



Applications for Colon Organoid Models in Cancer Research

Abstract
1. Introduction
2. Protocols for Colon Organoid Culture
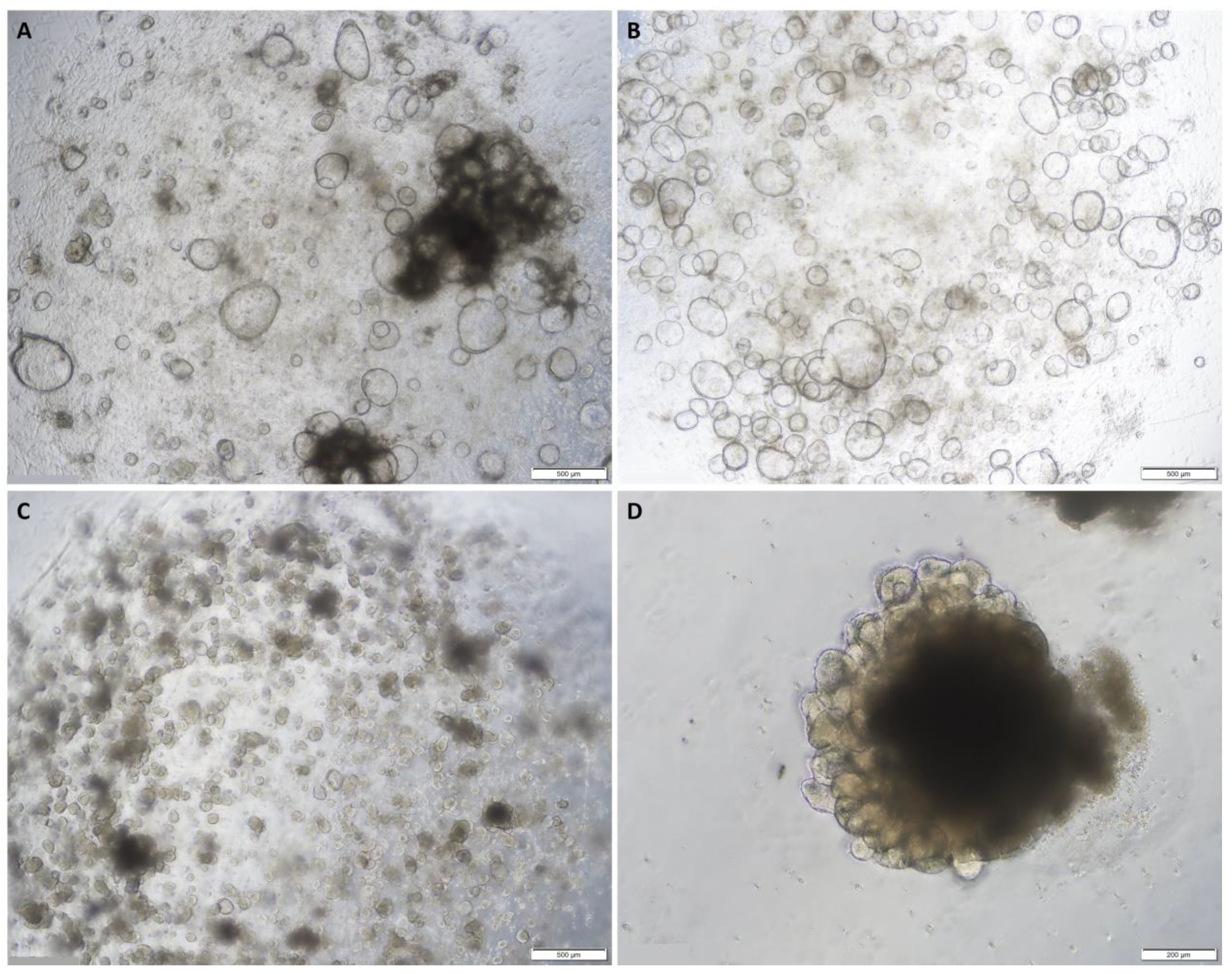
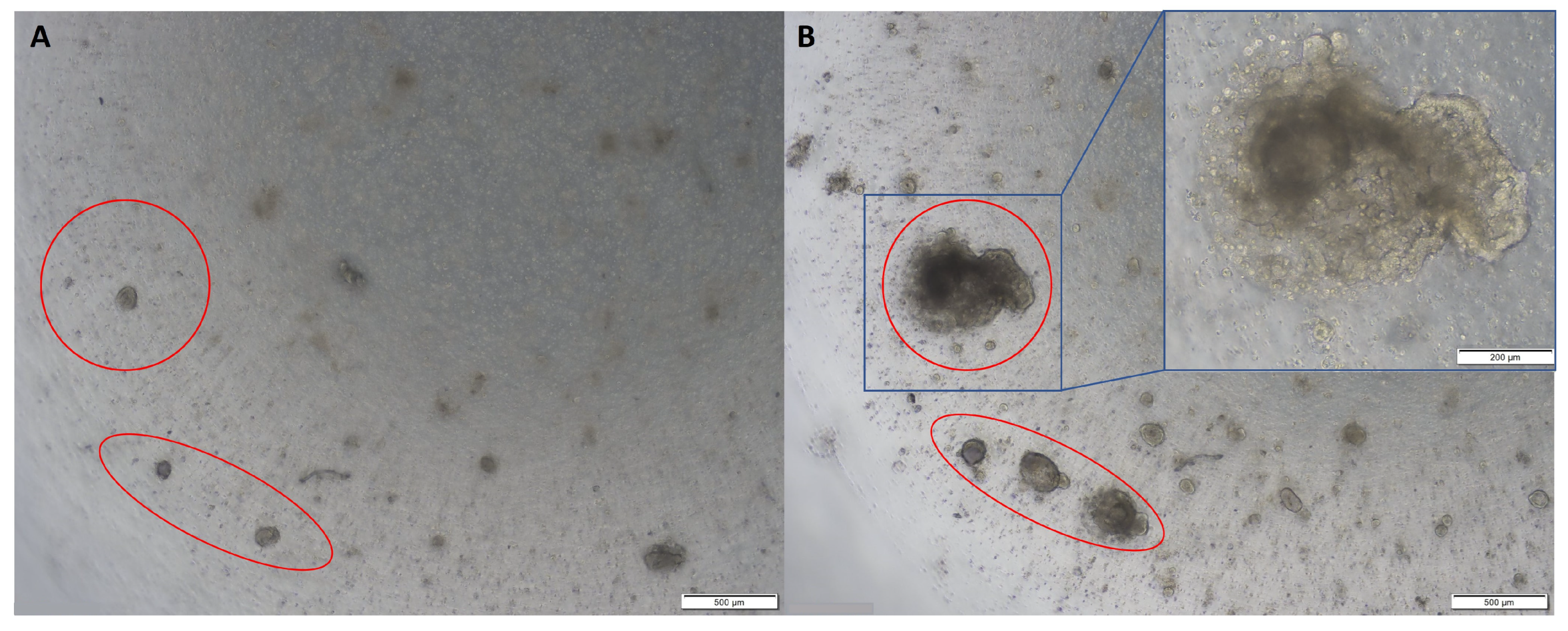
2.1. Establishing Normal and Tumour Organoids from Colon Tissues
2.2. Organoid Characterisation and Phenotype
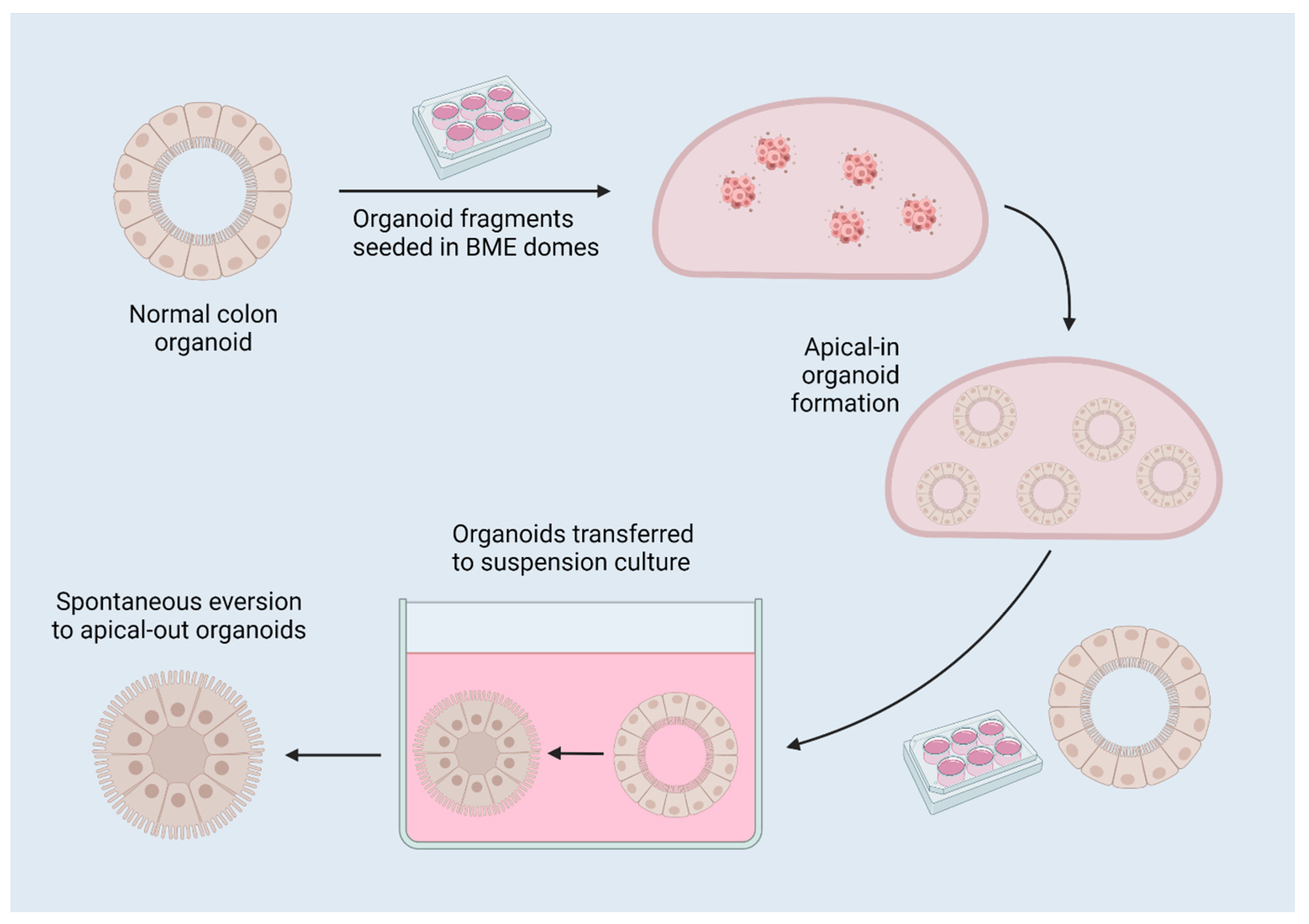
2.3. Reversed-Polarity Organoids
2.4. Cryopreservation and Recovery
3. Applications of Colon Organoids
3.1. Disease Modelling
3.2. Drug Screening
4. Experimental and Statistical Considerations
5. Organoid Libraries and Biobanks
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zheng, S.; Xin, L.; Liang, A.; Fu, Y. Cancer stem cell hypothesis: A brief summary and two proposals. Cytotechnology 2013, 65, 505–512. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Seaberg, R.M.; van der Kooy, D. Stem and progenitor cells: The premature desertion of rigorous definitions. Trends Neurosci. 2003, 26, 125–131. [Google Scholar] [CrossRef]
- Hayat, H.; Hayat, H.; Dwan, B.F.; Gudi, M.; Bishop, J.O.; Wang, P. A Concise Review: The Role of Stem Cells in Cancer Progression and Therapy. OncoTargets Ther. 2021, 14, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, E.; Silva-Vargas, V.; Doetsch, F. Eyes wide open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 2011, 8, 486–498. [Google Scholar] [CrossRef]
- De Angelis, M.L.; Francescangeli, F.; Nicolazzo, C.; Signore, M.; Giuliani, A.; Colace, L.; Boe, A.; Magri, V.; Baiocchi, M.; Ciardi, A.; et al. An organoid model of colorectal circulating tumor cells with stem cell features, hybrid EMT state and distinctive therapy response profile. J. Exp. Clin. Cancer Res. 2022, 41, 86. [Google Scholar] [CrossRef]
- Han, Y.; Duan, X.; Yang, L.; Nilsson-Payant, B.E.; Wang, P.; Duan, F.; Tang, X.; Yaron, T.M.; Zhang, T.; Uhl, S.; et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 2021, 589, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Miwa, H.; Kawachi, T.; Kume, S.; Takahashi, K. Generation of intestinal organoids derived from human pluripotent stem cells for drug testing. Sci. Rep. 2020, 10, 5989. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Sato, T. Somatic cell-derived organoids as prototypes of human epithelial tissues and diseases. Nat. Mater. 2021, 20, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Barbachano, A.; Fernandez-Barral, A.; Bustamante-Madrid, P.; Prieto, I.; Rodriguez-Salas, N.; Larriba, M.J.; Munoz, A. Organoids and Colorectal Cancer. Cancers 2021, 13, 2657. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett′s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Miyoshi, H.; Stappenbeck, T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013, 8, 2471–2482. [Google Scholar] [CrossRef]
- VanDussen, K.L.; Sonnek, N.M.; Stappenbeck, T.S. L-WRN conditioned medium for gastrointestinal epithelial stem cell culture shows replicable batch-to-batch activity levels across multiple research teams. Stem Cell Res. 2019, 37, 101430. [Google Scholar] [CrossRef]
- Boehnke, K.; Iversen, P.W.; Schumacher, D.; Lallena, M.J.; Haro, R.; Amat, J.; Haybaeck, J.; Liebs, S.; Lange, M.; Schafer, R.; et al. Assay Establishment and Validation of a High-Throughput Screening Platform for Three-Dimensional Patient-Derived Colon Cancer Organoid Cultures. J. Biomol. Screen 2016, 21, 931–941. [Google Scholar] [CrossRef]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef]
- Co, J.Y.; Margalef-Catala, M.; Li, X.; Mah, A.T.; Kuo, C.J.; Monack, D.M.; Amieva, M.R. Controlling Epithelial Polarity: A Human Enteroid Model for Host-Pathogen Interactions. Cell Rep. 2019, 26, 2509–2520.e4. [Google Scholar] [CrossRef]
- Co, J.Y.; Margalef-Catala, M.; Monack, D.M.; Amieva, M.R. Controlling the polarity of human gastrointestinal organoids to investigate epithelial biology and infectious diseases. Nat. Protoc. 2021, 16, 5171–5192. [Google Scholar] [CrossRef]
- Wilson, S.S.; Mayo, M.; Melim, T.; Knight, H.; Patnaude, L.; Wu, X.; Phillips, L.; Westmoreland, S.; Dunstan, R.; Fiebiger, E.; et al. Optimized Culture Conditions for Improved Growth and Functional Differentiation of Mouse and Human Colon Organoids. Front. Immunol. 2020, 11, 547102. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Puschhof, J.; Pleguezuelos-Manzano, C.; Martinez-Silgado, A.; Akkerman, N.; Saftien, A.; Boot, C.; de Waal, A.; Beumer, J.; Dutta, D.; Heo, I.; et al. Intestinal organoid cocultures with microbes. Nat. Protoc. 2021, 16, 4633–4649. [Google Scholar] [CrossRef]
- Bartfeld, S.; Clevers, H. Organoids as Model for Infectious Diseases: Culture of Human and Murine Stomach Organoids and Microinjection of Helicobacter Pylori. J. Vis. Exp. 2015, 105, e53359. [Google Scholar] [CrossRef]
- Han, S.H.; Shim, S.; Kim, M.J.; Shin, H.Y.; Jang, W.S.; Lee, S.J.; Jin, Y.W.; Lee, S.S.; Lee, S.B.; Park, S. Long-term culture-induced phenotypic difference and efficient cryopreservation of small intestinal organoids by treatment timing of Rho kinase inhibitor. World J. Gastroenterol. 2017, 23, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Wang, Y.; Chiang, I.L.; Ohara, T.E.; Fujii, S.; Cheng, J.; Muegge, B.D.; Ver Heul, A.; Han, N.D.; Lu, Q.; Xiong, S.; et al. Long-Term Culture Captures Injury-Repair Cycles of Colonic Stem Cells. Cell 2019, 179, 1144–1159.e15. [Google Scholar] [CrossRef]
- In, J.G.; Foulke-Abel, J.; Clarke, E.; Kovbasnjuk, O. Human Colonoid Monolayers to Study Interactions Between Pathogens, Commensals, and Host Intestinal Epithelium. J. Vis. Exp. 2019, 146, e59357. [Google Scholar] [CrossRef] [PubMed]
- Richiardone, E.; Van den Bossche, V.; Corbet, C. Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges. Organoids 2022, 1, 8. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.; Amato, F.; Braconi, C. Patient-Derived Organoids as a Model for Cancer Drug Discovery. Int. J. Mol. Sci. 2021, 22, 3483. [Google Scholar] [CrossRef] [PubMed]
- Toshimitsu, K.; Takano, A.; Fujii, M.; Togasaki, K.; Matano, M.; Takahashi, S.; Kanai, T.; Sato, T. Organoid screening reveals epigenetic vulnerabilities in human colorectal cancer. Nat. Chem. Biol. 2022, 18, 605–614. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Schnalzger, T.E.; de Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Roder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019, 38, e100928. [Google Scholar] [CrossRef]
- Betge, J.; Rindtorff, N.; Sauer, J.; Rauscher, B.; Dingert, C.; Gaitantzi, H.; Herweck, F.; Srour-Mhanna, K.; Miersch, T.; Valentini, E.; et al. The drug-induced phenotypic landscape of colorectal cancer organoids. Nat. Commun. 2022, 13, 3135. [Google Scholar] [CrossRef]
- Schurch, N.J.; Schofield, P.; Gierlinski, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef]
- Botti, G.; Di Bonito, M.; Cantile, M. Organoid biobanks as a new tool for pre-clinical validation of candidate drug efficacy and safety. Int. J. Physiol. Pathophysiol. Pharmacol. 2021, 13, 17–21. [Google Scholar] [PubMed]
- Mendy, M.; Caboux, E.; Lawlor, R.T.; Wright, J.; Wild, C.P. Common Minimum Technical Standards and Protocols for Biobanks Dedicated to Cancer Research; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Hallmans, G.; Vaught, J.B. Best practices for establishing a biobank. Methods Mol. Biol. 2011, 675, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Shabihkhani, M.; Lucey, G.M.; Wei, B.; Mareninov, S.; Lou, J.J.; Vinters, H.V.; Singer, E.J.; Cloughesy, T.F.; Yong, W.H. The procurement, storage, and quality assurance of frozen blood and tissue biospecimens in pathology, biorepository, and biobank settings. Clin. Biochem. 2014, 47, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Perrone, F.; Zilbauer, M. Biobanking of human gut organoids for translational research. Exp. Mol. Med. 2021, 53, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Engel, R.M.; Jarde, T.; Oliva, K.; Kerr, G.; Chan, W.H.; Hlavca, S.; Nickless, D.; Archer, S.K.; Yap, R.; Ranchod, P.; et al. Modeling colorectal cancer: A bio-resource of 50 patient-derived organoid lines. J. Gastroenterol. Hepatol. 2022, 37, 898–907. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Schutte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef]
- Sidorenkov, G.; Nagel, J.; Meijer, C.; Duker, J.J.; Groen, H.J.M.; Halmos, G.B.; Oonk, M.H.M.; Oostergo, R.J.; van der Vegt, B.; Witjes, M.J.H.; et al. The OncoLifeS data-biobank for oncology: A comprehensive repository of clinical data, biological samples, and the patient′s perspective. J. Transl. Med. 2019, 17, 374. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef]
- Kim, H.J.; Li, H.; Collins, J.J.; Ingber, D.E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. USA 2016, 113, E7–E15. [Google Scholar] [CrossRef]
- Kasendra, M.; Tovaglieri, A.; Sontheimer-Phelps, A.; Jalili-Firoozinezhad, S.; Bein, A.; Chalkiadaki, A.; Scholl, W.; Zhang, C.; Rickner, H.; Richmond, C.A.; et al. Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Sci. Rep. 2018, 8, 2871. [Google Scholar] [CrossRef] [PubMed]
- Pinho, D.; Santos, D.; Vila, A.; Carvalho, S. Establishment of Colorectal Cancer Organoids in Microfluidic-Based System. Micromachines 2021, 12, 497. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Normal Colon Organoid Media Formulations | Colon Tumour Organoid Media Formulations | ||||
|---|---|---|---|---|---|
| Advanced DMEM/F12 | 50% | 1× | 1× | 1× | 1× |
| L-WRN Conditioned Medium | 50% | - | - | - | - |
| Human recombinant Wnt3a | - | 100 ng/mL | - | - | - |
| Human recombinant R-spondin-1 | - | 1 ug/mL | - | - | - |
| Human recombinant Noggin | - | 100 ng/mL | - | 100 ng/mL | - |
| HEPES | 10 mM | 10 mM | 10 mM | 10 mM | 10 mM |
| Anti-Anti | 2× | 2× | 1× | 1× | 1× |
| GlutaMAX | 1× | 1× | 1× | 1× | 1× |
| B27 | 1× | 1× | - | 1× | 1× |
| N2 | - | - | - | - | 1× |
| EGF | 50 ng/mL | 50 ng/mL | 50 ng/mL | 50 ng/mL | 20 ng/mL |
| Gastrin | 10 nM | 10 nM | 10 nM | 10 nM | - |
| N-acetyl-L-cysteine | 1.25 mM | 1 mM | 1 mM | 1 mM | - |
| Nicotinamide | 10 mM | 10 mM | 10 mM | 10 mM | - |
| A83-01 (Alk4,5,7 inhibitor) | 500 nM | 500 nM | 500 nM | 500 nM | - |
| SB202190 (p38 inhibitor) | - | 10 uM | - | - | - |
| IGF-1 | 100 ng/mL | - | - | - | - |
| FGF-2 | 50 ng/mL | - | - | - | 10 ng/mL |
| Heparin | - | - | - | - | 4 ng/mL |
| Y-27632 (ROCKi)—first 48 h | 10 mM | 10 mM | 10 mM | 10 mM | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munro, M.J.; Tan, S.T.; Gray, C. Applications for Colon Organoid Models in Cancer Research. Organoids 2023, 2, 37-49. https://doi.org/10.3390/organoids2010003
Munro MJ, Tan ST, Gray C. Applications for Colon Organoid Models in Cancer Research. Organoids. 2023; 2(1):37-49. https://doi.org/10.3390/organoids2010003
Chicago/Turabian StyleMunro, Matthew J., Swee T. Tan, and Clint Gray. 2023. "Applications for Colon Organoid Models in Cancer Research" Organoids 2, no. 1: 37-49. https://doi.org/10.3390/organoids2010003
APA StyleMunro, M. J., Tan, S. T., & Gray, C. (2023). Applications for Colon Organoid Models in Cancer Research. Organoids, 2(1), 37-49. https://doi.org/10.3390/organoids2010003