
The Rapid Generation of Cell-Laden, FACS-Compatible Collagen Gels

 ,
,  ,
,
Abstract
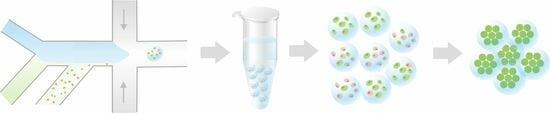
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
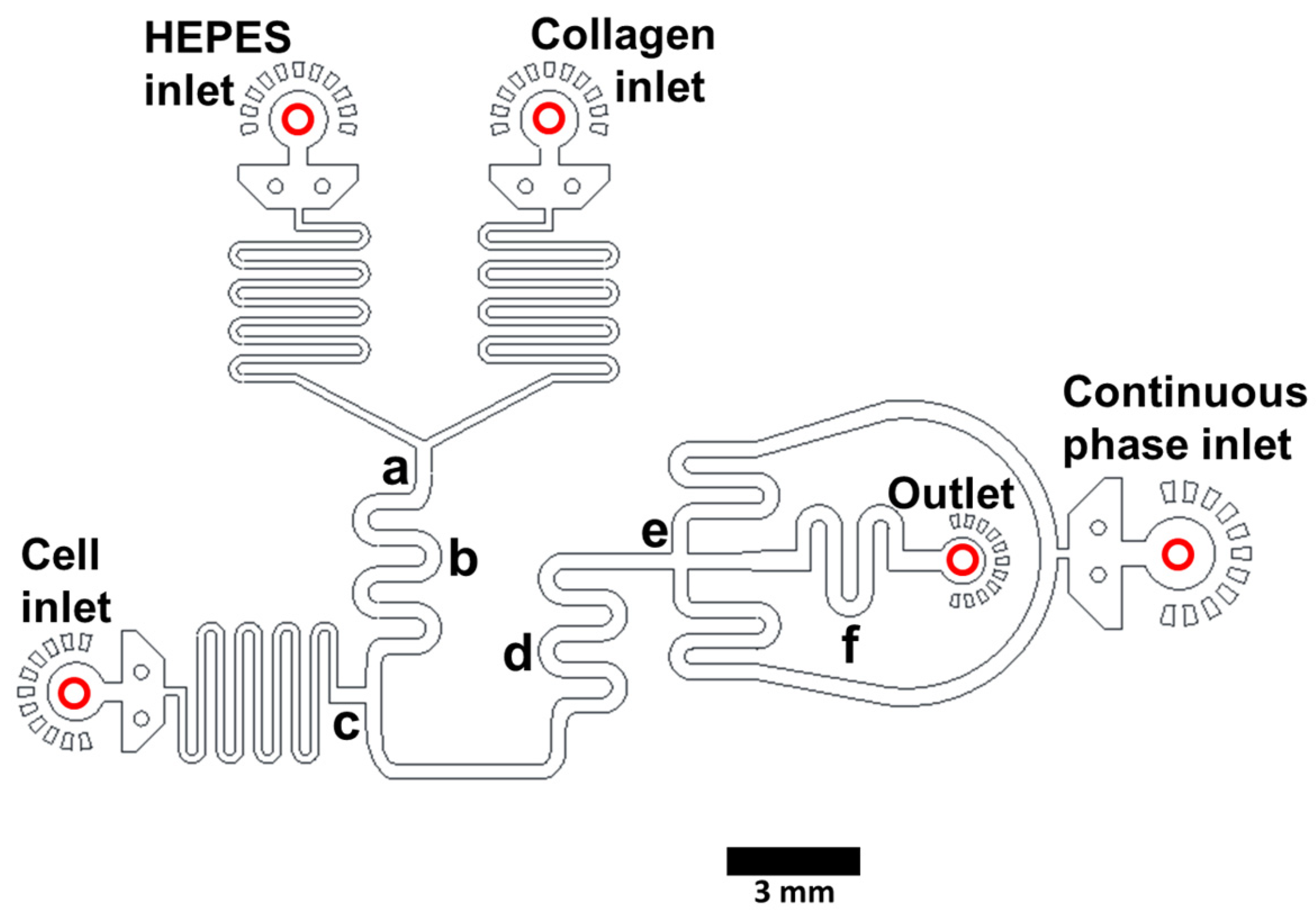
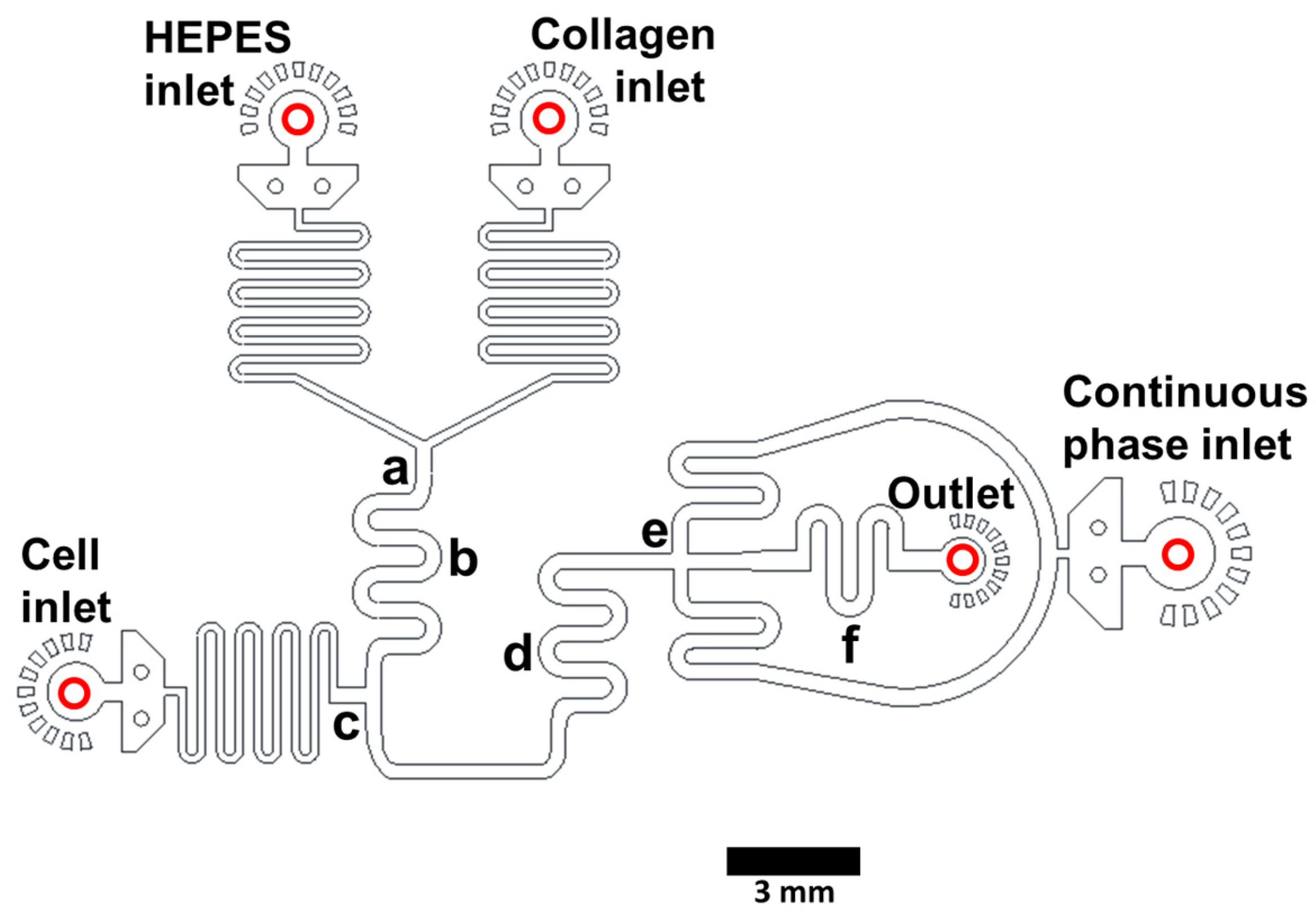
2.1. Microfluidic Device Design and Fabrication
2.2. Culture of Cell Lines
2.3. Generation of Collagen Gels and Transfer to Growth Medium
2.4. Characterization of Collagen Gels
2.5. Measurement of Cell Viability
2.6. Gel Sorting
3. Results and Discussion
3.1. Microfluidic Device Design and Fabrication and Collagen Hydrogel Generation
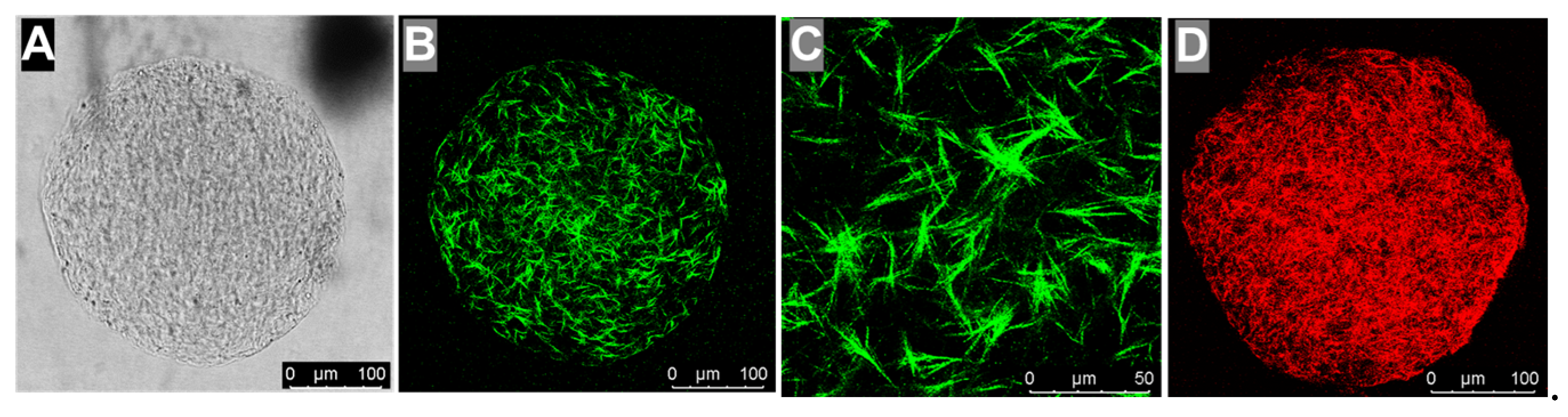
3.2. Collagen Hydrogel Characterization
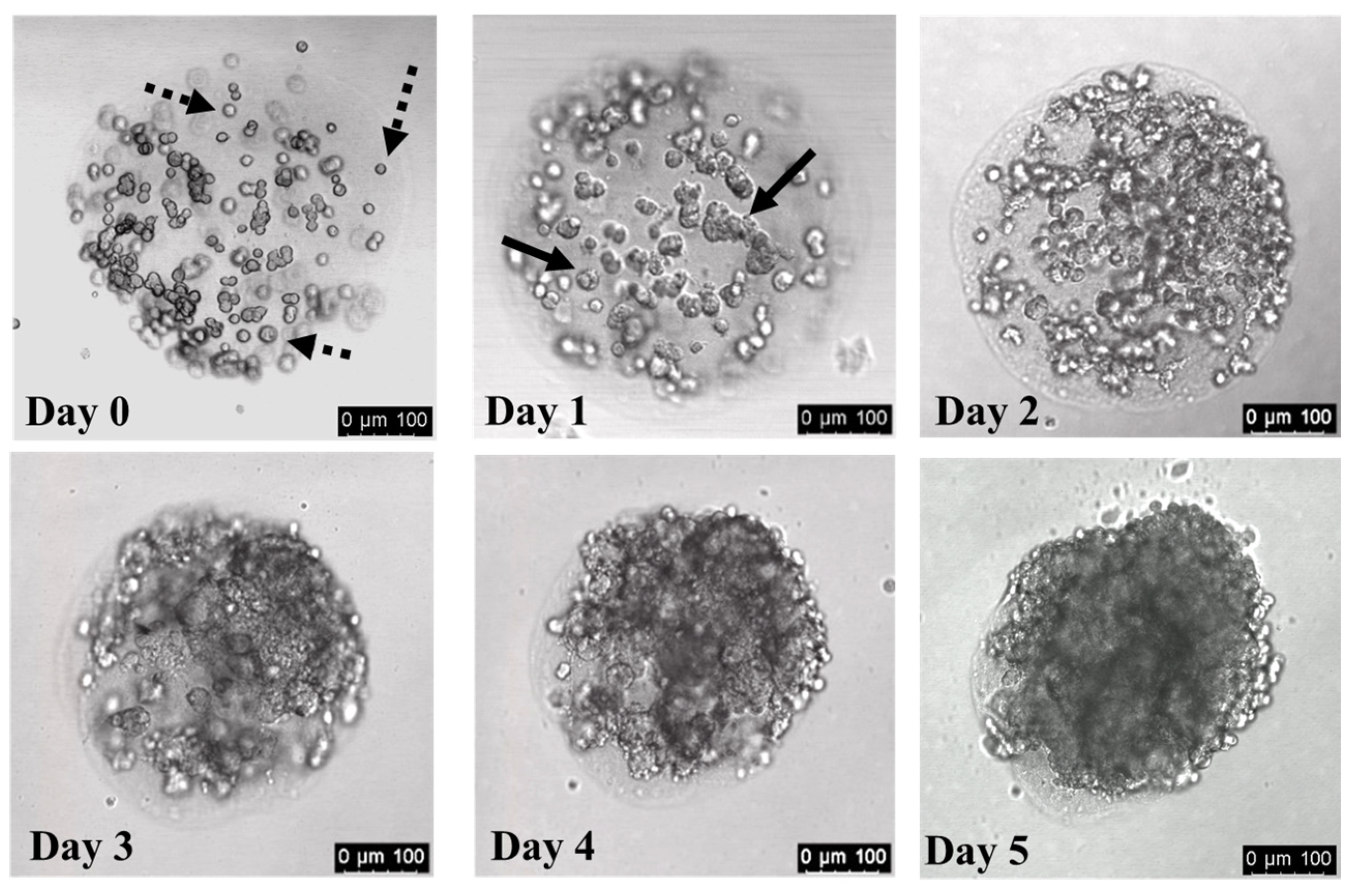
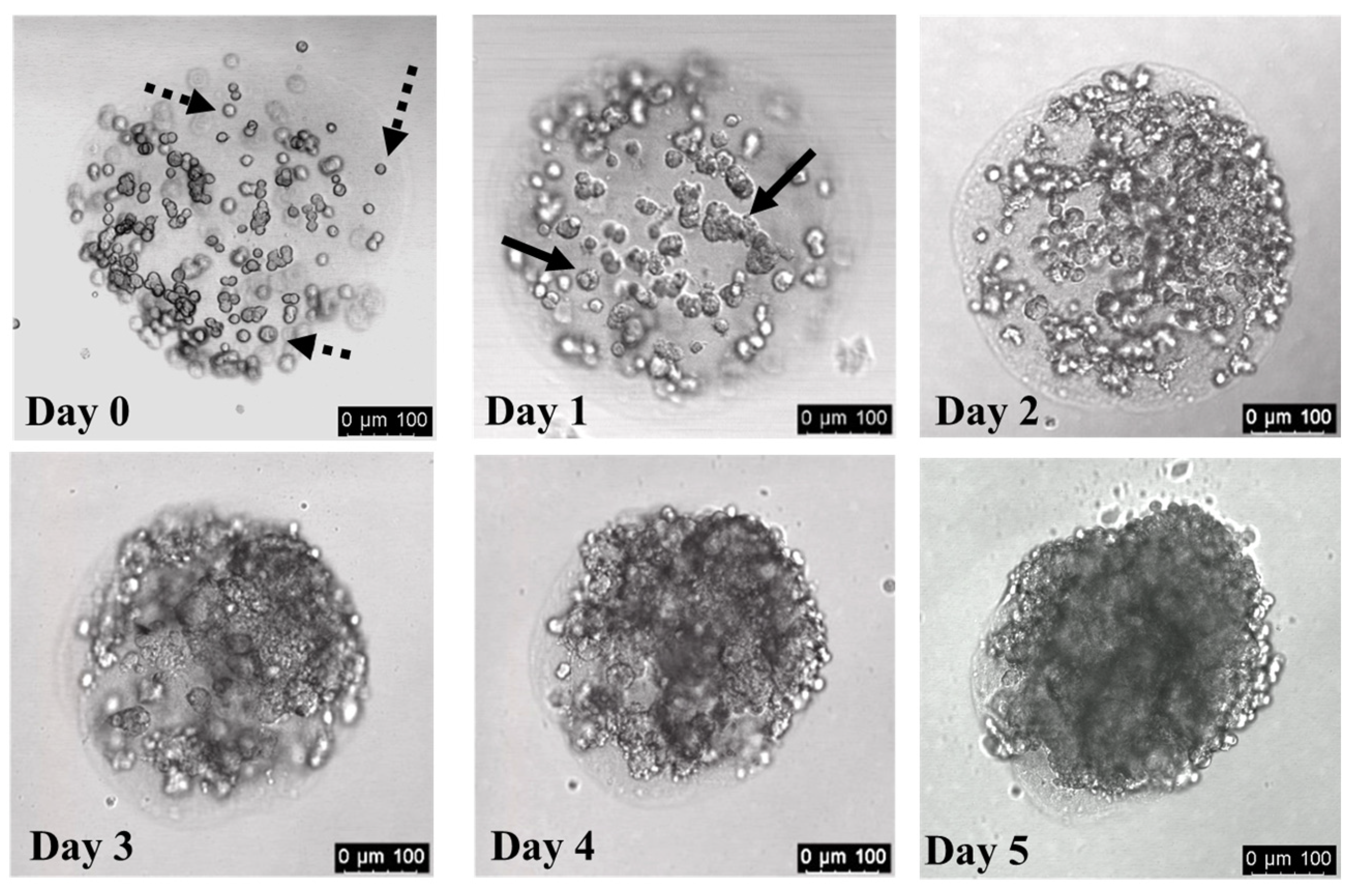
3.3. Cells Divide within Collagen Hydrogels
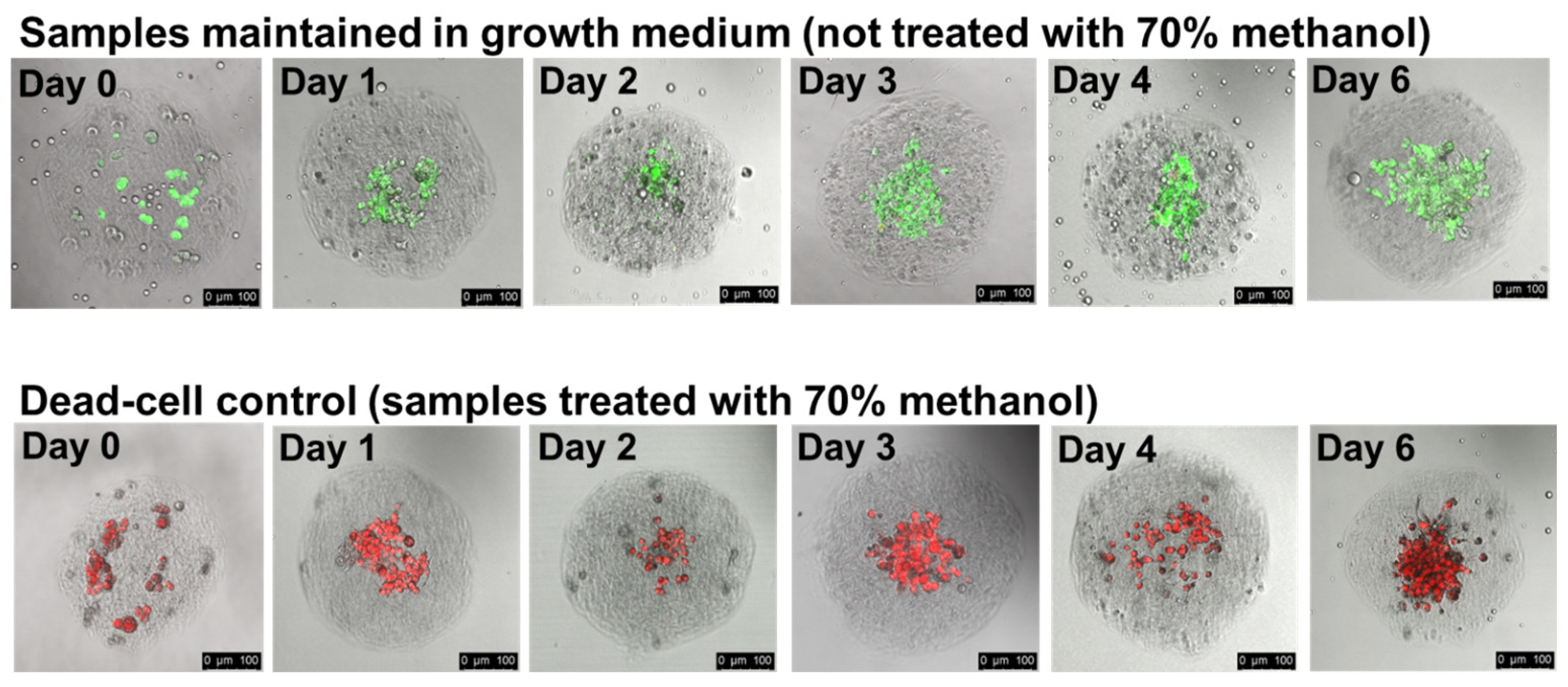
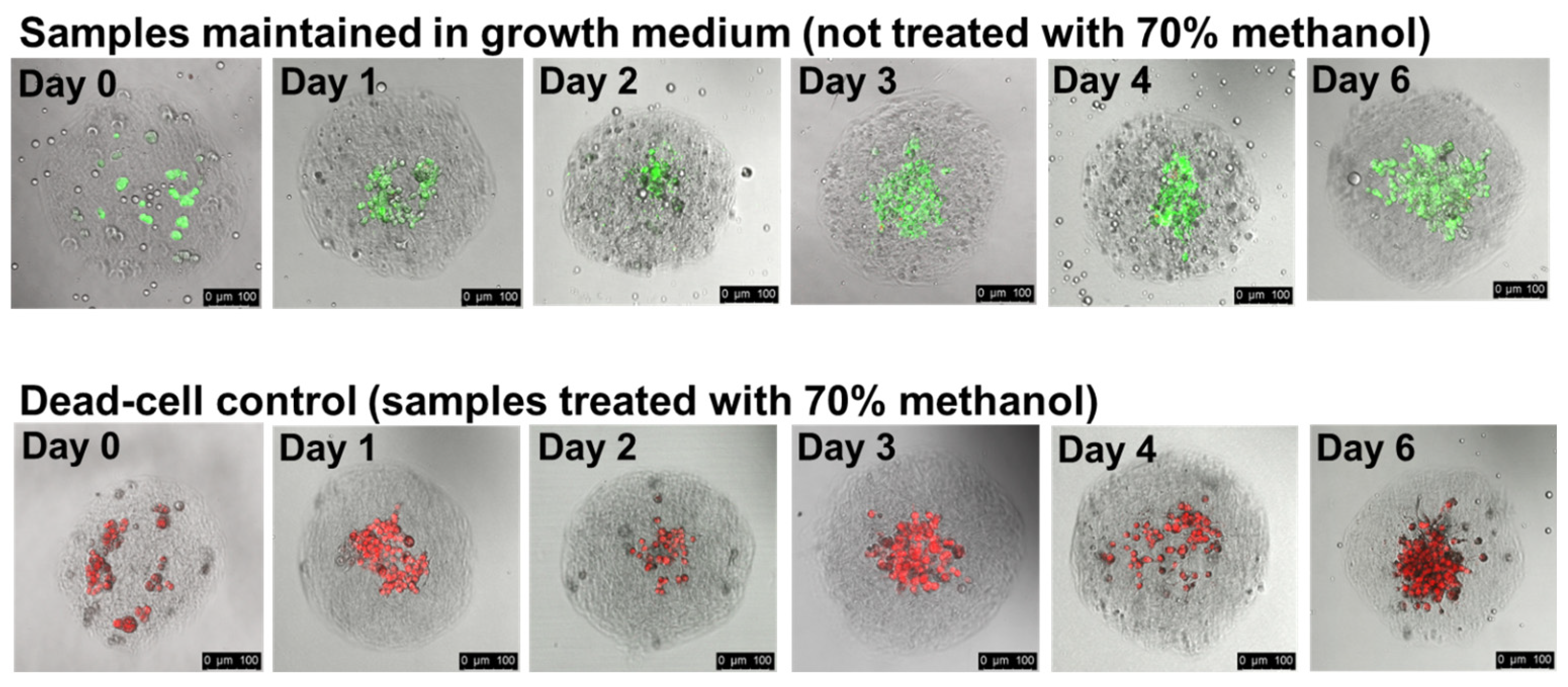
3.4. Cells Remain Viable within Collagen Hydrogels for Several Days
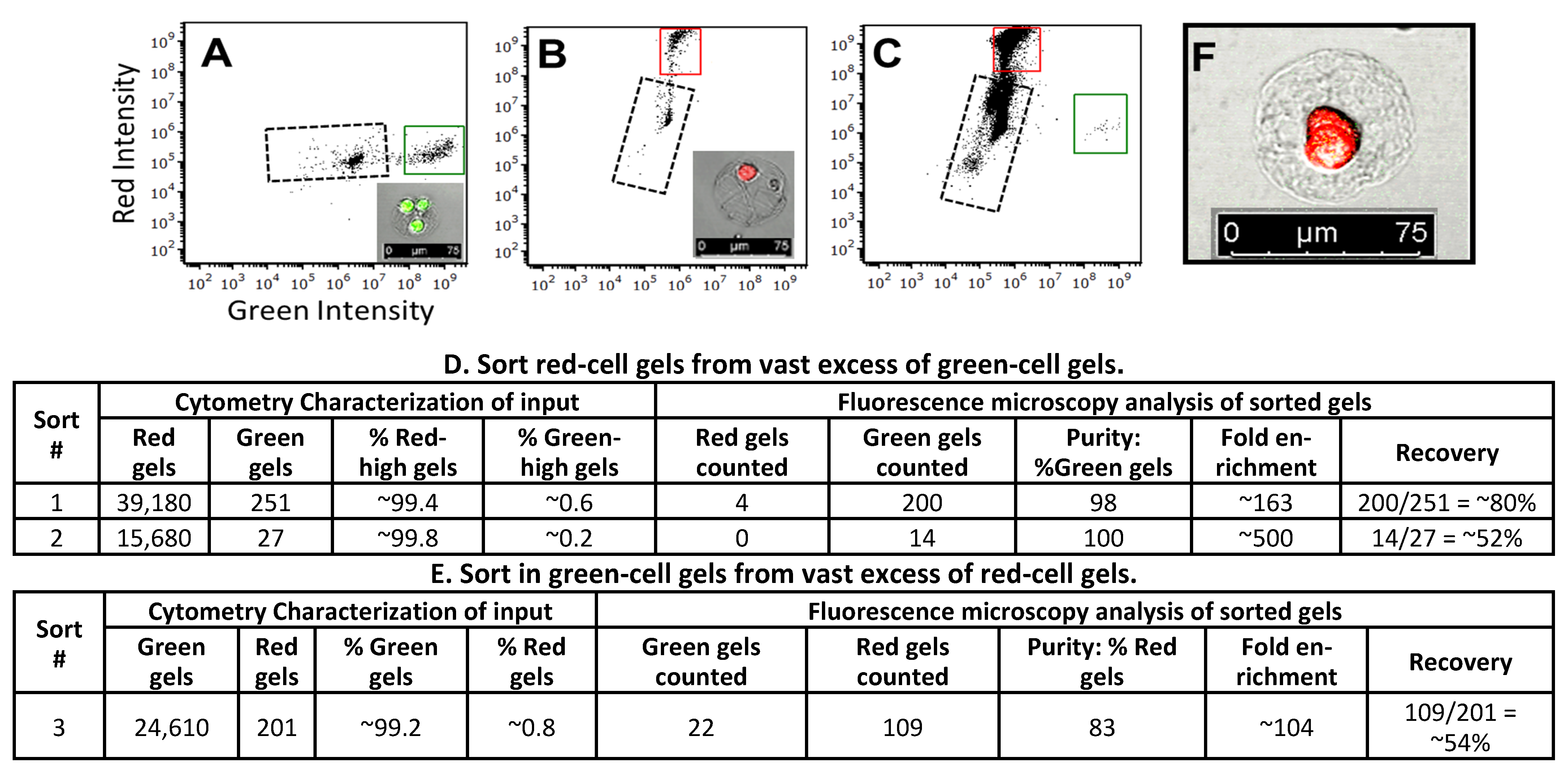
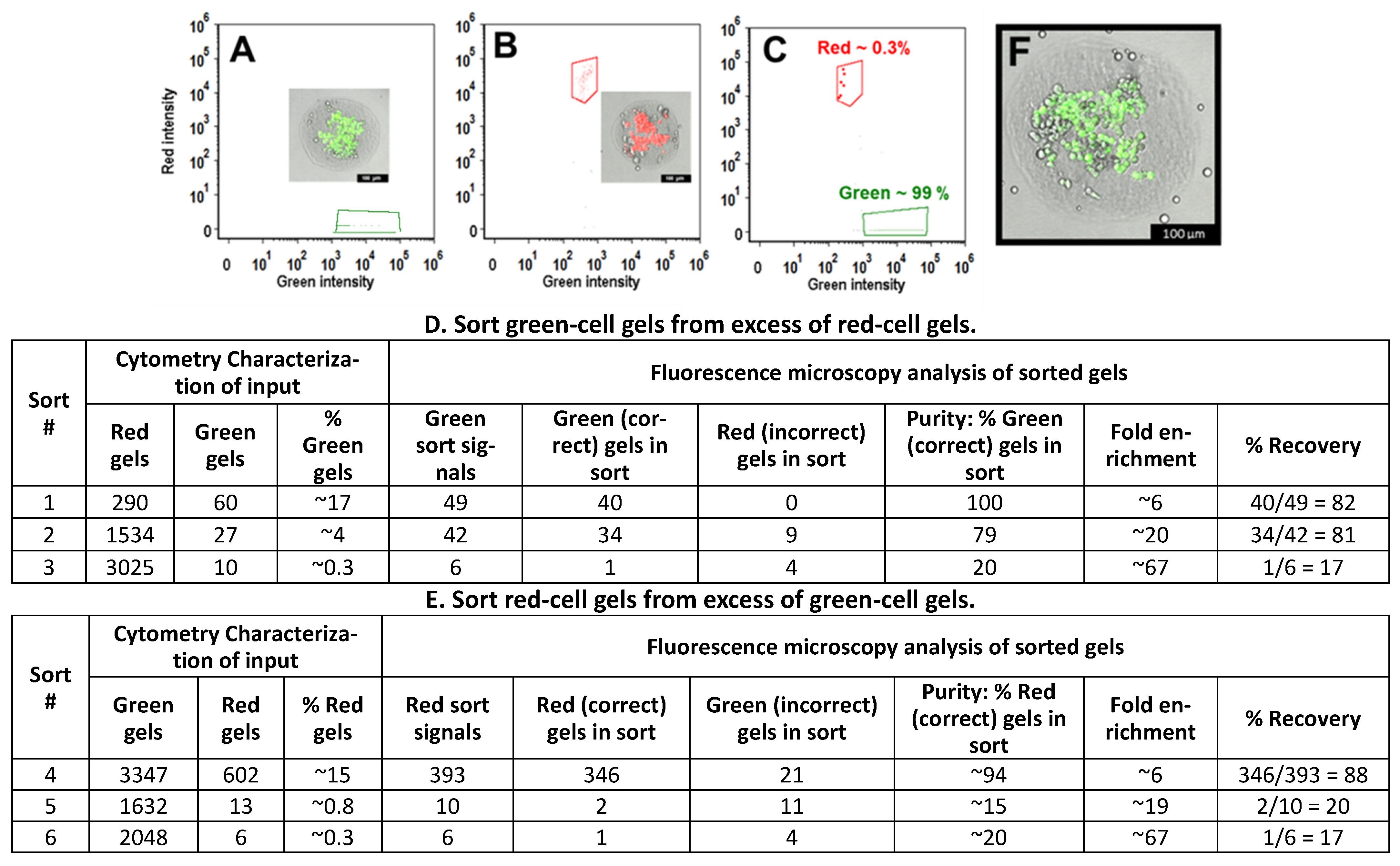
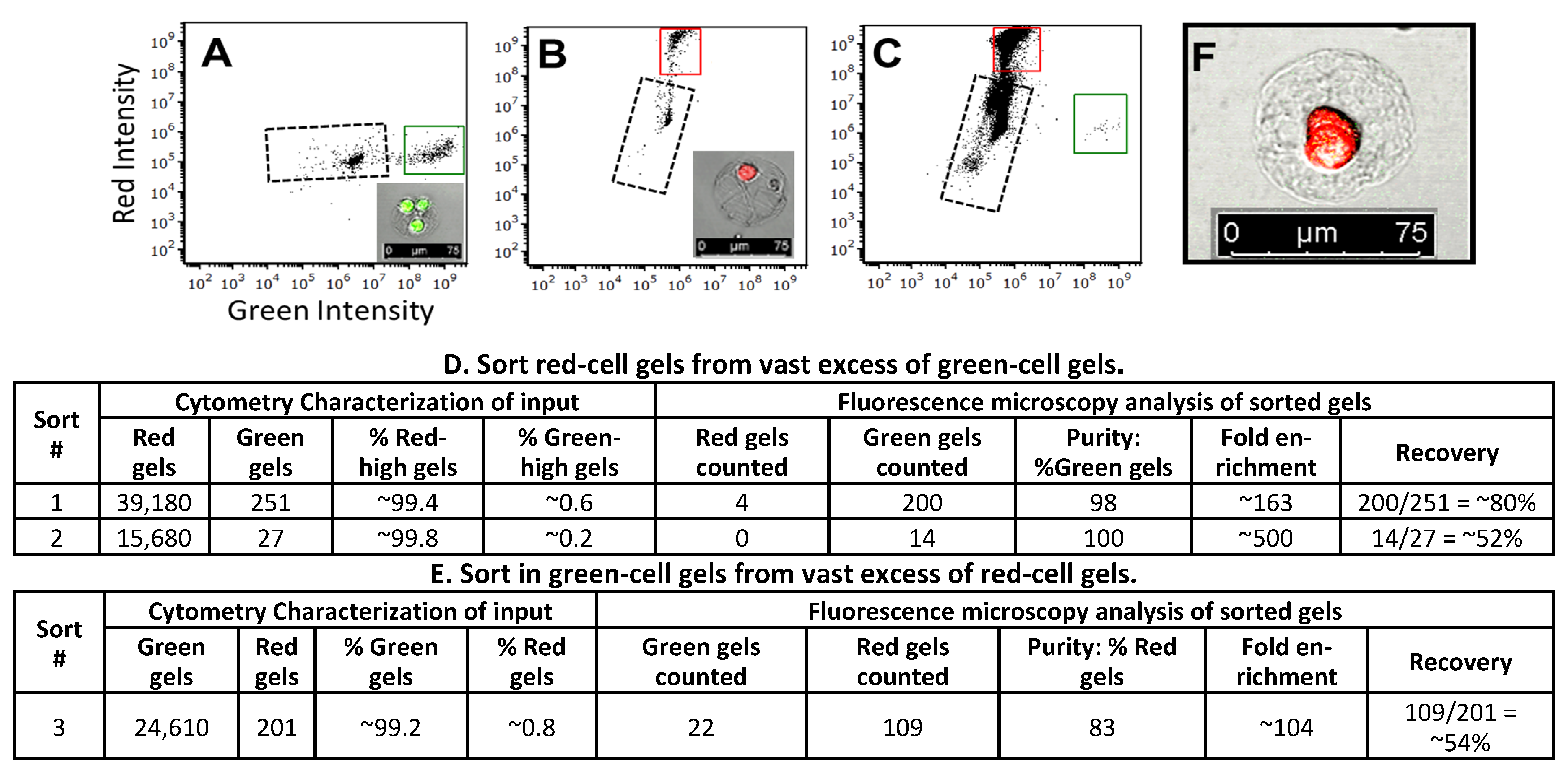
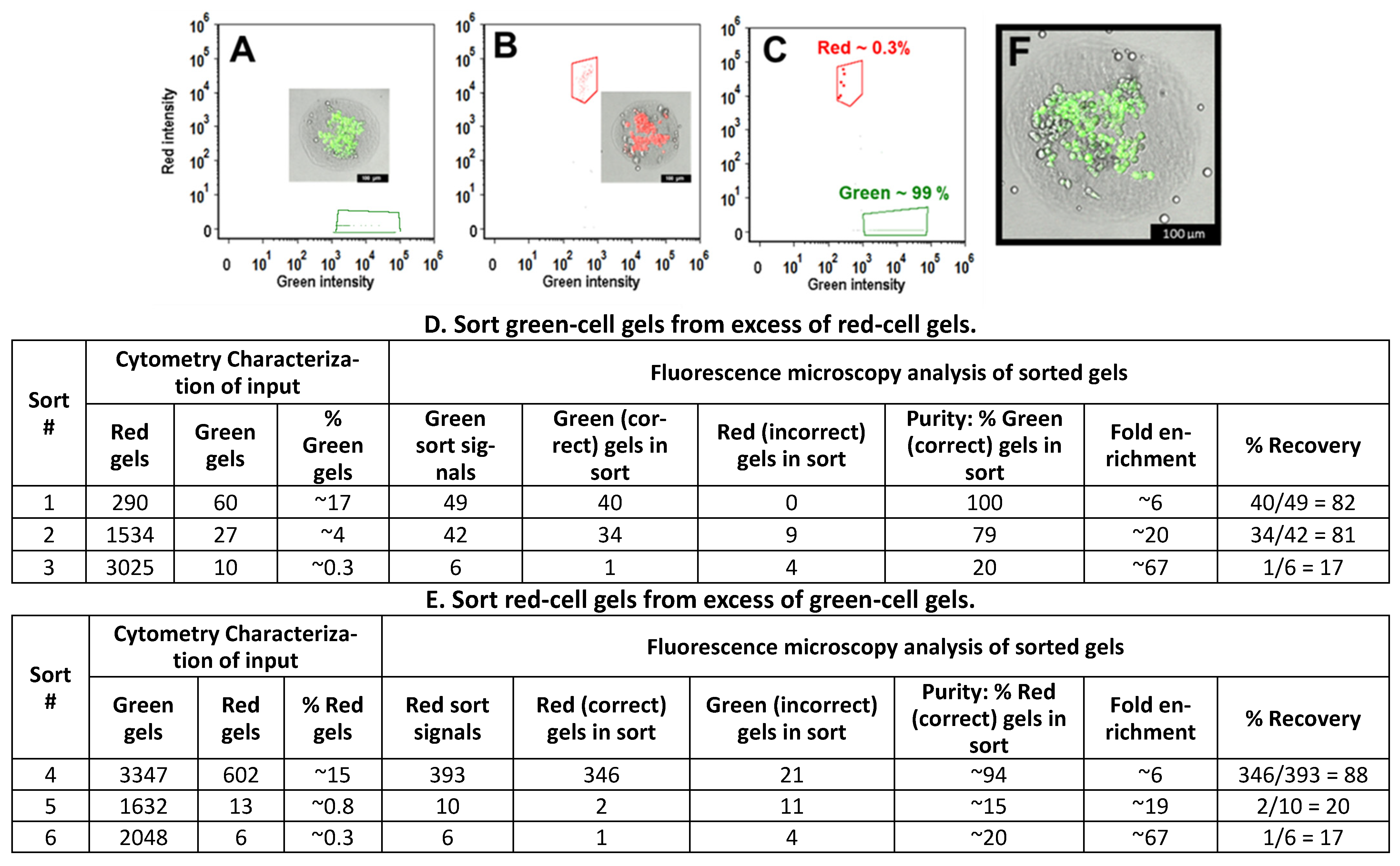
3.5. Sorting of Collagen Hydrogels
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bajaj, P.; Schweller, R.M.; Khademhosseini, A.; West, J.L.; Bashir, R. 3D biofabrication strategies for tissue engineering and regenerative medicine. Annu. Rev. Biomed. Eng. 2014, 16, 247–276. [Google Scholar] [CrossRef] [PubMed]
- LeSavage, B.L.; Suhar, R.A.; Broguiere, N.; Lutolf, M.P.; Heilshorn, S.C. Next-generation cancer organoids. Nat. Mater. 2022, 21, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Barrs, R.W.; Jia, J.; Ward, M.; Richards, D.J.; Yao, H.; Yost, M.J.; Mei, Y. Engineering a Chemically Defined Hydrogel Bioink for Direct Bioprinting of Microvasculature. Biomacromolecules 2021, 22, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Dawson, C.; Lamb, M.; Mueller, E.; Stefanek, E.; Akbari, M.; Hoare, T. Hydrogels for Tissue Engineering: Addressing Key Design Needs Toward Clinical Translation. Front. Bioeng. Biotechnol. 2022, 10, 849831. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, Y.T.; Morimoto, Y.; Takeuchi, S. Molding Cell Beads for Rapid Construction of Macroscopic 3D Tissue Architecture. Adv. Mater. 2011, 23, H90–H94. [Google Scholar] [CrossRef]
- Xia, B.; Krutkramelis, K.; Oakey, J. Oxygen-Purged Microfluidic Device to Enhance Cell Viability in Photopolymerized PEG Hydrogel Microparticles. Biomacromolecules 2016, 17, 2459–2465. [Google Scholar] [CrossRef]
- Jiang, Z.; Xia, B.; McBride, R.; Oakey, J. A microfluidic-based cell encapsulation platform to achieve high long-term cell viability in photopolymerized PEGNB hydrogel microspheres. J. Mater. Chem. B 2017, 5, 173–180. [Google Scholar] [CrossRef]
- Rossow, T.; Heyman, J.A.; Ehrlicher, A.J.; Langhoff, A.; Weitz, D.A.; Haag, R.; Seiffert, S. Controlled Synthesis of Cell-Laden Microgels by Radical-Free Gelation in Droplet Microfluidics. J. Am. Chem. Soc. 2012, 134, 4983–4989. [Google Scholar] [CrossRef]
- Steinhilber, D.; Rossow, T.; Wedepohl, S.; Paulus, F.; Seiffert, S.; Haag, R. A Microgel Construction Kit for Bioorthogonal Encapsulation and pH-Controlled Release of Living Cells. Angew. Chem. Int. Ed. 2013, 52, 13538–13543. [Google Scholar] [CrossRef]
- Rossow, T.; Bayer, S.; Albrecht, R.; Tzschucke, C.C.; Seiffert, S. Supramolecular Hydrogel Capsules Based on PEG: A Step Toward Degradable Biomaterials with Rational Design. Macromol. Rapid Commun. 2013, 34, 1401–1407. [Google Scholar] [CrossRef]
- Mao, A.S.; Shin, J.-W.; Utech, S.; Wang, H.; Uzun, O.; Li, W.; Cooper, M.; Hu, Y.; Zhang, L.; Weitz, D.A.; et al. Deterministic encapsulation of single cells in thin tunable microgels for niche modelling and therapeutic delivery. Nat. Mater. 2016, 16, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.M.; Koshy, S.T.; Hilderbrand, S.A.; Mooney, D.J.; Joshi, N.S. Versatile click alginate hydrogels crosslinked via tetrazine–norbornene chemistry. Biomaterials 2015, 50, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Yanakieva, D.; Elter, A.; Bratsch, J.; Friedrich, K.; Becker, S.; Kolmar, H. FACS-Based Functional Protein Screening via Microfluidic Co-encapsulation of Yeast Secretor and Mammalian Reporter Cells. Sci. Rep. 2020, 10, 10182. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, G.; Musicò, A.; Frigerio, R.; Strada, A.; Pizzi, A.; Talone, B.; Ghezzi, J.; Gautieri, A.; Chiari, M.; Metrangolo, P.; et al. Composite Peptide–Agarose Hydrogels for Robust and High-Sensitivity 3D Immunoassays. ACS Appl. Mater. Interfaces 2022, 14, 4811–4822. [Google Scholar] [CrossRef]
- Rosiak, P.; Latanska, I.; Paul, P.; Sujka, W.; Kolesinska, B. Modification of Alginates to Modulate Their Physic-Chemical Properties and Obtain Biomaterials with Different Functional Properties. Molecules 2021, 26, 7264. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.W.; Bush, J.A.; Plopper, G.E.; Stegemann, J.P. Osteogenic differentiation of mesenchymal stem cells in defined protein beads. J. Biomed. Mater. Res. B Appl. Biomater. 2008, 87, 213–221. [Google Scholar] [CrossRef]
- Mulas, C.; Hodgson, A.C.; Kohler, T.N.; Agley, C.C.; Humphreys, P.; Kleine-Brüggeney, H.; Hollfelder, F.; Smith, A.; Chalut, K.J. Microfluidic platform for 3D cell culture with live imaging and clone retrieval. Lab Chip 2020, 20, 2580–2591. [Google Scholar] [CrossRef]
- Loessner, D.; Meinert, C.; Kaemmerer, E.; Martine, L.C.; Yue, K.; Levett, P.A.; Klein, T.J.; Melchels, F.P.W.; Khademhosseini, A.; Hutmacher, D.W. Functionalization, preparation and use of cell-laden gelatin methacryloyl–based hydrogels as modular tissue culture platforms. Nat. Protoc. 2016, 11, 727–746. [Google Scholar] [CrossRef]
- Zhang, Y.; Ouyang, H.; Lim, C.T.; Ramakrishna, S.; Huang, Z.-M. Electrospinning of gelatin fibers and gelatin/PCL composite fibrous scaffolds. J. Biomed. Mater. Res. 2005, 72B, 156–165. [Google Scholar] [CrossRef]
- Ullah, F.; Othman, M.B.H.; Javed, F.; Ahmad, Z.; Akil, H.M. Classification, processing and application of hydrogels: A review. Mater. Sci. Eng. C 2015, 57, 414–433. [Google Scholar] [CrossRef]
- Li, Z.; Leung, M.; Hopper, R.; Ellenbogen, R.; Zhang, M. Feeder-free self-renewal of human embryonic stem cells in 3D porous natural polymer scaffolds. Biomaterials 2010, 31, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, B.; Engelbrecht, L.K.; Fernandez, P.; Hutterer, F.P.; Raich, M.K.; Scheel, C.H.; Bausch, A.R. Mechanical plasticity of collagen directs branch elongation in human mammary gland organoids. Nat. Commun. 2021, 12, 2759. [Google Scholar] [CrossRef] [PubMed]
- Rajan, N.; Habermehl, J.; Coté, M.-F.; Doillon, C.J.; Mantovani, D. Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nat. Protoc. 2006, 1, 2753–2758. [Google Scholar] [CrossRef] [PubMed]
- Mazutis, L.; Gilbert, J.; Ung, W.L.; Weitz, D.A.; Griffiths, A.D.; Heyman, J.A. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 2013, 8, 870–891. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Hung, K.-C.; Mitra, A.; Ung, L.W.; Lightwood, D.; Tu, R.; Starkie, D.; Cai, L.; Mazutis, L.; Chong, S.; et al. Rapid isolation of antigen-specific B-cells using droplet microfluidics. RSC Adv. 2020, 10, 27006–27013. [Google Scholar] [CrossRef]
- Yadavalli, V.K.; Svintradze, D.V.; Pidaparti, R.M. Nanoscale measurements of the assembly of collagen to fibrils. Int. J. Biol. Macromol. 2010, 46, 458–464. [Google Scholar] [CrossRef]
- Jiang, F.; Hörber, H.; Howard, J.; Müller, D.J. Assembly of collagen into microribbons: Effects of pH and electrolytes. J. Struct. Biol. 2004, 148, 268–278. [Google Scholar] [CrossRef]
- Yan, M.; Li, B.; Zhao, X.; Qin, S. Effect of concentration, pH and ionic strength on the kinetic self-assembly of acid-soluble collagen from walleye pollock (Theragra chalcogramma) skin. Food Hydrocoll. 2012, 29, 199–204. [Google Scholar] [CrossRef]
- Cheung, A.S.; Zhang, D.K.; Koshy, S.T.; Mooney, D.J. Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat. Biotechnol. 2018, 36, 160–169. [Google Scholar] [CrossRef]
- Brightman, A.; Rajwa, B.; Sturgis, J.; McCallister, M.; Robinson, J.; Voytik-Harbin, S. Time-lapse confocal reflection microscopy of collagen fibrillogenesis and extracellular matrix assembly in vitro. Biopolym. Orig. Res. Biomol. 2000, 54, 222–234. [Google Scholar] [CrossRef]
- Camacho, P.; Fainor, M.; Seims, K.B.; Tolbert, J.W.; Chow, L.W. Fabricating spatially functionalized 3D-printed scaffolds for osteochondral tissue engineering. J. Biol. Methods 2021, 8, e146. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tristan, C.A.; Chen, L.; Jovanovic, V.M.; Malley, C.; Chu, P.-H.; Ryu, S.; Deng, T.; Ormanoglu, P.; Tao, D.; et al. A versatile polypharmacology platform promotes cytoprotection and viability of human pluripotent and differentiated cells. Nat. Methods 2021, 18, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Grolman, J.M.; Zhang, D.; Smith, A.M.; Moore, J.S.; Kilian, K.A. Rapid 3D Extrusion of Synthetic Tumor Microenvironments. Adv. Mater. 2015, 27, 5512–5517. [Google Scholar] [CrossRef]
- Tian, Y.F.; Ahn, H.; Schneider, R.S.; Yang, S.N.; Roman-Gonzalez, L.; Melnick, A.M.; Cerchietti, L.; Singh, A. Integrin-specific hydrogels as adaptable tumor organoids for malignant B and T cells. Biomaterials 2015, 73, 110–119. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Y.; Huang, Q.; Collins, J.W.; Brouchon, J.; Nelson, J.A.; Niziolek, Z.; O’Neil, A.; Ye, F.; Weitz, D.A.; Heyman, J.A. The Rapid Generation of Cell-Laden, FACS-Compatible Collagen Gels. Organoids 2023, 2, 204-217. https://doi.org/10.3390/organoids2040016
Xiao Y, Huang Q, Collins JW, Brouchon J, Nelson JA, Niziolek Z, O’Neil A, Ye F, Weitz DA, Heyman JA. The Rapid Generation of Cell-Laden, FACS-Compatible Collagen Gels. Organoids. 2023; 2(4):204-217. https://doi.org/10.3390/organoids2040016
Chicago/Turabian StyleXiao, Yi, Qiaoling Huang, Jesse W. Collins, Julie Brouchon, Jeffery A. Nelson, Zachary Niziolek, Alison O’Neil, Fangfu Ye, David A. Weitz, and John A. Heyman. 2023. "The Rapid Generation of Cell-Laden, FACS-Compatible Collagen Gels" Organoids 2, no. 4: 204-217. https://doi.org/10.3390/organoids2040016
APA StyleXiao, Y., Huang, Q., Collins, J. W., Brouchon, J., Nelson, J. A., Niziolek, Z., O’Neil, A., Ye, F., Weitz, D. A., & Heyman, J. A. (2023). The Rapid Generation of Cell-Laden, FACS-Compatible Collagen Gels. Organoids, 2(4), 204-217. https://doi.org/10.3390/organoids2040016