Abstract
Organoid-based research has made significant discoveries and contributions to our understanding of human organ function in both health and disease. To continue making progress, it is crucial to acknowledge the crucial role of the immune system in all organs. Various immune cells, such as macrophages, T cells, and neutrophils, are resident in almost all human tissues and play essential roles in organ homeostasis, function, and disease. Using diverse methods, researchers have begun integrating immune cells into organoid models, leading to more physiologically relevant models that better represent various aspects of human disease. These methods range from immune cell injection to co-culture and tissue expansion with existing immune cells. Immune cells can be sourced from mature patients or generated from stem cells as immature immune cells. The successful incorporation of immune cells into organoids will enhance our understanding of organ function and provide a more accurate approximation of human disease.
1. Introduction
The immense number of failed clinical trials every year highlights the need to optimize models used in foundational and translational sciences for studying human disease [1]. Current in vitro approaches with human cells are either based on cell suspensions of individual cell types or homogenous monolayers, neither of which are best suited to recapitulate the complex structure and cell-cell interactions found in whole tissues. The alternative, in vivo work in animal models, provides a more comprehensive environment with physiological elements like blood flow, metabolism, and systemic signals (e.g., nerve impulses or systemic cytokines), as well as a complex mix of cell types and tissue-specific phenotypes. However, species differences still exist, even in mice genetically modified to express humanized receptors [2]. The use of organoids overcomes the challenges of conventional in vitro cell work using human cells or in vivo work with animal models. Organoids allow for complex cell structures, including a variety of cell phenotypes derived from human cells, and have quickly gained traction as an extremely useful tool to model human disease [3].
Two main sources for human organoids are tissue samples from an organ of interest or induced pluripotent stem cells (iPSC) [3]. Both of these sources can be used to model human disease, but they also have limitations. Biopsies provide various cell types, and if the sample is obtained from patients, then there is the additional advantage of those cells potentially exhibiting the desired human disease phenotype. However, the generation of organoids from organ biopsies is limited by the availability of the sampling of these organs, which can be invasive and risky for patients. In contrast, iPSC cells can be generated from any patient cell and differentiated into specific cell types or organoids, making them an applicable source with genetic diversity [4]. Nonetheless, it is currently expensive and time-consuming to generate and validate new iPSC cell lines.
Presently, human organoid models include the gut [5], lung [6], kidney [7], heart [8], liver [9], and blood vessels [10], with ongoing development in other areas. In these established organoids, derived from stem cells, a variety of cells can be generated to give structure and function to a given tissue. However, a major limitation in the field is the lack of consideration for the immune system. Incorporating immune cells into organoid models presents challenges, such as the short half-life of immune cells in culture and the difficulty of deriving certain immune phenotypes. In addition to these challenges, there is also the consideration of graft-versus-host disease if the sources of immune cells are not compatible with the target organ. In the body, each tissue contains a unique population of resident immune cells. Even the most basic immune cells exist in primordial species, such as the coelomocytes in sea urchins, which have a coagulatory and phagocytic function [11]. Thus, tissue physiology has developed close synergistic interactions with the immune system. The unique population of resident immune cells in each tissue plays a crucial role in organ function by defending against pathogens, maintaining homeostasis, and directing inflammatory responses [12]. Therefore, the inclusion of immune cells is crucial to better mimick human disease in organoid models.
This review aims to demonstrate the benefits of incorporating immune cells into organoids. First, it will provide an overview of the major resident immune cells in human organs and their main functional contributions. While the organoid research field is well established, the inclusion of the immune system into heterogenous organ models is a complex task due to issues with cell viability, phenotype, and integration. The review will explore several methodologies that researchers are exploring to address these challenges. Finally, as immense progress has been made in the area of tumor organoid-immune cell co-cultures, the review will discuss the key discoveries in this emerging field. By incorporating immune cells into existing organoid systems, researchers can gain valuable insight into how organs function under stress or disease. Enhancing the organoid system with immune cells will lead to a deeper understanding of human organ function and disease mechanisms.
2. Resident Immune Cells
Tissue-resident immune cells are found throughout the body. Some sites are heavily populated by immune cells such as the thymus, spleen, and lymph nodes, which constitute the central and peripheral immune systems [13]. Other sites have a clear defense purpose, such as at barrier sites or mucous membranes, including the skin [14], mouth and nose [15], gut [16], or lung [17]. However, there are also resident immune cells in non-barrier tissue organs such as the liver [18], kidney [19], and heart [20]. This next section will give a brief overview of the main resident immune cell types. Some cell types, such as neutrophils, are still a contentious topic in terms of their residency in certain tissues, such as the lymph node [21]. As it would be impossible to cover each type of resident immune cell found within every tissue, we discuss here the highlights of the known origins and functions of the major cell types (Figure 1).
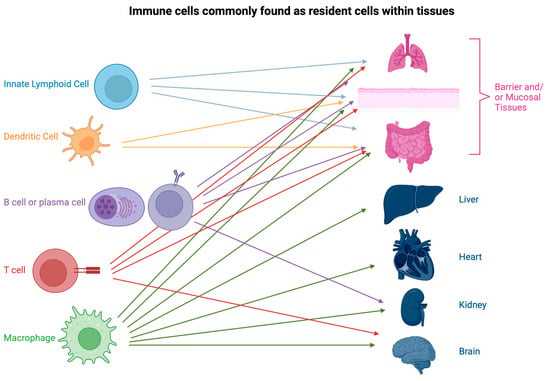
Figure 1.
Examples of immune cells commonly found as resident cells within key tissues and organs. Barrier and muscosal tissues, such as the lung, skin, and gut, are home to the highest variety and frequency of immune cells. Innate lymphoid cells (ILC) tend to be restricted to barrier tissues. Macrophages are found in every organ, often contributing to the essential functioning of the organ. These few examples are by no means exhaustive and only serve as a demonstration of the diversity of resident immune cells. Created with BioRender.com.
2.1. Macrophages
Macrophages are characterized by their ability to phagocytose pathogens, debris, and apoptotic cells [22]. Additionally, macrophages can release a plethora of signaling molecules that will drive the environment of a tissue into an inflammatory response, repair, or maintain homeostasis [22]. Macrophages are extremely heterogeneous and develop specific characteristics based on the organ where they are located. In organs like the heart or the brain, macrophages are seeded early during embryological development and become highly specialized in that tissue. Microglia in the brain are distinct from alveolar macrophages, which are distinct from macrophages (Kupffer cells) in the liver. In these organs, macrophages maintain homeostasis [23], control metabolism [24], direct inflammatory responses, and control tissue repair [25].
There are essentially three sources of tissue macrophages. In early embryological development, macrophages come from the yolk sac and the fetal liver [26]. It is well established that microglia develop from the embryonic yolk sac and migrate into the brain. However, most other resident macrophages arise from the fetal liver around embryonic day 14. Postnatally, macrophages develop from monocytes, which originate in the bone marrow along with most other immune cells in adults [27]. Even if resident macrophages are replaced by circulating monocytes that mature into tissue-resident monocytes, their functional phenotype will never completely replicate that of the embryonically seeded macrophages. There is also extensive evidence that resident macrophages are able to self-renew in adulthood to maintain the original resident population [27]. An understanding of the origin of tissue resident macrophages may help with the incorporation of macrophages into a target organoid.
In every organ, there is a dedicated population of macrophages specialized for the functioning of that particular organ. A key role of resident macrophages is to serve as the first line of defense against pathogens and prevent uncontrolled inflammatory responses [28]. Additionally, the clearance of proteins and apoptotic material by macrophages is important to maintain tissue homeostasis and prevent the development of autoimmunity. In the lung, in addition to the expected role of pathogen surveillance [29], specialized alveolar macrophages clear surfactant proteins to maintain homeostasis in the lung [30]. In the liver and spleen, macrophages are essential for erythrocyte clearance and iron recycling [31]. In the bone, macrophage-lineage progenitors differentiate into osteoclasts involved in bone remodelling driven by macrophage colony stimulating factor (M-CSF) and Receptor activator of nuclear factor kappa-Β ligand (RANKL) [32,33]. In adipose tissue, peroxisome proliferator-activated receptor (PPAR)γ-activated resident macrophages maintain insulin sensitivity as compared to recruited inflammatory macrophages, which promote obesity, insulin resistance, and glucose intolerance [24]. In the heart, cardiac macrophages modulate electrical conduction and release insulin-like growth factor (IGF)-1 to promote cardiomyocyte survival [34]. In the brain, microglia have been shown to prune neural synapses, improve synaptic transmission, and release factors to promote the growth of neurons and glial cells [34]. Furthermore, in development, macrophages play a role in ductal branching, which is relevant for those aiming to study breast, kidney, or pancreatic development with organoids [35]. All these examples show an important role for macrophages in the proper function of organs. Attempting to study human organ responses to disease in organoids without incorporating macrophages may limit discoveries.
2.2. Dendritic Cells
Dendritic cells (DCs) are cells of the myeloid lineage optimized for a process called antigen presentation to coordinate local and systemic immunity [36]. Antigen presentation involves the uptake of material from the environment and the subsequent display of peptides on MHC molecules that are on the cell surface to activate antigen-specific T cells. DCs are professional antigen presenting cells, which means they are extremely effective at this process [36]. In addition to direct interactions with T cells through antigen presentation, DCs can also direct immune responses through the secretion of mediators such as type-1 interferon (IFN) [37]. DCs are found not only in lymphoid organs and barrier tissues but also throughout the liver, heart, or kidney [38]. More specifically, DCs appear to be constantly replaced in most of these tissues from the circulating pool of monocytes, or common DC progenitors [39]. Based on their central regulatory role in immunity, DCs are an important backbone of tissue homeostasis, autoimmunity, allergy, or cancer, making it paramount to consider including DCs in defined organoid models.
2.3. T Cells
Naïve T cells circulate through the blood to travel to lymph nodes and the spleen, where they may encounter their cognate antigen and undergo activation. After activation, T cells are directed to target tissues and carry out their function. Some T cells will remain in the tissue, becoming resident memory cells, or Trm cells [40]. Trm cells are a subset of non-circulating memory T cells that reside in peripheral tissues for prolonged periods of time [40]. These cells are transcriptionally, phenotypically, and functionally distinct from circulating T cells and provide potent effector functions specific for pathogens that are encountered in epithelial barrier tissues such as the skin, gut, lung, and reproductive tracts [40,41]. Trm cells have been shown to be protective against viral [42], bacterial [43], and parasitic infections [44]. Improved immunity against these pathogens can be attributed to Trm cells actively patrolling the tissue to quickly release cytokines like IFN-γ, TNF-α, and interleukin (IL)-2, which activate surrounding stromal cells and resident immune cells, thereby boosting the tissue’s overall resistance to infection [42]. Trm cells also retain the capacity to proliferate [45].
However, autoreactive Trm cells may contribute to the development of autoimmune and inflammatory diseases such as psoriasis, type 1 diabetes mellitus, and inflammatory bowel disease [46]. Consequently, Trm cells play a diverse role in human health in the context of infection, autoimmunity, cancer, or transplantation [46]. If not properly controlled, Trm cells may offer immune protection against local pathogens at the expense of causing adverse pro-inflammatory responses [47]. In the context of tumor immunity, evidence from several human studies has identified Trm-like tumor-infiltrating lymphocytes (TILs) in melanoma, non-small cell lung cancer, breast, colorectal, bladder, ovarian, and cervical cancer [46]. Furthermore, depending on the specific marker and the tumor type, it has been estimated that 25–75% of TILs display a Trm cell-like phenotype [47]. As the presence of a high proportion of total TILs is a strong indicator of improved cancer prognosis and survival, CD8+ Trm cells may play an important role as targets of immune therapies [46,47].
T regulatory cells (Tregs) are a subset of CD4 T cells with a phenotype driven by FoxP3 expression. They use various mechanisms to suppress inflammation, such as the production of IL-10 and transforming growth factor (TGF)-β, sequestration of IL-2, and directly through CLTA-4 engagement on target T cells [48]. Tregs are resident in lymphoid and non-lymphoid tissues. They were first identified in the visceral adipose tissue (VAT) [49], then in the skin, central nervous system (CNS), and other tissues [48]. In obese patients, the number of Tregs in the VAT is significantly reduced, and the level of inflammation is increased [50]. Further, Tregs have even been implicated in the control of thermogenesis by promoting adipocyte beiging (i.e., becoming thermogenic adipocytes) [51]. In the VAT, Tregs have been shown to be regulated by different mechanisms in female and male mice [52].
Further, since the skin is exposed to the microbiome, the number of Tregs is proportionally high in this tissue. Their role here has mainly been shown to be homeostatic, including normal hair growth [53] and tissue repair [54]. Tregs are also found in the lung, skeletal muscle, and gut, carrying out modulatory functions during infection [48]. Deficiencies in Treg populations are common in chronic inflammatory diseases [48].
Although rare in the brain, Tregs are protective against many neurological diseases through their interaction with the glial cells of the brain. In particular, brain-resident Treg IL-10 modulates microglia to an anti-inflammatory phenotype in endotoxin challenge [55]. In a mouse model of Alzheimer’s Disease, supplemental IL-2 increased Treg numbers and resulted in improved outcomes [56]. The role of Tregs in the brain emphasizes that for studying CNS diseases, it is necessary to consider even this small population of immune cells.
2.4. Innate Lymphoid Cells
Mucosal tissues such as the gut and lung contain high numbers of innate lymphoid cells (ILCs). These cells are closely related to T cells but lack the ability to recognize antigens, so most of their function lies in their ability to secrete innate cytokines [57]. While ILCs are present throughout the body, their residency is concentrated in barrier sites and occurs perinatally [57]. ILCs are maintained by IL-7 and other locally secreted factors, which differ slightly depending on the tissue, resulting in tissue-specific phenotypes [57].
ILCs can be divided into three subtypes: ILC1, 2, and 3. These divisions match up with their secretory function and T helper cell paradigm (Th1, Th2, etc.). ILC1 releases IFN-γ to drive anti-viral and anti-bacterial immunity but does not have a strong cytotoxic function itself [37]. They can be found in the liver, small intestine, and spleen [37]. ILC2s have anti-parasitic and pro-healing functions [37]. They release IL-4, IL-5, and IL-13 to drive the innate immune response and repair damaged tissues [37]. ILC2s are usually found in the lung and adipose tissue, where their function is homeostatic [37]. In the gut, ILC2s can be more inflammatory, for example, responding to infections with Nippostrongylus brasiliensis [58]. ILC3s tends to be found in large numbers at mucosal sites, such as the intestine, and mainly produces IL-22, a homeostatic cytokine [59]. With the growing interest in microbiome interactions with gut function, gut organoid co-cultures with microbes have been developed [60]. However, as ILCs are intrinsic to gut function, especially epithelial integrity [61], it will be beneficial to consider including ILCs, in particular ILC2 and ILC3 populations, in gut organoid experiments.
2.5. Resident B Cells
Similar to T cells, B cells circulate through the blood, lymphatics, and secondary lymphoid organs and undergo activation after encountering their cognate antigen. After activation, B cells will take up residency in a tissue either as plasma cells (antibody producing cells) or memory B cells. Plasma cells are usually resident in secondary lymphoid organs, but following chronic inflammation, they can become resident in many tissues, including the joint, nervous system, kidneys, and barrier tissues [62]. At these sites, plasma cells will secrete antibodies that have undergone somatic hypermutation, meaning their affinity and isotype have been optimized. Resident B cells in the skin will increase with autoimmune diseases, such as pemphigus [63]. In this case, antibody secretion drives disease [41]. In the intestine, antibody secretion by resident B cells is important for maintaining tolerance to the gut microbiome [64]. As noted above, when designing gut organoid experiments to study microbiome interactions, B cells would be another valuable cell to include for this reason. Functionally, B memory cells can act as antigen presenting cells in tissues, promoting local immunity through interactions with T cells. In addition to quick reactivation and antibody production, B cells can also help maintain homeostasis by acting as regulatory B cells, such as in contact hypersensitivity [65]. The diverse roles of resident B cells is widespread in a variety of organs, so for establishing an accurate disease model in organoids B cells may be important to include.
3. Incorporating Immune Cells into Organoids: Methods and Discoveries
For this review, we have chosen to include a wide variety of “organoid” models, ranging from air-liquid interface (ALI) models to 3D spheroids to scaffold-based models. This is because the concept of incorporating immune cells into organoids is still a fledgling field, and limited studies exist where researchers have been successful in developing such co-culture systems. Overall, there are currently four main methodologies that have been used to incorporate immune cells into organoid system models (Figure 2). This includes (a) seeding immune cells and stromal cells together into a scaffold; (b) co-culture of isolated immune cells with an established organoid; (c) injecting immune cells directly into the lumen of an organoid, such as in lung organoids; or (d) expansion of an explant tissue sample complete with resident immune cells. Table 1 summarizes the highlighted studies.
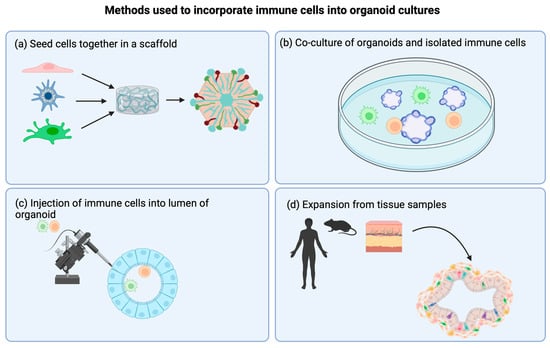
Figure 2.
Methods used to incorporate immune cells into organoid cultures. (a) Seeding cells together in a scaffold. Various cell types are seeded at the same time onto a scaffold. Often, researchers will choose to include parenchyma/stromal cells as well as immune cells of interest. (b) Direct co-culture of preformed organoids with isolated immune cells. After organoids are successfully generated, immune cells of interest are added to the system to mimic interactions with the organ. (c) Injection of immune cells into the lumen of the organoid. In order to promote infiltration of immune cells into the organoid, injecting into the lumen is a good option. This is an especially appropriate method when studying organs where immune cells are resident in the lumen, such as alveolar macrophages in the lumen of alveoli in the lung. (d) Expansion of endogenous cells from tissue samples. This methodology has been used mostly in tumor spheroid studies. It has the advantage of expanding and maintaining endogenous immune cells that retain their phenotype and proportions from the original tumor sample. Created with BioRender.com.

Table 1.
Methods for immune cell incorporation and discoveries.
3.1. Scaffold Systems
The advantage of seeding a scaffold is controlling what cells are incorporated into an organoid [80]. This means that a variety of immune cells and parenchymal cells can be included from various sources in order to best mimic the target tissue. In addition, as both these cell types create the tissue organoid at the same time, immune cells can be readily incorporated in a manner that results in functional interactions between immune cells and tissue parenchymal cells.
A current challenge is finding the optimal scaffold system [81]. A good scaffold allows cells to grow in a way that mimics the target tissue. Some scaffolds provide growth factors that will also direct cells to expand in a structure that is most similar to the organ of interest. As one example, Collin de l’Hortet et al. capitalized on the advantages of scaffold systems while also solving one of the limitations of most organoid systems. To overcome the lack of flow, the authors took a decellularized rat liver and used it as a scaffold [66]. They then seeded liver mesenchymal cells, liver fibroblasts, and human macrophages and obtained a high (almost 90%) engraftment efficiency [66]. This humanized liver model was then used to establish a steatosis model by knocking down sirtuin-1 expression in the hepatocytes [66]. They found an increase in inflammatory markers when macrophages were included in the scaffold system [66]. This study demonstrated the feasibility of creating an organoid model that includes immune cells using scaffolds and the effectiveness of this system to study a disease.
3.2. Co-Culture Models
A straightforward way to combine various cell types is a co-culture model. This offers flexibility in the choice of immune cell to add. Two groups used intestinal epithelial cells from mice to generate gut organoids and then co-cultured these together in Matrigel with intestinal epithelial lymphocytes [67,68]. The lymphocytes actively integrated into the organoid. In fact, the lymphocytes exhibited motility, moving towards the organoid, incorporating into the epithelial cell layer, and even exiting again [67]. Another group compared the motility and behavior of two types of intestinal epithelial lymphocytes, αβ T cells and γδ T cells [68]. Therefore, combined with live microscopy, this model allows the study of T cell behavior in an ex vivo system, providing new functional insight into the migration of these cells.
To study the brain microglia, Popova et al. generated cerebral organoids and subsequently used iPSC-derived human microglia to seed these brain organoids [69]. Interestingly, no factors required for microglial survival were needed, as the cerebral organoid environment itself provided growth and survival factors such as IL-34, CSF-1, and TGF-β [69]. Microglia invaded deep into the organoids and took on a ramified shape, indicative of a resting state. Using this system, the authors found that these microglia were able to make contact with neurons and carry out phagocytosis (two normal functions of microglia in the brain). After co-culturing, they completed RNA sequencing and found similar gene expression to primary human microglia [69]. Further, the presence of microglia in the cerebral organoid co-culture regulated the gene expression of both glia and neurons. Specifically, the type 1 IFN response was attenuated, and there was reduced DNA damage in radial glia. Additionally, the presence of microglia improved the maturation of neural networks [69], demonstrating the importance of including immune cell components in organoid experiments.
Rather than keeping human intestinal enteroids in 3D culture, Noel et al. found that seeding enteroids into a monolayer system was optimal for resident macrophages studies [70]. These monolayer models took advantage of the predeveloped enteroid morphology with a basal and apical layer that was maintained upon transplant onto a membrane [70]. Monocytes from human blood were differentiated into human macrophages and then seeded on the basolateral side [70]. Macrophages secreted cytokines such as IL-8 and TGF-β and changed their morphology to extend dendrites through the monolayer [70]. The presence of macrophages enhanced the barrier function of the intestinal epithelium monolayer, as measured by transepithelial electrical resistance [70]. Further, upon the addition of bacteria on the apical side, macrophages were able to extend protrusions through the epithelial monolayer to phagocytose bacteria [70]. Thus, this model offers a method to study the response of human gut tissue to bacterial infections.
3.3. Direct Injection of Immune Cells into Organoids
To overcome the challenge of ensuring co-cultured cells infiltrate into the organoid, immune cells can be directly injected into the lumen. This approach is especially appropriate for studying alveolar macrophages, which reside on the luminal surface of alveoli. To accomplish this, human bronchoalveolar lavage (BAL)-isolated macrophages were injected directly into individual mature bronchioalveolar organoids [71]. Not only did the macrophages survive up to 28 days within the organoid, they were also able to carry out some physiological functions, such as surfactant uptake and interaction with alveolar cells, through the expression of the tight junction protein Cx43 [71]. Following infection with influenza A virus, IL-6 and IL-1β release was significantly increased in organoids that included resident macrophages [71]. Another study used a similar approach; however, rather than using primary macrophages isolated from human BAL, they generated macrophages from iPSC [72]. Using this system, they found that organoids containing injected macrophages were able to respond to lipopolysaccharide stimulation by secretion of IL-1β and TNF-α [72]. This methodology to inject cells directly into the lumen of an organoid has the advantage of mimicking human biology, with the limitation that it can only be applied to certain tissues.
4. Organoids to Study Tumor Immunology
Another field where the immune cell contribution is essential is tumor biology. Growing cancer cells in 2D cell cultures in vitro or as xenograft models in vivo both fail to sufficiently model the complexity of human cancers as they lack human-specific tumor microenvironments (TME) and tumor-infiltrating immune cells [82]. In addition, cancer-derived cell lines may acquire considerable genetic mutations as a result of long-term maintenance and passages in vitro [83]. In contrast, tumor organoids can recapitulate features of the primary tissues both genetically and histologically and can even mimic responses to therapy [84].
The complex TME contains a complicated network of surrounding fibroblasts, pericytes, blood vessels, and an innate and adaptive immune cellular network that consists of lymphocytes (T and B cells), natural killer (NK) cells, macrophages, DCs, eosinophils, mast cells, and myeloid-derived suppressor cells (MDSCs) [85]. The tumor TME plays important roles in carcinogenesis, tumor progression, and metastases of malignant cells [86]. Current approaches to organoid and immune cell co-culture models either consist of the addition of immune cells directly to organoid cultures or maintaining and expanding native immune cells in tumoroids explanted from patients.
4.1. Co-Culture Models
As discussed above, co-culture models offer flexibility in choosing which immune cells to include, and experiments can be designed to use autologous patient-derived immune cells. Using co-cultures of murine-derived tumor organoids, dendritic cells, and cytotoxic T cells, the effects of PD1 blockade and immunotherapies were explored and revealed a role of hedgehog signaling in the regulation of PD-L1 expression [73]. Similarly, mouse and human-derived autologous pancreatic ductal carcinoma (PDAC) organoids and immune cell co-cultures were established to investigate the immunosuppressive function of MDSCs and PDL1-PD-1 blockade [74]. Collectively, the authors found that a combinatorial treatment that requires both the depletion of MDSCs and inhibition of PD-1/PD-L1 is necessary to enhance T cell effector function in target PD-L1-expressing PDAC cells.
Recently, autologous cancer organoid-immune cell co-cultures were developed to investigate the immunosuppressive function of MDSCs and PDL1-PD-1 blockade to test therapeutic approaches for gastric cancer [75]. Organoids and immune cells were either cultured together in Matrigel or the organoids were generated using AggreWell Microwell plates (a high-density array of pyramid-shaped microwells) [75]. By using these gastric cancer organoid/immune cell co-cultures, the authors were able to show that cabozantinib treatment, a tyrosine inhibitor, resulted in a targeted depletion of MDSCs and enhanced the efficacy of checkpoint inhibitor therapy by increasing cytotoxic T cell proliferation and effector functions. In addition, activating the mTOR signaling pathway increased PD-L1 expression, providing a new potential strategy for gastric cancer therapy [75]. Adding exogenous cells cannot fully recapitulate the in vivo environment; however, this can sometimes be an advantage if the goal of the experiment is to only characterize one particular cell type. Additionally, tumor organoids co-cultured with autologous peripheral blood T cells can be used to enrich for tumor-specific T cells [76].
4.2. Expansion of the Tissue Sample
Expanding endogenous cells already present in the source tissue has the advantage of possibly better recapitulating the relevant microenvironment. For example, one of the first studies that successfully did this used explanted tumor tissue to generate murine and patient-derived tumor spheroids that retained both lymphoid and myeloid immune cells [77]. Using these explants, proportions of immune cells were maintained in the tumor spheroids as compared to the source tissue [77]. Further, the complex organoids also demonstrated sensitivity or resistance to PD-1 blockade as seen in vivo [77]. Immune profiling of these spheroids following PD-1 blockade in vitro identified features that predicted responses to immune checkpoint blockade in vivo [77].
Patient-derived colorectal or lung cancer 3D air-liquid interface (ALI) tumor spheroids were used to study the infiltration of immune cell populations in vitro [78]. These tumor spheroids, resected from patients, included immune cells that persisted up to 8 days in culture. The authors then used this system to assess the efficacy of several FDA-approved drugs and found that colorectal cancer patient tumoroids are particularly sensitive to treatment with the already approved drugs 5-fluoruracil (5-FU) and trametinib [78]. Similar results were found using ALI models with 3D tumor spheroids, where the T cell receptor repertoire was conserved [79]. In addition to assessing immunotherapies and differences in the tumor microenvironment, explanting 3D tumor cultures from patients might also be feasible to test personalized therapeutics.
5. Sources of Immune Cells
There are essentially three sources from which immune cells can be derived for immune-organoid systems: iPSC cells, differentiation of blood or bone marrow cells, or direct isolation from the tissue of interest. iPSC cells may arguably be the best option, with a few practical caveats such as cost, access to personalized patient iPSC, and the difficulty in generating certain cell types [87]. While monocyte differentiation is well established with available kits and several published protocols, the generation of T cells from iPSC cells has not been as easy due to the complicated maturation steps involved in T cell development [88]. Current differentiation procedures take several weeks and may result in low yield [89]. Finally, working with iPSCs may involve some ethical barriers, requiring additional documentation. However, there are also multiple benefits to working with iPSCs, mainly the flexibility to generate target cells of choice and direct applicability to patient samples [88]. Additionally, immune cells derived from iPSC may have increased viability due to their immature state and can be used and generated “indefinitely”.. As certain immune cells become resident in organs during embryonic development [27], the immature state of iPSC-derived immune cells may better mimic this phenotype. For example, this is the case with microglia in the brain [27].
Differentiation of blood [90] or bone marrow-derived cells offers the benefit of easy access and the possibility to upscale to a larger amount of cells, which could be useful if immune cell-seeded organoids were employed for a screen. This is also a much cheaper method than iPSC cells to obtain a particular cell of interest [91]. Directly isolating immune cells of interest from a target tissue will result in complex populations of freshly isolated cells that are phenotypically similar to those in vivo. However, if the target cell is only present in small numbers within a tissue, then it may not be feasible to get sufficient biopsy for such an isolation. Further, the digestion enzymes and conditions used during the isolation process may affect viability and the expression of some cell surface receptors [92], which needs to be considered for the experimental setups. Another limitation of primary isolated immune cells is that certain cell types may not be very long-lived [93], even when incorporated into organoids.
6. Future Outlook
Great strides have been made to incorporate immune components into organoid models. The main limitations are the sources of immune cells, their successful incorporation, and the maintenance of their phenotype and longevity. There are multiple sources for immune cells, and the field will have to establish guidelines to reduce variability in experimental results derived from the various immune cell sources. Moreover, improved methods need to be developed to promote the incorporation of immune cells in a functional manner. Injecting macrophages into alveolar organoids is an example of successfully leveraging the biology of an organ to incorporate immune cells in the appropriate location [71]. Another notable model was the functional incorporation of microglia into human cerebral organoids by seeding iPSC-derived human microglia into an existing brain organoid [69]. These microglia did not require exogenous factors to be maintained, as the brain organoid provided all necessary physiological factors [69]. The final challenge is to maintain the viability and phenotypes of immune cells within organoids or tumor spheroids for long-term experiments.
While adding immune cells is an important step in further developing and exploring complex organoids, incorporating a functional blood supply and neurons has also been suggested to improve the growth, maturity, and longevity of organoids [94]. The intricate nature of vascular networks in these tissues presents hurdles in achieving robust and functional vasculature within the organoid models. Combining an organoid perfused with a microfluidic system would offer an opportunity to flow immune cells through the tissue. Successfully incorporating a blood supply and perfusable vasculature should improve the longevity of the organoid itself as well as that of the incorporated immune cells. Yip et al. recently reviewed the advances and challenges in the vascularization of organoids [95]. Vasculature would affect how nutrients, oxygen supply, or pH influence immune cell phenotypes. While resident in a tissue, immune cells can optimize their behavior and responses to maintain homeostasis and protect against pathogens without causing damage or disease.
The lack of uniformity in organoid fabrication within the scientific community poses a challenge for consistent comparison and reproducibility of results. Unfortunately, the necessary step of introducing immune cells will further complicate this problem. This inconsistency arises from the lack of standardized protocols and the inherent variation in donor-derived cells. This shortcoming can lead to technical bias, which may influence experimental results [73]. Some replicability issues can be attributed to the use of serum in culture media, which can vary between batches [74]. Similar concerns arise with the extracellular matrix, as it contains a diverse range of proteins that can have a variety of effects on the experimental outcome [74].
To address variation due to the stem cell source (e.g., donor cells), it is recommended to expand cells to reduce sampling bias. Even simple changes, such as using a specific enzyme mix to remove cells from a cell culture dish rather than a scraping tool, can improve uniformity [96]. Incorporating immune cells from different cell culture methods or sources will add complexity to this already challenging issue. In the future, researchers must engage in transparent dialogue and promote the detailed sharing of exact protocol details. This will be crucial for enhancing comparability and reproducibility in organoid studies.
The methods discussed in this review offer novel and unique approaches to dissecting the contribution of the immune system to various diseases. Many experimental possibilities exist once these protocols are established. For example, A “normal” tissue organoid could be co-cultured or perfused with patient-derived immune cells to see how much these new immune cells contribute to a disease phenotype. This could be performed with a pancreatic organoid perfused with monocytes from diabetes patients to measure the changes these monocytes can drive. Many iterations of this system could be devised for almost every disease. The exploration of resident immune cells should also contribute to a better understanding of normal physiology and complex disease processes, eventually leading to more rational and targeted therapeutic interventions.
7. Conclusions
Every single tissue in the body has an immune cell component. In many tissues, these immune cells are essential for the normal function of the organ. For instance, microglia have the ability to prune neurons, while osteoclasts maintain bone health. Organoid systems have shown great promise for studying human organs in vitro, but they will always be limited in their ability to fully recapitulate tissue complexities until a variety of cells are included, one of which is immune cells. Several examples of successful incorporation of immune cell populations into organoids or tumor spheroids exist, yet there are a plethora of avenues for expansion.
Organoid research is constantly evolving to better mimic the human system. Incorporating the complexity of the immune system enhances the potential for studying disease beyond model organisms. Without immune cells, the observed responses to diseases or drugs may be inaccurate or incomplete. Tissue organoids containing physiological immune populations represent the forefront of organoid research, facilitating rapid advancements in disease modeling.
Funding
A.B. is funded by a CIHR Banting Fellowship. M.A. is funded through CIHR. JMP holds CIHR grants: GR005784 CA150CHR 2018, GR004574 UBCMEDIC 2018.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fogel, D.B. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: A review. Contemp. Clin. Trials Commun. 2018, 11, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Nair, R.R.; Fisher, E.M.C.; Cunningham, T.J. Humanising the mouse genome piece by piece. Nat. Commun. 2019, 10, 1845. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2016, 22, 1392–1401. [Google Scholar] [CrossRef]
- Min, S.; Kim, S.; Cho, S.-W. Gastrointestinal tract modeling using organoids engineered with cellular and microbiota niches. Exp. Mol. Med. 2020, 52, 227–237. [Google Scholar] [CrossRef]
- Kong, J.; Wen, S.; Cao, W.; Yue, P.; Xu, X.; Zhang, Y.; Luo, L.; Chen, T.; Li, L.; Wang, F.; et al. Lung organoids, useful tools for investigating epithelial repair after lung injury. Stem Cell Res. Ther. 2021, 12, 95. [Google Scholar] [CrossRef]
- Romero-Guevara, R.; Ioannides, A.; Xinaris, C. Kidney Organoids as Disease Models: Strengths, Weaknesses and Perspectives. Front. Physiol. 2020, 11, 563981. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Israeli, Y.R.; Wasserman, A.H.; Aguirre, A. Heart Organoids and Engineered Heart Tissues: Novel Tools for Modeling Human Cardiac Biology and Disease. Biomolecules 2021, 11, 1277. [Google Scholar] [CrossRef]
- Harrison, S.P.; Baumgarten, S.F.; Verma, R.; Lunov, O.; Dejneka, A.; Sullivan, G.J. Liver Organoids: Recent Developments, Limitations and Potential. Front. Med. 2021, 8, 574047. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Wick, N.; Hantusch, B.; Novatchkova, M.; Taubenschmid, J.; Hämmerle, M.; Esk, C.; Bagley, J.A.; et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 2019, 565, 505–510. [Google Scholar] [CrossRef]
- Coffaro, K.A.; Hinegardner, R.T. Immune Response in the Sea Urchin Lytechinus pictus. Science 1977, 197, 1389–1390. [Google Scholar] [CrossRef]
- Rankin, L.C.; Artis, D. Beyond Host Defense: Emerging Functions of the Immune System in Regulating Complex Tissue Physiology. Cell 2018, 173, 554–567. [Google Scholar] [CrossRef]
- Thompson, E.C. Focus issue: Structure and function of lymphoid tissues. Trends Immunol. 2012, 33, 255. [Google Scholar] [CrossRef]
- Kabashima, K.; Honda, T.; Ginhoux, F.; Egawa, G. The immunological anatomy of the skin. Nat. Rev. Immunol. 2019, 19, 19–30. [Google Scholar] [CrossRef]
- Moutsopoulos, N.M.; Konkel, J.E. Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends Immunol. 2018, 39, 276–287. [Google Scholar] [CrossRef]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, E.Y.; Lauzon-Joset, J.-F.; Debley, J.S.; Ziegler, S.F. Cross-Talk Between Alveolar Macrophages and Lung Epithelial Cells is Essential to Maintain Lung Homeostasis. Front. Immunol. 2020, 11, 583042. [Google Scholar] [CrossRef]
- Robinson, M.W.; Harmon, C.; O’farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.J. Immune topology of the human kidney. Nat. Rev. Nephrol. 2019, 15, 729. [Google Scholar] [CrossRef]
- Sansonetti, M.; Waleczek, F.J.G.; Jung, M.; Thum, T.; Perbellini, F. Resident cardiac macrophages: Crucial modulators of cardiac (patho)physiology. Basic Res. Cardiol. 2020, 115, 77. [Google Scholar] [CrossRef] [PubMed]
- Bogoslowski, A.; Wijeyesinghe, S.; Lee, W.-Y.; Chen, C.-S.; Alanani, S.; Jenne, C.; Steeber, D.A.; Scheiermann, C.; Butcher, E.C.; Masopust, D.; et al. Neutrophils Recirculate through Lymph Nodes to Survey Tissues for Pathogens. J. Immunol. 2020, 204, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2018, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Hamidzadeh, K.; Goncalves, R. Macrophages and the maintenance of homeostasis. Cell. Mol. Immunol. 2021, 18, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma; controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Gentek, R.; Molawi, K.; Sieweke, M.H. Tissue macrophage identity and self-renewal. Immunol. Rev. 2014, 262, 56–73. [Google Scholar] [CrossRef]
- Epelman, S.; LaVine, K.J.; Randolph, G.J. Origin and Functions of Tissue Macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef]
- Maus, U.A.; Koay, M.A.; Delbeck, T.; Mack, M.; Ermert, M.; Ermert, L.; Blackwell, T.S.; Christman, J.W.; Schlöndorff, D.; Seeger, W.; et al. Role of resident alveolar macrophages in leukocyte traffic into the alveolar air space of intact mice. Am. J. Physiol. Cell. Mol. Physiol. 2002, 282, L1245–L1252. [Google Scholar] [CrossRef]
- Carey, B.; Trapnell, B.C. The molecular basis of pulmonary alveolar proteinosis. Clin. Immunol. 2010, 135, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Macrophages and Systemic Iron Homeostasis. J. Innate Immun. 2012, 4, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Kodama, H.; Nose, M.; Niida, S.; Yamasaki, A. Essential role of macrophage colony-stimulating factor in the osteoclast differentiation supported by stromal cells. J. Exp. Med. 1991, 173, 1291–1294. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.-Y.; Yoshida, H.; Sarosi, I.; Tan, H.-L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-Dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Nicolás-Ávila, J.A.; Hidalgo, A.; Ballesteros, I. Specialized functions of resident macrophages in brain and heart. J. Leukoc. Biol. 2018, 104, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef]
- Lipscomb, M.F.; Masten, B.J. Dendritic Cells: Immune Regulators in Health and Disease. Physiol. Rev. 2002, 82, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Matta, B.M.; Castellaneta, A.; Thomson, A.W. Tolerogenic plasmacytoid DC. Eur. J. Immunol. 2010, 40, 2667–2676. [Google Scholar] [CrossRef]
- Chen, K.; Wang, J.M.; Yuan, R.; Yi, X.; Li, L.; Gong, W.; Yang, T.; Li, L.; Su, S. Tissue-resident dendritic cells and diseases involving dendritic cell malfunction. Int. Immunopharmacol. 2016, 34, 1–15. [Google Scholar] [CrossRef]
- Merad, M.; Manz, M.G. Dendritic cell homeostasis. Blood 2009, 113, 3418–3427. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef]
- Paik, D.H.; Farber, D.L. Anti-viral protective capacity of tissue resident memory T cells. Curr. Opin. Virol. 2021, 46, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, A.; Mnich, M.E.; Clemente, B.; Cruz, A.R.; Tavarini, S.; Bagnoli, F.; Soldaini, E. Staphylococcus aureus-Specific Tissue-Resident Memory CD4+ T Cells Are Abundant in Healthy Human Skin. Front. Immunol. 2021, 12, 642711. [Google Scholar] [CrossRef]
- Glennie, N.D.; Volk, S.W.; Scott, P. Skin-resident CD4+ T cells protect against Leishmania major by recruiting and activating inflammatory monocytes. PLoS Pathog. 2017, 13, e1006349. [Google Scholar] [CrossRef]
- Mueller, S.N.; Gebhardt, T.; Carbone, F.R.; Heath, W.R. Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol. 2013, 31, 137–161. [Google Scholar] [CrossRef]
- Clark, R.A. Resident memory T cells in human health and disease. Sci. Transl. Med. 2015, 7, 269rv1. [Google Scholar] [CrossRef] [PubMed]
- Sasson, S.C.; Gordon, C.L.; Christo, S.N.; Klenerman, P.; Mackay, L.K. Local heroes or villains: Tissue-resident memory T cells in human health and disease. Cell. Mol. Immunol. 2020, 17, 113–122. [Google Scholar] [CrossRef]
- Dhodapkar, K.M. Role of Tissue-Resident Memory in Intra-Tumor Heterogeneity and Response to Immune Checkpoint Blockade. Front. Immunol. 2018, 9, 1655. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, D.; Min, B. Tissue Resident Foxp3+ Regulatory T Cells: Sentinels and Saboteurs in Health and Disease. Front. Immunol. 2022, 13, 865593. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- Eller, K.; Kirsch, A.; Wolf, A.M.; Sopper, S.; Tagwerker, A.; Stanzl, U.; Wolf, D.; Patsch, W.; Rosenkranz, A.R.; Eller, P. Potential Role of Regulatory T Cells in Reversing Obesity-Linked Insulin Resistance and Diabetic Nephropathy. Diabetes 2011, 60, 2954–2962. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Deng, Z.; Benadjaoud, F.; Yang, D.; Yang, C.; Shi, G.P. Regulatory T cells promote adipocyte beiging in subcutaneous adipose tissue. FASEB J. 2020, 34, 9755–9770. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumar, A.; Chisanga, D.; Blume, J.; Gloury, R.; Britt, K.; Henstridge, D.C.; Zhan, Y.; Torres, S.V.; Liene, S.; Collins, N. Sex-specific adipose tissue imprinting of regulatory T cells. Nature 2020, 579, 581–585. [Google Scholar] [CrossRef]
- Ali, N.; Zirak, B.; Rodriguez, R.S.; Pauli, M.L.; Truong, H.-A.; Lai, K.; Ahn, R.; Corbin, K.; Lowe, M.M.; Scharschmidt, T.C.; et al. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell 2017, 169, 1119–1129. [Google Scholar] [CrossRef]
- Nosbaum, A.; Prevel, N.; Truong, H.-A.; Mehta, P.; Ettinger, M.; Scharschmidt, T.C.; Ali, N.H.; Pauli, M.L.; Abbas, A.K.; Rosenblum, M.D. Cutting Edge: Regulatory T Cells Facilitate Cutaneous Wound Healing. J. Immunol. 2016, 196, 2010–2014. [Google Scholar] [CrossRef]
- Shemer, A.; Scheyltjens, I.; Frumer, G.R.; Kim, J.-S.; Grozovski, J.; Ayanaw, S.; Dassa, B.; Van Hove, H.; Chappell-Maor, L.; Boura-Halfon, S.; et al. Interleukin-10 Prevents Pathological Microglia Hyperactivation following Peripheral Endotoxin Challenge. Immunity 2020, 53, 1033–1049. [Google Scholar] [CrossRef]
- Dansokho, C.; Ahmed, D.A.; Aid, S.; Toly-Ndour, C.; Chaigneau, T.; Calle, V.; Cagnard, N.; Holzenberger, M.; Piaggio, E.; Aucouturier, P.; et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 2016, 139, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mao, K.; Germain, R.N. Thinking differently about ILCs-Not just tissue resident and not just the same as CD4+ T-cell effectors. Immunol. Rev. 2018, 286, 160–171. [Google Scholar] [CrossRef]
- Miller, M.M.; Reinhardt, R.L. The Heterogeneity, Origins, and Impact of Migratory iILC2 Cells in Anti-helminth Immunity. Front. Immunol. 2020, 11, 1594. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Puschhof, J.; Pleguezuelos-Manzano, C.; Clevers, H. Organoids and organs-on-chips: Insights into human gut-microbe interactions. Cell Host Microbe 2021, 29, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Wang, A.; Wang, Y.; Sun, Y.; Han, J.; Chen, W.; Wang, S.; Wu, Y.; Lu, Y. Innate Lymphoid Cells: Regulators of Gut Barrier Function and Immune Homeostasis. J. Immunol. Res. 2019, 2019, 2525984. [Google Scholar] [CrossRef]
- Woodruff, M.C.; Heesters, B.A.; Herndon, C.N.; Groom, J.R.; Thomas, P.G.; Luster, A.D.; Turley, S.J.; Carroll, M.C. Trans-nodal migration of resident dendritic cells into medullary interfollicular regions initiates immunity to influenza vaccine. J. Exp. Med. 2014, 211, 1611–1621. [Google Scholar] [CrossRef]
- Sitaru, C.; Mihai, S.; Zillikens, D. The relevance of the IgG subclass of autoantibodies for blister induction in autoimmune bullous skin diseases. Arch. Dermatol. Res. 2007, 299, 1–8. [Google Scholar] [CrossRef]
- Allie, S.R.; Randall, T.D. Resident Memory B Cells. Viral Immunol. 2020, 33, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Fujimoto, M.; Ishiura, N.; Kuwano, Y.; Nakashima, H.; Yazawa, N.; Okochi, H.; Sato, S.; Tedder, T.F.; Tamaki, K. CD19 expression in B cells is important for suppression of contact hypersensitivity. Am. J. Pathol. 2007, 171, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Solchaga, L.A.; Tognana, E.; Penick, K.; Baskaran, H.; Goldberg, V.M.; Caplan, A.I.; Welter, J.F. A rapid seeding technique for the assembly of large cell/scaffold composite construct. Tissue Eng. 2006, 12, 1851–1863. [Google Scholar] [CrossRef]
- Rana, D.; Arulkumar, S.; Vishwakarma, A.; Ramalingam, M. Considerations on Designing Scaffold for Tissue Engineering. In Stem Cell Biology and Tissue Engineering in Dental Sciences; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- de L’hortet, A.C.; Takeishi, K.; Guzman-Lepe, J.; Morita, K.; Achreja, A.; Popovic, B.; Wang, Y.; Handa, K.; Mittal, A.; Meurs, N.; et al. Generation of Human Fatty Livers Using Custom-Engineered Induced Pluripotent Stem Cells with Modifiable SIRT1 Metabolism. Cell Metab. 2019, 30, 385–401. [Google Scholar] [CrossRef]
- Rogoz, A.; Reis, B.S.; Karssemeijer, R.A.; Mucida, D. A 3-D enteroid-based model to study T-cell and epithelial cell interaction. J. Immunol. Methods 2015, 421, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Mochizuki, W.; Matsumoto, Y.; Matsumoto, T.; Fukuda, M.; Mizutani, T.; Watanabe, M.; Nakamura, T. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J. Gastroenterol. 2016, 51, 206–213. [Google Scholar] [CrossRef]
- Popova, G.; Soliman, S.S.; Kim, C.N.; Keefe, M.G.; Hennick, K.M.; Jain, S.; Li, T.; Tejera, D.; Shin, D.; Chhun, B.B.; et al. Human microglia states are conserved across experimental models and regulate neural stem cell responses in chimeric organoids. Cell Stem Cell 2021, 28, 2153–2166. [Google Scholar] [CrossRef]
- Noel, G.; Baetz, N.W.; Staab, J.F.; Donowitz, M.; Kovbasnjuk, O.; Pasetti, M.F.; Zachos, N.C. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci. Rep. 2017, 7, 45270. [Google Scholar] [CrossRef]
- Vazquez-Armendariz, A.I.; Heiner, M.; El Agha, E.; Salwig, I.; Hoek, A.; Hessler, M.C.; Shalashova, I.; Shrestha, A.; Carraro, G.; Mengel, J.P.; et al. Multilineage murine stem cells generate complex organoids to model distal lung development and disease. EMBO J. 2020, 39, e103476. [Google Scholar] [CrossRef]
- Seo, H.-R.; Han, H.-J.; Lee, Y.; Noh, Y.-W.; Cho, S.-J.; Kim, J.-H. Human Pluripotent Stem Cell-Derived Alveolar Organoid with Macrophages. Int. J. Mol. Sci. 2022, 23, 9211. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef]
- Huang, L.; Bockorny, B.; Paul, I.; Akshinthala, D.; Frappart, P.-O.; Gandarilla, O.; Bose, A.; Sanchez-Gonzalez, V.; Rouse, E.E.; Lehoux, S.D.; et al. PDX-derived organoids model in vivo drug response and secrete biomarkers. J. Clin. Investig. 2020, 5, e135544. [Google Scholar] [CrossRef]
- Xu, H.; Jiao, D.; Liu, A.; Wu, K. Tumor organoids: Applications in cancer modeling and potentials in precision medicine. J. Hematol. Oncol. 2022, 15, 58. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Xia, T.; Du, W.; Chen, X.; Zhang, Y. Organoid models of the tumor microenvironment and their applications. J. Cell. Mol. Med. 2021, 25, 5829–5841. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, J.; Holokai, L.; Syu, L.; Steele, N.G.; Chang, J.; Wang, J.; Ahmed, S.; Dlugosz, A.; Zavros, Y. Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget 2018, 9, 37439–37457. [Google Scholar] [CrossRef]
- Holokai, L.; Chakrabarti, J.; Lundy, J.; Croagh, D.; Adhikary, P.; Richards, S.S.; Woodson, C.; Steele, N.; Kuester, R.; Scott, A.; et al. Murine- and Human-Derived Autologous Organoid / Immune Cell Co-Cultures as Pre-Clinical Models of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 3816. [Google Scholar] [CrossRef] [PubMed]
- Koh, V.; Chakrabarti, J.; Torvund, M.; Steele, N.; Hawkins, J.A.; Ito, Y.; Ito, Y.; Wang, J.; Helmrath, M.A.; Merchant, J.L.; et al. Hedgehog transcriptional effector GLI mediates mTOR-Induced PD-L1 expression in gastric cancer organoids. Cancer Lett. 2021, 518, 59–71. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; Van De Haar, J.; Fanchi, L.F.; Slagter, M.; Van Der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef]
- Finnberg, N.K.; Gokare, P.; Lev, A.; Grivennikov, S.I.; MacFarlane, I.V.A.W.; Campbell, K.S.; Winters, R.M.; Kaputa, K.; Farma, J.M.; Abbas, A.E.-S.; et al. Application of 3D tumoroid systems to define immune and cytotoxic therapeutic responses based on tumoroid and tissue slice culture molecular signatures. Oncotarget 2017, 8, 66747–66757. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef]
- Chun, Y.S.; Byun, K.; Lee, B. Induced pluripotent stem cells and personalized medicine: Current progress and future perspectives. Anat. Cell Biol. 2011, 44, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Jaenisch, R. Technical Challenges in Using Human Induced Pluripotent Stem Cells to Model Disease. Cell Stem Cell 2009, 5, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Nianias, A.; Themeli, M. Induced Pluripotent Stem Cell (iPSC)–Derived Lymphocytes for Adoptive Cell Immunotherapy: Recent Advances and Challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef]
- Raulf-Heimsoth, M. T Cell—Primary Culture from Peripheral Blood. Allergy Methods Protoc. 2008, 138, 17–30. [Google Scholar] [CrossRef]
- Raab, S.; Klingenstein, M.; Liebau, S.; Linta, L. A Comparative View on Human Somatic Cell Sources for iPSC Generation. Stem Cells Int. 2014, 2014, 768391. [Google Scholar] [CrossRef] [PubMed]
- Autengruber, A.; Gereke, M.; Hansen, G.; Hennig, C.; Bruder, D. Impact of enzymatic tissue disintegration on the level of surface molecule expression and immune cell function. Eur. J. Microbiol. Immunol. 2012, 2, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Blanter, M.; Gouwy, M.; Struyf, S. Studying Neutrophil Function in vitro: Cell Models and Environmental Factors. J. Inflamm. Res. 2021, 14, 141–162. [Google Scholar] [CrossRef]
- Little, M.H.; Combes, A.N. Kidney organoids: Accurate models or fortunate accidents. Genes Dev. 2019, 33, 1319–1345. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Wang, N.; Sugimura, R. Give them vasculature and immune cells—How to fill the gap of organoids. Cells Tissues Organs 2023, 1–14. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; et al. Organoids. Nat. Rev. Methods Prim. 2022, 2, 94. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).