a saline-BSA solution, in the context of clinical diagnosis [46]. The mechanisms underlying BSA’s beneficial binding effect are not fully understood [47], but could rely on hydrophobic interactions between the enzyme and BSA [43]. Our optimization of an LDH-preservation buffer, mixed with CM at a 1:1 ratio, extends LDH stability for up to a month at −20 °C, as evidenced by consistent LDH levels prior to a decline after one month of storage, compared to samples without the buffer. Notably, loss of LDH activity in CM without buffer is already observable after one week at −20 °C, followed by a drastic decrease until total loss of LDH activity after three months. Furthermore, it should be noticed that the buffer not only enables the extension of LDH activity up to one month, but that the subsequent decline was identical between the samples, and that an LDH activity was still detectable after four months of storage. This suggests the potential for comparing samples stored for more than one month by taking into account a global decrease in LDH activity levels in the CM.
While the preservation buffer extends LDH stability, its impact on signal linearity (absorbance or luminescence) and consistency between actual and measured values (total protein concentration and normalized ratios) necessitates detailed scrutiny. Indeed, the presence of bovine serum albumin (BSA) in the preservation buffer, in addition to the high protein and amino acid content present in the organoid culture medium, tend to saturate the absorbance signals, requiring adapted dilutions. Bicinchoninic assay (BCA) with CMB samples diluted in distilled water at various dilution factors indicate that the preservation buffer does not disrupt the proportionality between protein concentration and absorbance, implying maintained linearity. However, discrepancies emerge when converting absorbance to protein concentration using a BSA standard curve. Additionally, luminescence-based LDH detection reveals signal saturation at the most concentrated dilutions. Our findings suggest that CMB dilutions ranging from 1/2.5 to 1/4 with distilled water maintain measurement consistency. Thus, it is crucial to optimize dilution factors tailored to their experimental setup and analytical techniques to ensure consistent and reliable measurements.
Utilization of a BCA assay for protein quantification may not be optimal, given observed discrepancies in the conversion from absorbance to protein concentration. Exploring alternative protein quantification methods could improve accuracy in this regard. Similarly, other non-invasive quantification methodologies than protein dosage, such as measurement of glucose consumption in conditioned medium as presented by Fertan and colleagues [48], could stand as alternative LDH normalization methods.
Validation of our methodology with exposures of cerebral organoids to valproic acid (VPA) demonstrates its efficacy in detecting changes in LDH release into the CM, in accordance with the exposure doses. Exposure to 10 mM VPA significantly elevates LDH levels compared to controls, indicating high cytotoxicity, as expected. However, exposure to 1 mM VPA shows no significant impact on LDH levels, which is also in accordance with the literature. Indeed, VPA is known to act as a developmental neurotoxin, impairing cortical development and cytoarchitectures in early stage cerebral organoids at a 1 mM concentration, but without affecting cerebral organoid morphological integrity [33,34,35]. It is plausible that the 10 mM concentration induced cell death, whereas the 1 mM concentration likely affected cytoarchitectures without triggering increased cell death. This underscores the precision of the luminescent detection technique and the methodology’s ability to discriminate between experimental conditions.
Furthermore, this optimized LDH assay paves the way for more complex experimental settings to be analyzed, such as comparisons of different culture conditions, either in distinct culture supports involving various volumes of culture medium, or with different timepoints for medium renewal, requiring distinct timepoint samplings and thus variations in time spent with 3D cultures in the CM. Consequently, this protein-based normalized LDH assay appears to be adapted to the emerging field of organs-on-chips, requiring accurate normalization to be compared with conventional cell culture supports.
Although our methodology serves as a framework to efficiently and reliably assess cellular viability in 3D cultures, it should be noticed that this reliability appears to reach a limit when comparing highly similar experimental conditions. Indeed, the results of VPA exposure on cerebral organoids suggest that this method is better suited for comparing and quantifying viability in experimental conditions with significant differences of cytotoxicity levels, rather than for discerning smaller discrepancies in effect size between closely related conditions. Overall, to comprehensively evaluate cellular viability in 3D cultures, it is advisable to employ multiple assays and markers [49,50]. Similarly to the LDH assay, additional non-invasive studies based on conditioned medium analysis, notably including the detection of apoptosis and oxidative stress markers, as well as evaluation of metabolic activity, might be considered. Additionally, endpoint methods such as immunohistochemical analyses (e.g., detection of apoptosis via cleaved-caspase3 [51] or using TUNEL assay [52], and DNA damage markers such as yH2AX [53]) and transcriptomic analyses (e.g., assessment of pro- and anti-apoptotic markers BAX and BCL2 [54]) can provide valuable insights and help in obtaining a more comprehensive characterization of cytotoxicity levels.
The development of this refined LDH assay represents an advancement in addressing the challenges associated with longitudinal monitoring of 3D culture viability. By tailoring the standard LDH assay to accommodate the specific constraints of 3D systems, our proposed normalization and long-term storage methodologies facilitate the transition of this routine test from 2D to 3D. This enhanced LDH assay not only paves the way for the development of quantification techniques adapted to the analysis of conditioned media from 3D cultures, but also broadens the scope of cellular viability assessment in 3D. Moving forward, integration of this method in routine characterization of 3D cultures holds great promises for enhancing the versatility of these models and for advancing various biomedical and toxicological applications that rely on cytotoxicity evaluations.
5. Conclusions
The development of this refined LDH release assay addresses critical challenges in assessing cellular viability in 3D cultures. By optimizing the assay for 3D systems, including the use of BSA for enzyme stabilization and a methodology for LDH activity quantification and normalization, we facilitate accurate and long-term 3D viability assessments. This innovative approach not only eases the transition of routine LDH assays from 2D to 3D models, but also ensures reliable LDH measurements for longitudinal studies. Consequently, this assay enhances the capacity for detailed cytotoxicity evaluations in complex 3D cultures, paving the way for advanced preclinical and toxicological applications.
Author Contributions
Conceptualization, H.C. and P.-A.V.; methodology, H.C., T.L. and P.-A.V.; validation, J.-P.D., F.Y. and P.-A.V.; formal analysis, H.C., L.M., T.L. and P.-A.V.; investigation, H.C. and L.M.; resources, J.-P.D. and F.Y.; writing—original draft preparation, H.C. and P.-A.V.; writing—review and editing, H.C., L.M., T.L., J.-P.D., F.Y. and P.-A.V.; supervision, P.-A.V.; project administration, F.Y.; funding acquisition, J.-P.D. and F.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
Acknowledgments
The authors would like to thank Jessica Rontard from NETRI company for her help in conceptualizing this work and Simon Benetiere for his help in samples processing.
Conflicts of Interest
Héloïse Castiglione was employed by the company NETRI. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The Third Dimension Bridges the Gap between Cell Culture and Live Tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- Cacciamali, A.; Villa, R.; Dotti, S. 3D Cell Cultures: Evolution of an Ancient Tool for New Applications. Front. Physiol. 2022, 13, 836480. [Google Scholar] [CrossRef]
- Simian, M.; Bissell, M.J. Organoids: A Historical Perspective of Thinking in Three Dimensions. J. Cell Biol. 2017, 216, 31–40. [Google Scholar] [CrossRef]
- Corrò, C.; Novellasdemunt, L.; Li, V.S.W. A Brief History of Organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Garreta, E.; Kamm, R.D.; Chuva de Sousa Lopes, S.M.; Lancaster, M.A.; Weiss, R.; Trepat, X.; Hyun, I.; Montserrat, N. Rethinking Organoid Technology through Bioengineering. Nat. Mater. 2021, 20, 145–155. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primers 2022, 2, 94. [Google Scholar] [CrossRef]
- Suarez-Martinez, E.; Suazo-Sanchez, I.; Celis-Romero, M.; Carnero, A. 3D and Organoid Culture in Research: Physiology, Hereditary Genetic Diseases and Cancer. Cell Biosci. 2022, 12, 39. [Google Scholar] [CrossRef]
- Grün, C.; Altmann, B.; Gottwald, E. Advanced 3d Cell Culture Techniques in Micro-Bioreactors, Part I: A Systematic Analysis of the Literature Published between 2000 and 2020. Processes 2020, 8, 1656. [Google Scholar] [CrossRef]
- Altmann, B.; Grün, C.; Nies, C.; Gottwald, E. Advanced 3D Cell Culture Techniques in Micro-Bioreactors, Part II: Systems and Applications. Processes 2020, 9, 21. [Google Scholar] [CrossRef]
- Castiglione, H.; Vigneron, P.A.; Baquerre, C.; Yates, F.; Rontard, J.; Honegger, T. Human Brain Organoids-on-Chip: Advances, Challenges, and Perspectives for Preclinical Applications. Pharmaceutics 2022, 14, 2301. [Google Scholar] [CrossRef]
- Hetzel, L.A.; Ali, A.; Corbo, V.; Hankemeier, T. Microfluidics and Organoids, the Power Couple of Developmental Biology and Oncology Studies. Int. J. Mol. Sci. 2023, 24, 10882. [Google Scholar] [CrossRef]
- Rontard, J.; Maisonneuve, B.G.; Honegger, T. Expanding Human-Based Predictive Models Capabilities Using Organs-on-Chip: A Standardized Framework to Transfer and Co-Culture Human IPSCs into Microfluidic Devices. Arch. Pharm. Pharm. Sci. 2023, 7, 17–21. [Google Scholar]
- Ko, J.; Park, D.; Lee, J.; Jung, S.; Baek, K.; Sung, K.E.; Lee, J.; Jeon, N.L. Microfluidic High-Throughput 3D Cell Culture. Nat. Rev. Bioeng. 2024, 1–17. [Google Scholar] [CrossRef]
- Fang, G.; Lu, H.; Al-Nakashli, R.; Chapman, R.; Zhang, Y.; Ju, L.A.; Lin, G.; Stenzel, M.H.; Jin, D. Enabling Peristalsis of Human Colon Tumor Organoids on Microfluidic Chips. Biofabrication 2022, 14, 015006. [Google Scholar] [CrossRef]
- Leung, C.M.; de Haan, P.; Ronaldson-Bouchard, K.; Kim, G.A.; Ko, J.; Rho, H.S.; Chen, Z.; Habibovic, P.; Jeon, N.L.; Takayama, S.; et al. A Guide to the Organ-on-a-Chip. Nat. Rev. Methods Primers 2022, 2, 33. [Google Scholar] [CrossRef]
- Kratochvil, M.J.; Seymour, A.J.; Li, T.L.; Paşca, S.P.; Kuo, C.J.; Heilshorn, S.C. Engineered Materials for Organoid Systems. Nat. Rev. Mater. 2019, 4, 606–622. [Google Scholar] [CrossRef]
- Hofer, M.; Lutolf, M.P. Engineering Organoids. Nat. Rev. Mater. 2021, 6, 402–420. [Google Scholar] [CrossRef]
- Nachlas, M.M.; Margulies, S.I.; Goldberg, J.D.; Seligman, A.M. The Determination of Lactic Dehydrogenase with a Tetrazolium Salt; Elsevier: Amsterdam, The Netherlands, 1960; Volume 1, pp. 317–326. [Google Scholar]
- Cook, J.A.; Mitchel, J.B. Viability Measurements in Mammalian Cell Systems. Anal. Biochem. 1989, 179, 1–7. [Google Scholar] [CrossRef]
- Kaja, S.; Payne, A.J.; Naumchuk, Y.; Koulen, P. Quantification of Lactate Dehydrogenase for Cell Viability Testing Using Cell Lines and Primary Cultured Astrocytes. Curr. Protoc. Toxicol. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A Single-Cell Type Transcriptomics Map of Human Tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Farhana, A.; Lappin, S.L. Biochemistry, Lactate Dehydrogenase; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Chan, F.K.M.; Moriwaki, K.; De Rosa, M.J. Detection of Necrosis by Release of Lactate Dehydrogenase Activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar]
- Korzeniewski, C.; Callewaert, D.M. An Enzyme-Release Assay for Natural Cytotoxicity 1. J. Oflmmunologicalmethods 1983, 64, 313–320. [Google Scholar] [CrossRef]
- Allen, M.; Millett, P.; Dawes, E.; Rushton, N. Lactate Dehydrogenase Activity as a Rapid and Sensitive Test for the Quantification of Cell Numbers in Vitro. Clin. Mater. 1994, 16, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Gelein, R.; Corson, N.; Wade-Mercer, P.; Jiang, J.; Biswas, P.; Finkelstein, J.N.; Elder, A.; Oberdörster, G. Validation of an LDH Assay for Assessing Nanoparticle Toxicity. Toxicology 2011, 287, 99–104. [Google Scholar] [CrossRef]
- Decker, T.; Lohmann-Matthes, M.-L. A Quick and Simple Method for the Quantitation of Lactate Dehydrogenase Release in Measurements of Cellular Cytotoxicity and Tumor Necrosis Factor (TNF) Activity. J. Immunol. Methods 1988, 15, 61–69. [Google Scholar] [CrossRef]
- Burd, J.F.; Usategui-Gomez, M. A Colorimetric Assay for Serum Lactate Dehydrogenase. Clin. Chim. Acta 1973, 46, 223–227. [Google Scholar] [CrossRef]
- Babson, A.L.; Phillips, G.E. A Rapid Colorimetric Assay for Serum Lactate Dehydrogenase. Clin. Chim. Acta 1965, 12, 210–215. [Google Scholar] [CrossRef]
- Cox, M.C.; Mendes, R.; Silva, F.; Mendes, T.F.; Zelaya-Lazo, A.; Halwachs, K.; Purkal, J.J.; Isidro, I.A.; Félix, A.; Boghaert, E.R.; et al. Application of LDH Assay for Therapeutic Efficacy Evaluation of Ex Vivo Tumor Models. Sci. Rep. 2021, 11, 18571. [Google Scholar] [CrossRef]
- Zang, Z.; Yin, H.; Du, Z.; Xie, R.; Yang, L.; Cai, Y.; Wang, L.; Zhang, D.; Li, X.; Liu, T.; et al. Valproic Acid Exposure Decreases Neurogenic Potential of Outer Radial Glia in Human Brain Organoids. Front. Mol. Neurosci. 2022, 15, 1023765. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Wang, Y.; Zhu, Y.; Tao, T.; Yin, F.; Guo, Y.; Liu, H.; Li, F.; Wang, P.; Chen, Y.; et al. Neurodevelopmental Impairment Induced by Prenatal Valproic Acid Exposure Shown with the Human Cortical Organoid-on-a-Chip Model. Microsyst. Nanoeng. 2020, 6, 49. [Google Scholar] [CrossRef]
- Meng, Q.; Zhang, W.; Wang, X.; Jiao, C.; Xu, S.; Liu, C.; Tang, B.; Chen, C. Human Forebrain Organoids Reveal Connections between Valproic Acid Exposure and Autism Risk. Transl. Psychiatry 2022, 12, 130. [Google Scholar] [CrossRef] [PubMed]
- Nassor, F.; Jarray, R.; Biard, D.S.F.; Maïza, A.; Papy-Garcia, D.; Pavoni, S.; Deslys, J.P.; Yates, F. Long Term Gene Expression in Human Induced Pluripotent Stem Cells and Cerebral Organoids to Model a Neurodegenerative Disease. Front. Cell Neurosci. 2020, 14, 1–7. [Google Scholar] [CrossRef]
- Pavoni, S.; Jarray, R.; Nassor, F.; Guyot, A.C.; Cottin, S.; Rontard, J.; Mikol, J.; Mabondzo, A.; Deslys, J.P.; Yates, F. Small-Molecule Induction of Aβ-42 Peptide Production in Human Cerebral Organoids to Model Alzheimer’s Disease Associated Phenotypes. PLoS ONE 2018, 13, 1–15. [Google Scholar] [CrossRef]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Kang, Y.; Roselaar, N.; Cakir, B.; Kim, K.; Lombroso, A.P.; Hwang, M.; Zhong, M.; et al. Fusion of Regionally-Specified HPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stel Cell 2017, 21, 383–398. [Google Scholar] [CrossRef]
- Xiang, Y.; Tanaka, Y.; Cakir, B.; Patterson, B.; Kim, K.; Sun, P.; Kang, Y.; Zhong, M.; Liu, X.; Patra, P.; et al. HESC-Derived Thalamic Organoids Form Reciprocal Projections When Fused with Cortical Organoids. Cell Stem Cell 2019, 24, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Temple, J.; Velliou, E.; Shehata, M.; Lévy, R. Current Strategies with Implementation of Three-Dimensional Cell Culture: The Challenge of Quantification. Interface Focus. 2022, 12, 20220019. [Google Scholar] [CrossRef]
- Silva, C.; Martins, M.; Jing, S.; Fu, J.; Cavaco-Paulo, A. Practical Insights on Enzyme Stabilization. Crit. Rev. Biotechnol. 2018, 38, 335–350. [Google Scholar] [CrossRef]
- Chang, B.; Mahoney, R. Enzyme Thermostabilization by Bovine Serum Albumin and Other Proteins: Evidence for Hydrophobic Interactions. Biotechnol. Appl. Biochem. 1995, 22, 203–214. [Google Scholar] [PubMed]
- Finn, T.E.; Nunez, A.C.; Sunde, M.; Easterbrook-Smith, S.B. Serum Albumin Prevents Protein Aggregation and Amyloid Formation and Retains Chaperone-like Activity in the Presence of Physiological Ligands. J. Biol. Chem. 2012, 287, 21530–21540. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Voronina, P.A.; Shmurak, V.I.; Vovk, M.A.; Batalova, A.A.; Jenkins, R.O.; Goncharov, N.V. The Universal Soldier: Enzymatic and Non-enzymatic Antioxidant Functions of Serum Albumin. Antioxidants 2020, 9, 966. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Zhu, Z.; Wang, J.; Huang, W.; Zhang, C.; Zeng, J.; Zhao, H.; Qi, T.; Zhou, W.; Zhang, T.; et al. Preparation, Stability and Commutability of Candidate Reference Materials for Lactate Dehydrogenase (LDH). Clin. Biochem. 2021, 91, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Duskey, J.T.; da Ros, F.; Ottonelli, I.; Zambelli, B.; Vandelli, M.A.; Tosi, G.; Ruozi, B. Enzyme Stability in Nanoparticle Preparations Part 1: Bovine Serum Albumin Improves Enzyme Function. Molecules 2020, 25, 4593. [Google Scholar] [CrossRef] [PubMed]
- Fertan, E.; Böken, D.; Murray, A.; Danial, J.S.H.; Lam, J.Y.L.; Wu, Y.; Goh, P.A.; Alić, I.; Cheetham, M.R.; Lobanova, E.; et al. Cerebral Organoids with Chromosome 21 Trisomy Secrete Alzheimer’s Disease-Related Soluble Aggregates Detectable by Single-Molecule-Fluorescence and Super-Resolution Microscopy. Mol. Psychiatry 2024, 29, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Kari, S.; Subramanian, K.; Altomonte, I.A.; Murugesan, A.; Yli-Harja, O.; Kandhavelu, M. Programmed Cell Death Detection Methods: A Systematic Review and a Categorical Comparison. Apoptosis 2022, 27, 482–508. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, M.J. Cell Viability Assays: Introduction. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2011; Volume 740, pp. 1–6. [Google Scholar]
- Bressenot, A.; Marchal, S.; Bezdetnaya, L.; Garrier, J.; Guillemin, F.; Plénat, F. Assessment of Apoptosis by Immunohistochemistry to Active Caspase-3, Active Caspase-7, or Cleaved PARP in Monolayer Cells and Spheroid and Subcutaneous Xenografts of Human Carcinoma. J. Histochem. Cytochem. 2009, 57, 289–300. [Google Scholar] [CrossRef]
- Loo, D.T. In Situ Detection of Apoptosis by the TUNEL Assay: An Overview of Techniques. Methods Mol. Biol. 2011, 682, 3–13. [Google Scholar]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010, 2010, 1–9. [Google Scholar] [CrossRef]
- Basu, A.; Haldar, S. The Relationship between Bcl2, Bax and P53: Consequences for Cell Cycle Progression and Cell Death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).