Abstract
Hemophilia is an X-linked genetic disorder that predominantly affects males, with females typically serving as asymptomatic carriers. Hemophilia A results from a deficiency or dysfunction of coagulation factor VIII, while a deficiency in factor IX causes hemophilia B. A less common condition, factor XI deficiency (formerly hemophilia C), is categorized as a rare bleeding disorder. The severity of hemophilia is classified based on the activity concentration of factors VIII and IX: severe (<1 IU/dL), moderate (1–5 IU/dL), and mild (6–<40 IU/dL). One of the most prevalent complications of hemophilia is hemarthrosis, bleeding into joint cavities, which, if unrecognized or untreated, can lead to hemophilic arthropathy. The pathophysiology of hemophilic arthropathy involves two key mechanisms: the accumulation of iron from blood in synovial joints, which cannot be cleared due to repeated bleeding, and the inflammatory response, resulting in synovial hyperplasia and the progressive destruction of cartilage and bone. Hemophilic arthropathy significantly impairs quality of life, causing chronic pain, joint deformities, and sometimes requiring surgical intervention. This thesis will examine the pathophysiology and management strategies for hemophilic arthropathy.
1. Introduction
Hemophilia is an X-linked inherited bleeding disorder that predominantly affects males. One of the most debilitating long-term complications associated with this condition is hemophilic arthropathy, a progressive joint disease that significantly impairs quality of life [1]. Despite its clinical relevance, the available body of literature remains limited, with a relative scarcity of recent studies, notable heterogeneity in therapeutic approaches and patient presentations, and challenges inherent to researching a rare disorder. The primary objective of this review is to synthesize the current understanding of the key pathogenic mechanisms underlying hemophilic arthropathy, particularly the so-called “two-punch hypothesis”, and to explore emerging treatment strategies, with a particular focus on the potential of gene therapy [2].
2. Epidemiology
The global prevalence of hemophilia B is 1 in 30,000 male children, and that of hemophilia A is 1 in 5000 male children [3] or approximately 1 in 10,000 newborns [4]. Studies in certain countries, such as the Netherlands, have marked a significant increase in the median life expectancy of patients with hemophilia (PWH) to 77 years, which is 6 years shorter than the general male population [1]. Research performed in Sweden and the United Kingdom has similarly marked a rise in the median survival rate for patients with moderate and severe forms of the disease, attributing this to improved access alongside a global increase in the general male life expectancy [5]. Currently available data from 106 countries clearly show the prevalence of hemophilia A and B. Hemophilia A has a prevalence of 12.8 per 100,000 men in developed countries and 6.6 per 100,000 men in developing countries. Hemophilia B has a prevalence of 2.7 per 100,000 men in developed countries and 1.2 per 100,000 men in developing countries [4]. Hemophilic arthropathy is one of the most common symptoms of hemophilia. Arthropathy develops due to frequent bleeding into the joint cavity and manifests with pain in the joints, a sensation of tightness, and joint swelling. If left untreated, it can lead to significant joint destruction, disability, and reduced quality of life. While previously thought to occur only in individuals with severe or moderate hemophilia, it is now recognized that it can develop in all forms of the condition [4].
3. Causes
Hemophilic arthropathy presents various clinical pictures among patients with an identical coagulation factor concentration and prophylactic regimens. Multiple factors cause this heterogeneity in clinical outcomes [6].
3.1. Genetic Factors
One of the primary factors that influence the frequency of hemarthrosis and the development of hemophilic arthropathy is the plasma concentration of coagulation factors VIII and IX, upon which the basis of hemophilia classification is formed: severe (<1 IU/dL), moderate (1–5 IU/dL), and mild (6–<40 IU/dL) [1]. Genetic factors, such as thrombophilia and genetic polymorphisms, are associated with an increased expression of inflammatory cytokines, which may contribute to developing hemarthrosis [6].
3.2. Local Factors
The frequency of spontaneous hemarthrosis depends on several local factors such as the vascularization of the synovial membrane, mechanical stress upon the joints caused by body weight, and specific regulations of hemostasis in the joints. The synovial membrane of the joints in patients with hemophilic arthropathy is hypertrophied and exhibits reduced concentrations of tissue factor (TF) and an elevated concentration of tissue factor pathway inhibitors. Stimulated by hemarthrosis, synovial cells increase the expression of the urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitor-1 (PAI-1), leading to a higher plasmin concentration in the synovial fluid. Additionally, elevated concentrations of thrombomodulin have been detected in the synovial fluid of hemophilia patients. This local environment promotes early thrombus degradation and an increase in fibrinolytic activity. With recurrent bleeding, an elevated concentration of uPA enhances chemotaxis, angiogenesis, the proliferation of synovial cells, and damage to joint cartilage and bone [6].
3.3. Environmental Factors
Some of the environmental factors potentially influencing the clinical presentation of hemophilic arthropathy are BMI, lifestyle, and frequency of hemarthrosis, as well as usage, duration, and protocol of prophylactic therapy. Currently, there is no specifically defined goal value for coagulation factors needed to prevent joint injury; however, every 1% increase in the activity of coagulation factors above 15% lowers the chance of recurrent hemarthrosis by 18% [6]. Patients who enjoy greater physical activity have a greater chance of developing arthropathy. Additionally, trauma, impaired joint biomechanics, and increased body mass exert unfavorable stress on the joints and further promote the development of arthropathy.
4. Coagulation Cascade
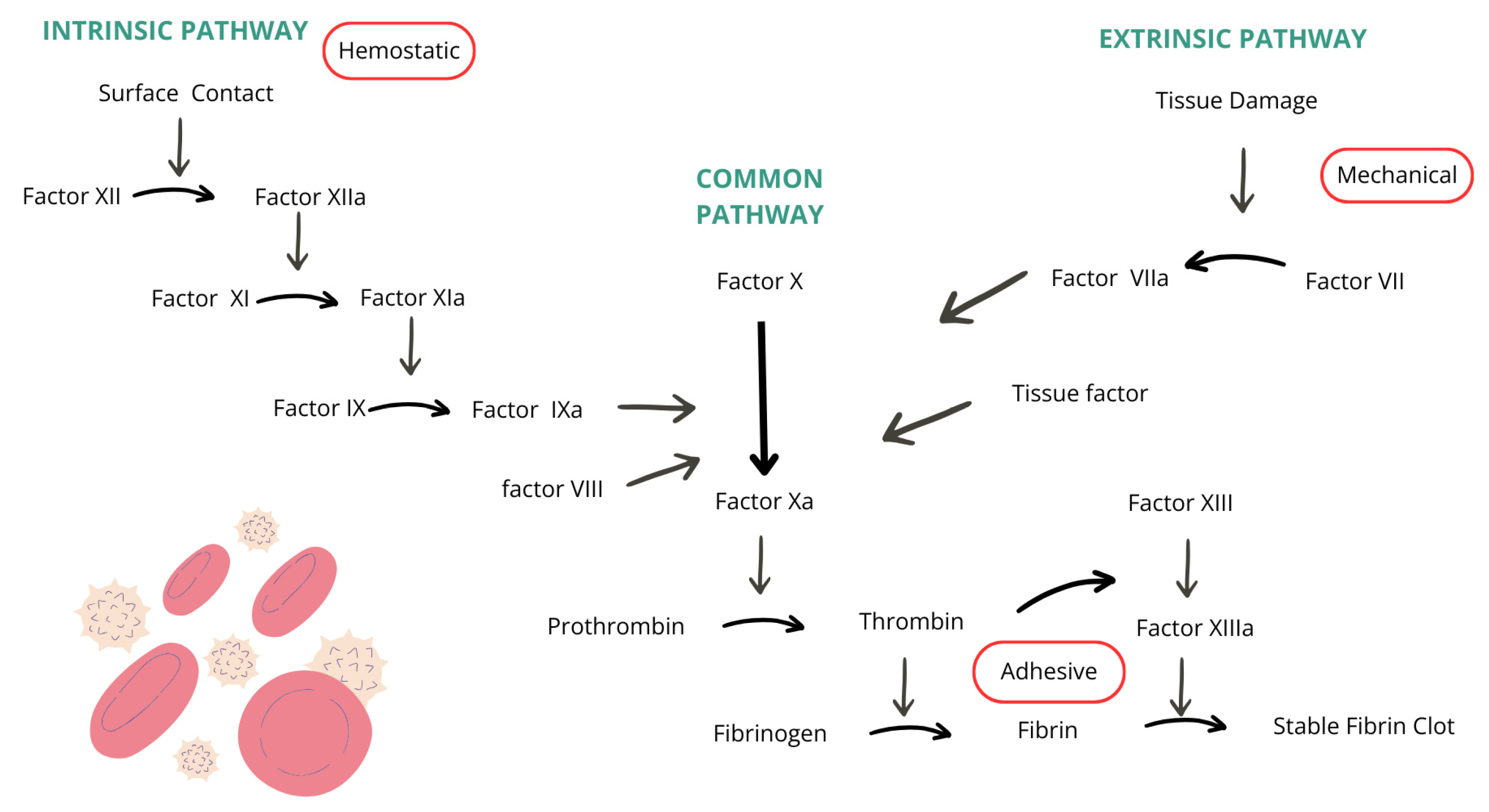
The function of the coagulation cascade is to create a prothrombin activator which activates prothrombin molecules in the presence of calcium ions by changing them to thrombin. Thrombin has an enzymatic function of converting fibrinogen into fibrin strands, essential components in thrombus formation. Blood coagulation is divided into two pathways: extrinsic and intrinsic. The extrinsic pathway begins when blood encounters damaged blood vessels or tissues. Tissue factor, phospholipids, and a lipoprotein complex with proteolytic activity are released from the injured tissue. The binding of lipoprotein complex with coagulation factor VII, in the presence of calcium ions, leads to the conversion of factor X into its active form, factor Xa. Factor Xa, combined with phospholipids of the tissue factor or platelets and with factor V, forms a prothrombin activator. Xa functions as a protease and causes the breakdown of prothrombin into thrombin, and active factor V, known as Va, accelerates the process and amplifies prothrombin production through positive feedback, resulting in an increasing thrombin concentration. Factors II, VII, IX, and X are vitamin K-dependent and are synthesized in the liver. The synthesization of prothrombin and fibrinogen also takes place in the liver, which is why liver diseases can interfere with the blood coagulation process [7]. The intrinsic pathway begins with the injury of the blood, specifically the exposure of blood components to the collagen in the damaged blood vessel wall. Coagulation factor XII encounters collagen and is converted into its active form, factor XIIa, which functions as a proteolytic enzyme. At the same time, platelets in contact with the collagen begin releasing phospholipids which contain factor 3. Factor XIIa, in the presence of high-molecular-weight kininogen and prekallikrein, activates factor XI into its active form, factor XIa. Factor XIa activates factors IX and X alongside VIIIa, platelet phospholipids, and platelet factor 3. Factor X combines with factor V and platelet or tissue phospholipids to form a prothrombin activator. A deficiency in factors IX or VIII, both critical components of the coagulation cascade and essential for clot formation, leads to hemophilia. The coagulation cascade described in the text is illustrated in Figure 1, where arrows represent the activation pathways of various coagulation factors [8].
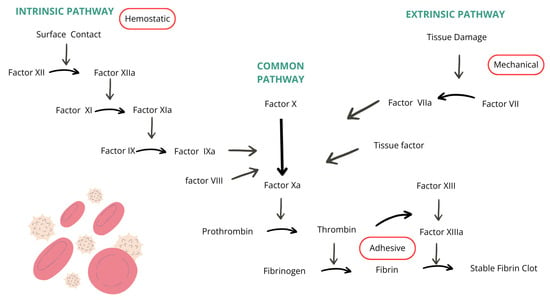
Figure 1.
Coagulation cascade [8].
Development of Inhibitors Against Coagulation Factors
Inhibitors are mononuclear antibody plasma cells produced as an immune response to replacement coagulation factors. They occur in a third of patients on replacement therapy, most commonly between 20 and 30 days from the beginning of treatment [9]. There are several mechanisms through which these inhibitors disrupt coagulation: blocking factor VIII from binding to phospholipids or von Willebrand factor (VWF) [10], as well as blocking it from binding to factor IX [11], increasing the degradation and clearance of factor VIII with their proteolytic activity [12]. The exact reasons for inhibitor development are not entirely clear; however, it is presumed they are multifactorial and dependent on patient-specific factors: genetic mutation predisposing to antibody production, the severity of hemophilia, and other genetic regulatory mechanisms. They are also dependent on treatment-related factors such as the use of plasma-derived factors, age at first exposure to therapy, dosage, and type of prophylaxis. Factors such as trauma and surgery may also lead to the development of inhibitors and thus lower the effectiveness of the treatment. The presence of inhibitors significantly impairs patients’ quality of life, worsens clinical outcomes, complicates treatment, and reduces life expectancy. [13]
5. Pathophysiology
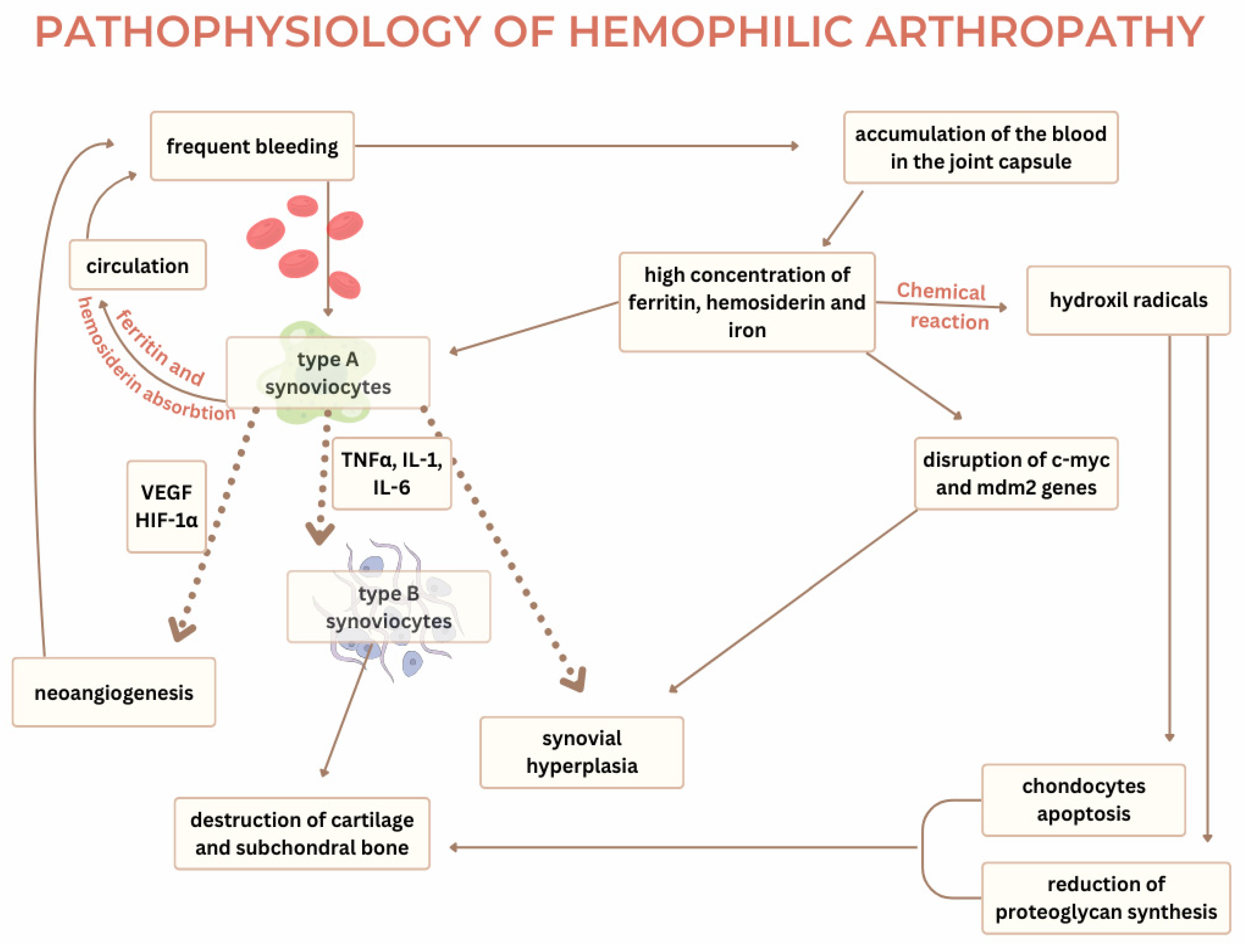
Current knowledge on the pathophysiology of hemophilic arthropathy explains two mechanisms required for developing the disease—the two-punch hypothesis. The first punch in this hypothesis appears due to an iron-induced chemical reaction. After an episode of hemarthrosis, hemosiderin from the blood promotes a chemical reaction in the synovial membrane, leading to inflammation or the second punch. The pathophysiological process of hemophilic arthropathy shares common ground with degenerative diseases such as osteoarthritis and inflammatory diseases of the joints such as rheumatoid arthritis [2]. Hemophilic arthropathy most commonly occurs in the synovial joints—ankle, knee, and elbow. The synovial joint consists of bone endings covered in cartilage that are connected with ligaments and a joint capsule. The synovial tissue is spread on the inner side of the capsule and produces synovial liquid which functions as a lubricant, reducing the friction between two joint bodies. It is yellowish in color, transparent, and viscous with a composition like blood plasma but lacking fibrinogen. The synovial membrane consists of the intima layer and the subintima layer. The intima layer contains type A synoviocytes and type B synoviocytes. Type A resembles macrophages and monocytes. Type B resembles fibrocytes and is responsible for synthesizing extracellular matrix protein and nutrients for chondrocytes [14]. The subintimal synovia consists of fibrous and fatty tissue and is highly vascularized with blood and lymphatic vessels. The synovial tissue’s reaction to blood in the joint capsule is considered a key factor in the development of hemophilic arthropathy [2]. In individuals without hemophilia, type A synoviocytes absorb hemosiderin and ferritin after bleeding in the joint cavity and recycle them back into circulation. However, due to frequent bleeding, the constantly elevated hemosiderin and ferritin in the synovial fluid exceed the capacity of the synoviocytes to absorb them and result in the accumulation of blood in the joint capsule [6]. The propensity for bleeding often stems from mechanical injuries and, depending on the severity of hemophilia and the nature of the injury, micro- and macro-bleeding occur. Low expression of tissue factors in the joint capsule, deficiency in coagulation factors, and increased fibrinolysis lead to the inadequate formation of thrombi, their accelerated degradation, and frequent hemarthrosis [14]. Current studies suggest that excess iron disrupts the regulation of key genes such as c-myc and mdm2, which are crucial in the proliferation of synoviocytes [15]. These genes promote synovial hypertrophy. C-myc is a proto-oncogene responsible for cell proliferation, while mdm2, in conjunction with p53, has a regulatory role in apoptosis of the cells [14]. The influence of hemosiderin on the dysregulated genes leads to the uncontrolled proliferation of the synovial membrane, its hypertrophy, and the development of villous projections. Concentrations of iron metabolites in the nuclei and cytoplasm of synovial cells have been recorded as higher in patients with hemophilic arthropathy compared to patients with rheumatoid arthritis and osteoarthritis. The chemical reactions of hemosiderin with synoviocytes increase the concentration of hydroxyl radicals, which triggers the apoptosis of the chondrocytes and reduces the synthesis of proteoglycans. The presence of hemosiderin and ferritin supports the excretion of inflammatory cytokines (TNF-alpha, IL-6, IL-1-beta) from type A synoviocytes, leading to inflammation, activation of type B synoviocytes, and synovial hyperplasia. Neongiogenesis caused by inflammation is one of the mechanisms which promotes the development of hemarthrosis. The inflammatory response within the hypertrophied synovial membrane leads to hypoxia [2] and, in the presence of inflammatory cytokines, leads to the production of hypoxia-inducible factor 1α (HIF-1α), which stimulates the production of VEGF, SDF-1, and MMP-9 [14]. VEGF binds to its receptors, VEGFR-1 and VEGFR-2, on the pannus and promotes neoangiogenesis [6]. The newly formed blood vessels are fragile and prone to rupture, perpetuating the cycle of bleeding, hypoxia, and angiogenesis. Elevated VEGF-A concentrations have been found in the blood of hemophilic patients, signaling their role in synovial proliferation [15]. Cytokines and proangiogenic molecules are the cause of permanent changes in the synovium, which leads to chronic inflammation [2]. Synovitis manifests with tissue hypertrophy, neoangiogenesis, and migration of inflammatory cells in the affected area. The inflammatory cytokines also activate type A synoviocytes in the synovium, which in turn activate tissue proteinases (matrix metalloproteinases). Alongside tPA and other matrix components, matrix metalloproteinases contribute to cartilage and subchondral bone destruction. IL-1β, secreted by macrophages, prompts chondrocytes to produce hydrogen peroxide, forming hydroxyl radicals, further enhancing chondrocyte apoptosis and degradation of the extracellular matrix [15]. New research suggests that synovitis is not necessary for the development of cartilage destruction. The cartilage of the joint consists of chondrocytes and their products and the extracellular matrix (type II collagen and proteoglycans). With age, the number of chondrocytes decreases and proliferation becomes rare, making chondrocyte apoptosis significantly disrupt the balance between the synthesis and degradation of the cartilage matrix. The damaged cartilage loses its mechanical properties.
The direct influence of hemosiderin from the blood on cartilage stimulates the release of inflammatory cytokines IL-1 and TNF-alpha, which are responsible for producing hydroxyl radicals with apoptotic effects on chondrocytes. Bleeding increases local plasmin production within the joint while acting as a catalyst on the cartilage and degrading fibrin, which complicates thrombus formation and further damages the cartilage [14]. The exposure of joint cartilage to blood for more than 4 days reduces the synthesis of proteoglycans by 50% essential structure parts of the joint cartilage [15]. Interleukin 1 and factor tumor necrosis α are considered primary cytokines of this pathophysiological process. As described in the text, Figure 2 depicts the underlying mechanisms of hemophilic arthropathy, with dotted arrows highlighting cytokine-mediated processes.
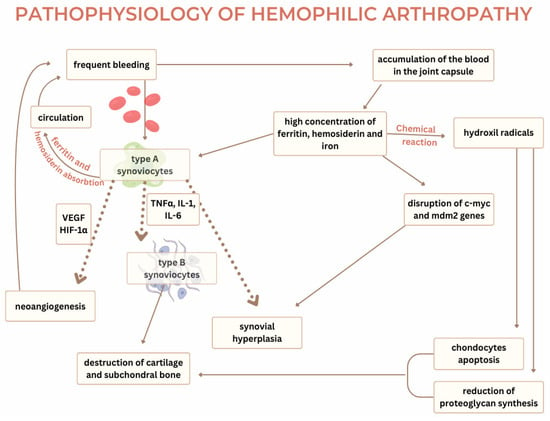
Figure 2.
The pathophysiology of hemophilic arthropathy [2,14,15].
5.1. Interleukin 1
IL-1 is one of the primary regulators of the inflammatory response. It has a catabolic effect on cells and amplifies the inflammatory response. Upon binding to its receptor, IL-1 triggers the NF-κB transcription factor, leading to the increased expression of genes responsible for producing inflammatory mediators (cytokines and chemokines), enzymes, and adhesion molecules. IL-1β enhances the absorption of hemosiderin into type B synoviocytes, causing hemosiderin to accumulate in the synovium. Through positive feedback, hemosiderin further stimulates the release of IL-1β, contributing to chronic inflammation of the synovial membrane. Histopathological analyses of the synovium in patients with hemophilia have shown higher concentrations of IL-1β compared to samples from individuals without hemophilia [2].
5.2. Factor Tumor Necrosis α (TNFα)
TNFα is a member of the tumor necrosis factor superfamily and one of the key factors in the development of hemophilic arthropathy. TNFα inhibits the synthesis of proteoglycans and type II collagen, which are essential components of the synovial membrane. It promotes the activation of metalloproteinases, which exert a catabolic effect on the synovium. Due to its influences on the synthesis of thrombomodulin in synoviocytes, TNFα facilitates joint bleeding. When thrombomodulin binds to thrombin, it activates protein C, which inhibits coagulation by degrading factors Va and VIIIa. As a mediator of inflammation, TNFα increases the expression of thrombomodulin in synovial cells, contributing to hemarthrosis formation [2]. Apart from chemical and inflammatory processes, studies have confirmed that degenerative changes also occur in joints affected by hemophilic arthropathy. Imaging techniques have revealed cystic changes in bones, subchondral sclerosis, osteophytes, epiphyseal expansion, and osteoporosis. It remains unclear whether these degenerative changes develop independently or as a consequence of cartilage and synovial damage.
In hemophilic patients, intraosseous microbleeds are more frequent, contributing to degenerative bone changes in the joints. Some changes, such as cystic ones, lack a clear origin. Osteoporosis in hemophilic arthropathy is believed to be a result of muscle atrophy, reduced mobility due to joint deformities and pain, lower BMI in hemophilic individuals, and frequent viral infections associated with coagulation factor transfusions [14].
Bone formation and resorption are regulated by the triad of OPG, RANK, and RANKL. The RANK ligand is a transmembrane protein expressed on osteoblasts and stromal cells of the bone. It is produced by lymphocytes and synovial cells while the inflammatory mediators stimulate its production, which promotes osteoclastogenesis or the reabsorption of the bone. RANKL, by binding to RANK, which is found on the osteoclast precursors, leads to the differentiation and maturation of osteoclast precursors into osteoclasts, which are responsible for bone resorption. OPG is a receptor that competes with RANK for RANKL binding. Its role is to inhibit bone resorption by preventing the differentiation of osteoclast precursors into mature osteoclasts. This triad of OPG/RANK/RANKL is responsible for bone remodeling, and deficiency in OPG in the synovial cells of patients with hemophilia with a greater expression of RANK and RANKL leads to disrupted homeostasis and promotes degeneration of the bones and the development of hemophilic arthropathy. Lower concentrations of OPG in serum and histological samples have been recorded in individuals with hemophilia A compared to those with hemophilia B. It is suggested that this may explain why individuals with hemophilia A, under similar conditions as those with hemophilia B, exhibit a more severe clinical presentation of arthropathy. Persons suffering from hemophilia A have lower concentrations of OPG in their serum and histological samples in comparison to people suffering from hemophilia B. This is assumed as the reason why hemophilia A patients, who share the same characteristics with hemophilia B patients, have more severe clinical presentations [15]. Currently, there is no clear evidence of why a difference in clinical presentation exists in patients with the same type and severity of the disease. There is a hypothesis that mechanical forces combined with weight-bearing stress promote bleeding, which may damage the cartilage and the development of arthropathy. Some studies have shown that persons with the HLA B27 phenotype alongside mutations in genes for pro- and anti-inflammatory cytokines, antifibrinolytic proteins, and heme oxygenase-1 are predisposed to more frequent bleeding episodes. Mutations in HFE, which affect hepcidin, have been linked to degenerative diseases in the general population and hemophilia patients. Some hypotheses suggest that the primary regulators for synovitis development differ from those causing bone and cartilage degeneration. TNF is the primary factor for the development of synovitis, while IL-1β is critical for cartilage damage. Thus, the presence of specific cytokines appears to drive distinct pathways in the progression of arthropathy. The amount of blood in the joint is a significant factor in arthropathy development; studies have demonstrated that one episode of severe hemarthrosis causes greater cartilage damage than frequent microbleeds [14].
5.3. Hemophilic Arthropathy—Clinical Presentation
The risk for bleeding in hemophilia patients depends primarily on the lack of factors for coagulations and the patient’s age, not on the type of hemophilia. In newborns, 1–4% of those who have a severe form of hemophilia (<1 IU/dL factors) will develop intracranial hematoma, which a cephalhematoma and a subgaleal hemorrhage may accompany. Most extracranial and intracranial bleeding in newborns happens during traumatic deliveries, e.g., the use of forceps or vacuum extraction. These may lead to complications such as neurological damage, developmental delays, or even death. Additionally, individuals with hemophilia, especially severe forms, may suffer excessive bleeding after surgeries, and frequent spontaneous bleeding in the lower extremities, gluteal, and iliopsoas muscle. Most infants between 6 and 8 months of age develop hematomas or spontaneous bleeding as they become more physically active [3]. Patients suffering from hemophilia have more frequent gastrointestinal bleeding, dental bleeding, and bleeding in organs such as a kidney or retina, which may lead to significant organ damage and require complicated treatment and cooperation with other specialists [16]. Hemarthrosis is bleeding in the joint, which happens spontaneously and frequently in hemophilia A and B patients. The most common location of the bleeding is the ankle, but the elbow and knee follow closely. The bleeding more often affects greater joints and those carrying greater weights [3]. Such joints also more often present with synovitis and hemophilic arthropathy which, if left untreated, may lead to joint deformity, muscle atrophy, and a reduced quality of life. Acute joint bleeding manifests with a subjective feeling of “an aura” or tingling in the joint, rapid swelling due to effusion, loss of range of motion, pain, and a feeling of warmth over the swollen joint. Patients often keep their joints flexed to reduce intra-articular pressure and alleviate pain. With the progression of the disease, the range of motion reduces even more, and the affected joints remain in a slightly flexed position. As a result of joint deformity, surrounding muscle atrophy, the ligaments, and the joint capsule become stretched, which leads to joint instability [17].
6. Diagnostics
6.1. X-Ray
One of the methods which may visualize the injured joint is the X-ray. In the beginning phase of the disease, an edematous soft tissue of the joint can be seen, while the progression of the disease presents with the depletion of the cartilage and bone damage. The method also shows joint space narrowing, subchondral cysts, bone erosions, and bone deformities [6].
Based on the X-ray findings, there are five phases of hemophilic arthropathy [18].
Stage One (soft tissue edema)—bleeding in the joint capsule and surrounding tissue leads to edema.
Stage Two (osteoporosis)—development of osteoporosis or synovial hyperplasia.
Stage Three (bone lesions)—an overgrowth of the epiphysis with widening of the joint notch and loss of joint congruence, while the cartilage remains intact.
Stage Four (cartilage destruction)—narrowing of the joint space due to cartilage damage.
Stage Five (joint destruction)—complete loss of joint space and development of bone erosion.
The World Hemophilia Federation suggests using Pettersson’s classification to determine the severity of hemophilic arthropathy on the X-ray findings. The classification is scored from 0 to 13 and includes the presence of subchondral cysts, joint space narrowing, irregular subchondral surfaces, osteoporosis, enlargement of epiphyses, erosions of joint body edges, joint body incongruence, and joint deformity. A higher score on the scale indicates a more severe form of hemophilic arthropathy [19].
6.2. Ultrasound
Ultrasonography is a method that, without radiation, can quickly and affordably visualize the anatomy of the joint. It permits the visualization of joint pathologies in the early stages of the disease while there is still no edema or pain present. This method is useful for identifying joint abnormalities in patients with hemophilic arthropathy. Ultrasound can confirm acute hemarthrosis and be used to follow the development of chronic disease. Its downside is the inability to visualize synovial hyperplasia and blood clots well enough, so MRI is a better diagnostic choice for such changes. Additionally, ultrasound is a subjective method, meaning its reading greatly depends on the reader and their abilities, knowledge of the technique, and joint pathologies. On the other hand, the advantage of this method is its wide applicability in children’s diagnostics since it does not require sedation or the use of a contrast. It is also affordable, can be repeated, is mobile, and is quicker than an MRI. The Power Doppler permits the visualization of joint vascularization and differentiation between a hemorrhage and other types of effusions in the joint [20]. The fact that children’s joints are anatomically different from adults’ is a possible obstacle for this diagnostic method since, currently, there are not enough ultrasound atlases covering the pathologies of children’s joints, which makes ultrasound a non-ideal diagnostic tool for children [6]. Another disadvantage is its inability to visualize joint anatomy when a massive effusion is present due to the inability of the sound waves to penetrate deep enough [20].
6.3. Magnetic Resonance Imaging
Magnetic Resonance Imaging (MRI) is considered a gold standard in visualizing joint pathologies. It enables early visualization of the damaged cartilage, edematous tissue, hemosiderin deposits, bone cysts, osteopenia, and joint effusion [20]. The negative side is the difficulty of differentiating the synovial fluid from blood collection in the joint capsule and other effusions. Additionally, MRI is not a widely available method, it has a higher cost, it is time-consuming, it can only capture one joint at a time, it involves the use of contrast to determine effusion etiology, and sometimes it demands sedation (e.g., in children) [6].
6.4. Laboratory Findings
When hemophilia is suspected, blood work should be performed to check for activated partial thromboplastin time (APTT), normal prothrombin time (PT), and platelet count. The findings most often show elongated APTT. Additionally, the concentration of coagulation factors VIII and IX should be measured. The coagulation factor titer can determine the type and severity of hemophilia [3].
6.5. Biomarkers
Biomarkers are molecules that serve as a tool for diagnosing and supervisioning disease. Due to the development of hemophilic arthropathy in individuals receiving prophylaxis, the goal is to identify highly specific biomarkers, correlate with disease severity, and indicate disease prediction and treatment outcomes. Currently, there is no ideal biomarker, but based on the pathophysiology of hemophilic arthropathy, those involved in cartilage and bone remodeling are being investigated. Some biomarkers that correlate with the pathology observed on radiographic images include CTX-II, COMP, and CS-846. These biomarkers are persistently present in individuals with hemophilia but also experience an acute, reversible increase in concentration during hemarthrosis. In addition to the previously mentioned markers, elevated concentrations of VEGF have been observed in the blood of individuals who have developed hemophilic arthropathy and/or synovitis. Table 1 presents the classification of potential biomarkers in hemophilic arthropathy. The table includes a selection of biomarkers, outlining their respective advantages, disadvantages, and limitations. This overview provides insight into the clinical relevance, diagnostic potential, and current challenges associated with each biomarker. [20].

Table 1.
Classification of potential biomarkers in hemophilic arthropathy [20].
6.6. Prophylaxis
The World Hemophilia Federation revised the terminology for prophylaxis in hemophilia patients in 2012. Since then, prophylaxis has been categorized as episodic, continuous (which may be primary, secondary, and tertiary), and intermittent. Studies have shown that receiving prophylactic therapy before the age of 4 helps preserve joint mobility [21].
- Episodic prophylaxis
A type of prophylaxis in which coagulation factors are received when bleeding is present.
- 2.
- Continuous prophylaxis
A type of prophylaxis received 85% of the year, at least 45 out of 52 weeks.
- Primary—received before the age of 3 and before the second major bleeding in the joint (hemarthrosis).
- Secondary—received after two or more episodes of hemarthrosis and before the development of hemophilic arthropathy.
- Tertiary—received after hemophilic arthropathy is already developed with an idea of stopping further progression of the disease.
- 3.
- Intermittent prophylaxis
A type of prophylaxis is received for only a short period to replace the role of coagulation factors before and after surgery [22].
7. Treatment
7.1. Conservative Treatment
7.1.1. The RICE Protocol
The RICE protocol represents four actions in the treatment of the joints: rest, cooling, pressure, and elevation of the injured joint (Rest, Ice, Compression, Elevation) [23]. The therapy aims to prevent further bleeding, pain, and inflammation. Alongside the RICE protocol, some cases require the POLICE protocol, in which P stands for Protection and OL for Optimal Loading. Different approaches depend on the patient’s state and the disease’s development [16]. Cooling the joint has a vasoconstricting effect, which will lessen swelling and inflammation but might cause frostbite, so the recommended maximum for cooling is 10 min every 2 h.
7.1.2. Intermittent Prophylaxis
This type of prophylaxis is received in the case of acute bleeding. In the case of hemarthrosis, the recommendation stands for VIII to be received three to four times weekly and for factor IX two times weekly to prevent further bleeding and joint injury [24].
7.1.3. Nonsteroidal Drugs
Nonsteroidal drugs such as cyclooxygenase inhibitor 2 have shown themselves to be useful in treating arthropathic pain in hemarthrosis. This group of drugs is effective due to their anti-inflammatory, analgesic, and anti-angiogenic effects, which reduce the likelihood of recurrent bleeding compared to other nonsteroidal drugs that have anti-aggregatory effects, increasing the risk of recurrent bleeding [25].
7.1.4. Desmopressin
Desmopressin is a synthetic analog of vasopressin, an antidiuretic hormone, which has wide use as a medication and is used in treating hemophilia A [26]. Its use is recommended for achieving hemostasis in patients with moderate hemophilia A. Desmopressin promotes the synthesis of factor VIII in the organism. Besides its antidiuretic effect, desmopressin binds to V2 receptors on the endothelium and triggers the exocytosis of Weibel–Palade bodies and alpha granules from platelets through cyclic adenosine monophosphate (cAMP), thus leading to the release of von Willebrand factor and tissue plasminogen activator (tPA) stored in Weibel–Palade bodies with the presumed simultaneous release of FVIII [27]. The von Willebrand factor is responsible for activating the cascade by linking to the Gp1b receptors on platelets and subendothelial collagen of the injured tissue [23]. Additionally, due to an increased FVIII concentration, activated partial thromboplastin time (APTT) is reduced. Administration is possible parenterally, subcutaneously, and intranasally. Regular laboratory monitoring of the drug concentration in the blood is required [27]. Desmopressin is recommended for use in persons with moderate hemophilia A and acute bleeding episodes. In the case of a massive bleed, desmopressin is not indicated due to the time required to achieve effective blood concentrations; instead, in such scenarios, concentrates of FVIII are recommended [28].
7.2. Replacement Therapy for Coagulation Factors
7.2.1. Plasma-Derived Factor VIII Concentrates
Plasma-derived factor VIII concentrates are used in treating hemophilia A. Factor is filtered from the donor’s plasma and categorized based on the concentration of purity. There are three types of factor VIII concentrates: the intermediate-purity factor (6–10 units of FVIII/mg protein), high-purity factor (50–150 units of FVIII/mg protein), and ultra-high-purity factor (3000 units of FVIII/mg protein) [29]. Some patients develop antibodies against FVIII, rendering the therapy ineffective [23]. Factor VIII concentrate is administered intravenously and has a half-life of approximately 12 h, which necessitates therapy up to three times a week. With current technology and viral inactivation methods, these products are safe to use without risk of infection transmission [29].
7.2.2. Recombinant Human Factor VIII
Recombinant factor VIII includes all generations of FVIII produced through the genetic modification process. It is categorized into four generations, depending on the cells from which it derives, exposure to human or animal proteins, and potential sources of infections. The genetic modification process allows for the deletion of the B structure of FVIII or modification in a one-chain molecule. These genetically modified factors have a longer half-life and coincidently a lower frequency of admittance. They are safe to use and also lower the number of bleeding episodes [30].
7.2.3. Extended Half-Life Recombinant Factor VIII
Efanesoctocog Alfa
Efanesoctocog Alfa is a molecule that contains recombinant factor VIII. Its stability and extended half-life are based on its connection to vWF. Previous studies have not demonstrated the development of antibodies against FVIII [31].
Fusion Factor VIII-Fc
Fusion factor VIII-Fc is a recombinant protein used to treat hemophilia A. The B domain of factor VIII is removed through genetic modification and is replaced by the Fc domain of human immunoglobulin G. The ability of the fusion protein to link with vWF and phospholipids as a part of the coagulation cascade is secured thanks to the structural and functional preservation of the FVIII domain. Factor VIII has a half-life of approximately 12 h, which demands the frequent administration of the factor, mostly three times a week, to keep the plasma concentration of FVIII above 1%. The Fc component prolongs the half-life by 1.5 times that of the recombinant protein FVIII. Longer half-life promotes less frequent application, such as 1 to 2 times per week. Alongside the Fc domain, an adequate amount of vWF is required, as demonstrated by studies showing that with patients who had lower concentrations of vWF, the factor half-life was shorter than in those with a higher vWF concentration [32].
PEGylated Factor VIII
PEGylated factor VIII is one of the drugs used in the therapy of hemophilia A. PEGylated molecules are molecules linked by polyethylene glycol. The PEGylation process enhances bioavailability, reduces the drug’s molecular mass, and increases the amount of protein. In this case, factor VIII can be administered in a single dose [33]. The PEGylated form of the factor has a half-life of 1.4 to 1.5% longer than recombinant FVIII and is administered intravenously. The preferred administration for this drug is as an intermittent prophylaxis, as demonstrated by research showing that 96.1% of participants had excellent results (complete absence of recurring bleeding episodes) if given 1 to 2 times per week during acute bleeding episodes [34].
7.2.4. Plasma-Derived Factor IX Derivatives
Plasma-derived factor IX concentrates are used in the therapy of hemophilia B. Factor is produced for intravenous administration. A monoclonal product is a purified monoclonal antibody with a high concentration of factor IX. It does not contain other vitamin K-dependent components, making its use associated with a lower risk of thromboembolic events. It is administered intravenously as continuous prophylaxis and has a half-life of 22 h. Current studies have not shown the development of antibodies against this medication [7].
7.2.5. Recombinant Factor IX
Recombinant factor IX is a molecule produced through genetic modification. It is safe to use in patients who have already received FIX therapy and those who have not received any. Administration of the drug is intravenous, and its predicted half-life is 16 to 17 h. Studies have shown a lower incidence of bleeding by 79% in patients who received this therapy than in those who did not receive any prophylaxis [35].
7.2.6. Extended Half-Life Recombinant Factor IX
Long-acting factors are genetically modified to extend their half-life and thus reduce the frequency of administration to every 2 to 4 weeks. By linking the Fc component to factor IX, the half-life of FIX has been extended from 18 to 90 h. The medication is designed for weekly intravenous administration and is safe for use in both children and adults [36]. It has a half-life of 102 h and can be used for continuous or perioperative prophylaxis [37]. Long-acting factors are safe for use, reduce the incidence of spontaneous bleeding, and decrease the frequency of administration, positively impacting patients’ quality of life.
7.3. New Medications
7.3.1. Emicizumab
Emicizumab is a recombinant humanized IgG antibody that functions like factor VIII in patients with hemophilia A and is administered subcutaneously. Its function is based on linking the activated factor IX with factor X, causing a catalytic reaction and activating FX, thereby maintaining coagulation homeostasis. Due to differences in the molecular structure between emicizumab and factor VIII, antibodies to factor VIII do not react to emicizumab therapy. This makes it safe to use in patients with developed antibodies to factor VIII. Furthermore, emicizumab has a half-life of 4 to 5 weeks, requiring less frequent dosing for hemophilia A patients [38].
7.3.2. Genetic Therapy
Genetic therapy is a potential therapy for treating hemophilia patients by introducing a “healthy” gene or cells to replace the defective one. Genetically modified and functional gene F8 or F9 is delivered intravenously with the use of a vector into the hepatocytes. The most suitable vector was shown to be AAV (adeno-associated virus). The virus is hepatotropic, which is essential since factor IX is synthesized in hepatocytes and factor VIII in liver sinusoidal cells. AAV is episomal, reducing the risk of integration into human DNA and potential mutagenesis [39]. Studies have shown reduced bleeding episodes and a minimal number of significant side effects after a single dose. Limitations, however, include its availability only in specialized hospital centers, variable gene expression among individuals, the asymptomatic elevation of alanine transaminase (requiring corticosteroid treatment), and the required use of contraception to prevent gene transfer. Long-term toxicity and oncogenic risks remain unknown. While expensive, gene therapy is cost-effective over time due to the reduced need for prophylaxis, treatment, and rehabilitation [39]. The approved genetic therapy for hemophilia A is valoctocogene roxaparvovec, available in Europe from 2022 and in the U.S. from 2023. It is composed of factor VIII with a deleted B domain contained within AAV serotype 5 and is administered intravenously at 4 or 6 × 1013 vg/kg (vector genome per kg body weight). Therapy has been shown to have an increased factor VIII concentration and reduced bleeding episodes. Side effects include elevated alanine transaminases, hypersensitivity, and rare anaphylactic reactions [40]. Approved genetic therapy for hemophilia B is etranacogene dezaparvovec, containing a form of F9 gene Padua with a missense mutation that promotes greater gene activity inserted into AAV serotype 5. The FDA approved the therapy in 2022, and the NHS (National Health Service) approved it in 2024 for use in patients suffering from mild to severe forms of hemophilia B. The therapy is administered in a single dose of 2 × 1013 vg/kg intravenously. So far, cases have shown its success in reducing the incidence of bleeding episodes in patients [41]. Side effects include an elevated concentration of alanine transferase in the blood and one case of hepatocellular carcinoma. Fidanacogene elaparvovec is a gene therapy approved by the FDA in April 2024 which also contains a form of the F9 Padua gene. It is administered in a single dose of 5 × 1011 vg/kg intravenously and has been proven to reduce the frequency of bleeding episodes. The drug has shown a single side effect of elevated alanine transferase, which has been treated with glucocorticosteroids [8]. None of the approved gene therapies for hemophilia have reported thrombotic events, deaths related to the treatment, or the development of antibodies against the therapy to this date [40].
8. Invasive Treatment
8.1. Arthrocentesis
Arthrocentesis is an invasive method that extracts blood from the joint using a needle. It is used in the prevention of synovitis development, with acute and massive bleedings to allow for the shortest exposure of synovium to blood, thus lowering the possibility of injury [42].
8.2. Synovectomy
Synovectomy is a procedure in which the synovium is removed surgically or ablated using chemical agents or radioisotopes. The procedure is based on the pathophysiological mechanism of hemophilic arthropathy, where frequent joint bleeding and exposure of synovium to blood lead to its hypertrophy and increased vascularization, thus perpetuating the cycle [17].
8.2.1. Radiosynovectomy
Radiosynovectomy is a method of treating hemophilic arthropathy by localizing radiation to the joint using radiopharmaceuticals. It is most often used in patients with rheumatoid arthritis but may also be used in hemophilia patients with hemophilic arthropathy [43]. Radiosynovectomy is based on the use of radioisotopes.
8.2.2. Chemical Synovectomy
Chemical synovectomy, or chemical synoviorthesis, uses different chemical agents to destroy or inactivate inflamed and hypertrophic synovial tissue, lowering the incidence of bleeding episodes and joint pain. The most commonly used chemical in patients with hemophilic arthropathy is rifampicin [17].
8.2.3. Surgical Synovectomy
Surgical synovectomy is an invasive method that removes and destroys inflamed or hypertrophic synovium to reduce hemarthrosis episodes and preserve joint function. It is indicated in patients with subacute and chronic synovitis who have shown no improvement after 6 months of conservative therapy [17].
8.3. Joint Replacement
Therapies used in the prevention of hemarthrosis are not entirely effective, and so many individuals with hemophilia develop hemophilic arthropathy over time, requiring orthopedic surgery. The most common procedures include radial head resection, total hip replacement, open knee debridement, total knee replacement, and ankle joint arthrodesis [42]. Surgeries in hemophilic patients always come with a great risk and, as such, require a multidisciplinary team and specific protocols to ensure optimal patient care. The most frequently operated joints are the elbow, knee, and ankle [44].
9. Rehabilitation
Rehabilitation of the joint aims to restore its function to the state before hemarthrosis. Following an episode of hemarthrosis, coagulation factors should be administered, and once pain subsides, physical therapy should be initiated. This therapy includes passive and active exercises, proprioception training, and balance exercises to restore the joint’s functional capacity. Plans are tailored individually, progressively adding weight to achieve maximum functionality. Joint conditions must be monitored continuously, with adjustments to therapy or exercises in the case of deterioration [16]. For individuals with osteoporosis or osteopenia, supplementation with vitamin D, bisphosphonates, or RANK ligand inhibitors is recommended to prevent further bone remodeling [45]. Maintaining a healthy weight, engaging in regular physical activity suitable for the patient’s condition, and guidance from physiotherapists experienced in hemophilia are encouraged. COX-2 inhibitors are recommended for analgesia, and weak or strong opioids can be prescribed after consulting a pain specialist in the case of severe pain. After radiosynovectomy or chemical synovectomy, joint mobilization should begin within 48 h, followed by a three-week physical rehabilitation program. Before surgical procedures, preoperative physical therapy is advised with progressive postoperative therapy tailored to the patient’s response. The ultimate goal of postoperative rehabilitation is to maximize joint function recovery. Rehabilitation and treatment are best conducted in specialized centers with experienced professionals in hemophilia care [16].
10. Prognosis
The average life expectancy for hemophilia patients has been extended with the use of prophylaxis and new medications. However, it remains lower compared to the life expectancy of the general population, averaging 63 years for those with a severe form of the disease, hepatitis C, or AIDS. Patients with milder forms of the disease who receive regular prophylaxis have an average life expectancy of about 73 years [46]. Individuals with hemophilia have a lower incidence of cardiovascular diseases. Quality of life studies have shown that hemophilia patients have a worsened quality of life compared to the general population [47]. This is primarily due to their low self-confidence, frequent pain from bleeding episodes, development of hemophilic arthropathy causing mobility issues, and infections from inadequately purified factors [48]. Furthermore, this population of individuals shows higher rates of anxiety and depression, and chronic pain often limits their participation in sports activities [46]. The most common causes of death in hemophilia patients are liver failure from hepatitis C infection, bleeding, and cancer [49].
11. Discussion
The development of gene therapy for hemophilia marks a transformative shift in treatment, offering long-term solutions that could replace traditional factor replacement therapies. While factor VIII and IX infusions have long been the standard, they require frequent administration, are expensive, and carry risks of complications, such as inhibitors and viral infections. Gene therapy, by contrast, aims to address the root cause of the disorder by introducing functional genes that enable the body to produce the missing coagulation factors, potentially reducing or eliminating the need for regular infusions [39]. However, despite its promise, gene therapy for hemophilia presents several challenges. The variability in gene expression between individuals, especially given factors like liver function, immune response, and the nature of the viral vector, complicates its broad application. Additionally, side effects, such as elevated alanine transaminases (which require corticosteroid treatment) or rare but serious events like hepatocellular carcinoma, need to be carefully managed [40,41]. Long-term safety and efficacy remain key concerns, and ongoing studies are necessary to determine the best practices for patient selection, dosage, and monitoring. Furthermore, while gene therapy is a potential game-changer, its high cost and limited availability in specialized centers may restrict access for many patients. For it to become a truly widespread solution, healthcare systems will need to address these barriers, ensuring equitable access and affordability.
12. Conclusions
In conclusion, hemophilia is a complex X-linked genetic disorder primarily affecting males, with hemophilic arthropathy being one of its most debilitating complications. The pathophysiology of hemophilic arthropathy is driven by the accumulation of iron from repeated joint bleeds, leading to inflammation, synovial hyperplasia, and cartilage degradation. The two-punch hypothesis, involving both iron-induced inflammation and synovial cell proliferation, underscores the intricate mechanisms behind joint destruction. As hemophilic arthropathy progresses, chronic pain, joint deformities, and the need for surgical interventions often diminish patients’ quality of life [2,14,15]. Advancements in gene therapy, such as valoctocogene roxaparvovec for hemophilia A [40] and etranacogene dezaparvovec for hemophilia B [41], offer promising treatment alternatives. These therapies have shown significant success in reducing bleeding episodes and improving long-term outcomes by introducing functional clotting factors into patients’ systems. Despite some side effects, such as elevated alanine transaminases and potential risks like hepatocellular carcinoma, these gene therapies represent a revolutionary step forward in hemophilia care, moving beyond lifelong prophylactic treatment. As research continues, these therapies are expected to become more widely accessible, offering hope for better management and improved quality of life for individuals with hemophilia.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hassan, S.; Monahan, R.C.; Mauser-Bunschoten, E.P.; Van Vulpen, L.F.D.; Eikenboom, J.; Beckers, E.A.M.; Hooimeijer, L.; Ypma, P.F.; Nieuwenhuizen, L.; Coppens, M.; et al. Mortality, life expectancy, and causes of death of persons with hemophilia in the Netherlands 2001–2018. J. Thromb. Haemost. 2021, 19, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, I.; Iannuzzo, G.; Dell’Aquila, F.; Di Minno, M.N.D. Pathophysiological Role of Synovitis in Hemophilic Arthropathy Development: A Two-Hit Hypothesis. Front. Physiol. 2020, 11, 541. [Google Scholar] [CrossRef]
- Peyvandi, F.; Garagiola, I.; Young, G. The past and future of haemophilia: Diagnosis, treatments, and its complications. Lancet 2016, 388, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Berntorp, E.; Fischer, K.; Hart, D.P.; Mancuso, M.E.; Stephensen, D.; Shapiro, A.D.; Blanchette, V. Haemophilia. Nat. Rev. Dis. Primers 2021, 7, 45. [Google Scholar] [CrossRef]
- Shapiro, S.; Makris, M. Haemophilia and ageing. Br. J. Haematol. 2019, 184, 712–720. [Google Scholar] [CrossRef]
- Gualtierotti, R.; Solimeno, L.P.; Peyvandi, F. Hemophilic arthropathy: Current knowledge and future perspectives. J. Thromb. Haemost. 2021, 19, 2112–2121. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; McMillan, C.W.; White, G.C.; Bergman, G.E.; Horton, M.W.; Saidi, P. Purified factor IX using monoclonal immunoaffinity technique: Clinical trials in hemophilia B and comparison to prothrombin complex concentrates. Blood 1992, 79, 568–575. [Google Scholar] [CrossRef]
- Guyton, C.; Jehn, E.; Hall, J. Textbook of Medical Physiology, 13th ed.; Medicinska Naklada: Zagreb, Croatia, 2017. [Google Scholar]
- Peyvandi, F.; Ettingshausen, C.E.; Goudemand, J.; Jiménez-Yuste, V.; Santagostino, E.; Makris, M. New findings on inhibitor development: From registries to clinical studies. Haemophilia 2017, 23, 4–13. [Google Scholar] [CrossRef]
- Prescott, R.; Nakai, H.; Saenko, E.L.; Scharrer, I.; Nilsson, I.M.; Humphries, J.E.; Hurst, D.; Bray, G.; Scandella, D.; the Recombinate and Kogenate Study Groups. The inhibitor antibody response is more complex in hemophilia A patients than in most nonhemophiliacs with factor VIII autoantibodies. Recombinate and Kogenate Study Groups. Blood 1997, 89, 3663–3671. [Google Scholar] [CrossRef]
- Zhong, D.; Saenko, E.L.; Shima, M.; Felch, M.; Scandella, D. Some human inhibitor antibodies interfere with factor VIII binding to factor IX. Blood 1998, 92, 136–142. [Google Scholar] [CrossRef]
- Lacroix-Desmazes, S.; Bayry, J.; Misra, N.; Horn, M.P.; Villard, S.; Pashov, A.; Stieltjes, N.; d’Oiron, R.; Saint-Remy, J.M.; Hoebeke, J.; et al. The prevalence of proteolytic antibodies against factor VIII in hemophilia A. N. Engl. J. Med. 2002, 346, 662–667. [Google Scholar] [CrossRef]
- Garagiola, I.; Palla, R.; Peyvandi, F. Risk factors for inhibitor development in severe hemophilia A. Thromb. Res. 2018, 168, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Van Vulpen, L.F.D.; Mastbergen, S.C.; Lafeber, F.P.J.G.; Schutgens, R.E.G. Differential effects of bleeds on the development of arthropathy—Basic and applied issues. Haemophilia 2017, 23, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, D.; Manetti, M.; Matucci-Cerinic, M. Pathophysiology of Hemophilic Arthropathy. J. Clin. Med. 2017, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Santagostino, E.; Dougall, A.; Kitchen, S.; Sutherland, M.; Pipe, S.W.; Carcao, M.; Mahlangu, J.; Ragni, M.V.; Windyga, J.; et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia 2020, 26 (Suppl. S6), 1–158. [Google Scholar] [CrossRef]
- Van Vulpen, L.F.D.; Thomas, S.; Keny, S.A.; Mohanty, S.S. Synovitis and synovectomy in haemophilia. Haemophilia 2021, 27 (Suppl. S3), 96–102. [Google Scholar] [CrossRef]
- Lan, H.H.; Eustace, S.J.; Dorfman, D. Hemophilic arthropathy. Radiol. Clin. N. Am. 1996, 34, 446–450. [Google Scholar] [CrossRef]
- Pettersson, H.; Ahlberg, A.; Nilsson, I.M. A radiologic classification of hemophilic arthropathy. Clin. Orthop. 1980, 149, 153–159. [Google Scholar] [CrossRef]
- Wyseure, T.; Mosnier, L.O.; Von Drygalski, A. Advances and challenges in hemophilic arthropathy. Semin. Hematol. 2016, 53, 10–19. [Google Scholar] [CrossRef]
- Manco-Johnson, M.J.; Soucie, J.M.; Gill, J.C.; Joint Outcomes Committee of the Universal Data Collection, US Hemophilia Treatment Center Network. Prophylaxis usage, bleeding rates, and joint outcomes of hemophilia, 1999 to 2010: A surveillance project. Blood 2017, 129, 2368–2374. [Google Scholar] [CrossRef]
- Oldenburg, J. Optimal treatment strategies for hemophilia: Achievements and limitations of current prophylactic regimens. Blood 2015, 125, 2038–2044. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Rath, G.; Goyal, A.K. Advancement in the treatment of haemophilia. Int. J. Biol. Macromol. 2018, 118, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Aledort, L.M.; Haschmeyer, R.H.; Pettersson, H. A longitudinal study of orthopaedic outcomes for severe factor-VIII-deficient haemophiliacs. The Orthopaedic Outcome Study Group. J. Intern. Med. 1994, 236, 391–399. [Google Scholar] [CrossRef]
- Rattray, B.; Nugent, D.J.; Young, G. Celecoxib in the treatment of haemophilic synovitis, target joints, and pain in adults and children with haemophilia. Haemophilia 2018, 24, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Gualtierotti, R.; Tafuri, F.; Arcudi, S.; Solimeno, P.L.; Acquati, J.; Landi, L.; Peyvandi, F. Current and Emerging Approaches for Pain Management in Hemophilic Arthropathy. Pain Ther. 2022, 11, 1–15. [Google Scholar] [CrossRef]
- McCarty, T.S.; Patel, P. Desmopressin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK554582/ (accessed on 27 April 2024).
- Franchini, M.; Lippi, G. The use of desmopressin in acquired haemophilia A: A systematic review. Blood Transfus. 2011, 9, 377–382. [Google Scholar] [CrossRef]
- Mannucci, P.M. The choice of plasma-derived clotting factor concentrates. Baillieres Clin. Haematol. 1996, 9, 273–290. [Google Scholar] [CrossRef]
- Lusher, J.M. Recombinant clotting factor concentrates. Baillieres Clin. Haematol. 1996, 9, 291–303. [Google Scholar] [CrossRef]
- Konkle, B.A.; Shapiro, A.D.; Quon, D.V.; Staber, J.M.; Kulkarni, R.; Ragni, M.V.; Chhabra, E.S.; Poloskey, S.; Rice, K.; Katragadda, S.; et al. BIVV001 Fusion Protein as Factor VIII Replacement Therapy for Hemophilia A. N. Engl. J. Med. 2020, 383, 1018–1027. [Google Scholar] [CrossRef]
- Hermans, C.; Mancuso, M.E.; Nolan, B.; Pasi, K.J. Recombinant factor VIII Fc for the treatment of haemophilia A. Eur. J. Haematol. 2021, 106, 745–761. [Google Scholar] [CrossRef]
- Giorgi, M.E.; Agusti, R.; de Lederkremer, R.M. Carbohydrate PEGylation, an approach to improve pharmacological potency. Beilstein J. Org. Chem. 2014, 10, 1433–1444. [Google Scholar] [CrossRef]
- Konkle, B.A.; Stasyshyn, O.; Chowdary, P.; Bevan, D.H.; Mant, T.; Shima, M.; Engl, W.; Dyck-Jones, J.; Fuerlinger, M.; Patrone, L.; et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood 2015, 126, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Windyga, J.; Lissitchkov, T.; Stasyshyn, O.; Mamonov, V.; Rusen, L.; Lamas, J.L.; Oh, M.S.; Chapman, M.; Fritsch, S.; Pavlova, B.G.; et al. Pharmacokinetics, efficacy and safety of BAX326, a novel recombinant factor IX: A prospective, controlled, multicentre phase I/III study in previously treated patients with severe (FIX level < 1%) or moderately severe (FIX level ≤ 2%) haemophilia B. Haemophilia 2014, 20, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.D.; Ragni, M.V.; Valentino, L.A.; Key, N.S.; Josephson, N.C.; Powell, J.S.; Cheng, G.; Thompson, A.R.; Goyal, J.; Tubridy, K.L.; et al. Recombinant factor IX-Fc fusion protein (rFIXFc) demonstrates safety and prolonged activity in a phase 1/2a study in hemophilia B patients. Blood 2012, 119, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Santagostino, E.; Martinowitz, U.; Lissitchkov, T.; Pan-Petesch, B.; Hanabusa, H.; Oldenburg, J.; Boggio, L.; Negrier, C.; Pabinger, I.; von Depka Prondzinski, M.; et al. Long-acting recombinant coagulation factor IX albumin fusion protein (rIX-FP) in hemophilia B: Results of a phase 3 trial. Blood 2016, 127, 1761–1769. [Google Scholar] [CrossRef]
- Okaygoun, D.; Oliveira, D.D.; Soman, S.; Williams, R. Advances in the management of haemophilia: Emerging treatments and their mechanisms. J. Biomed. Sci. 2021, 28, 64. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C. Gene therapy for hemophilia. Hematol. Am. Soc. Hematol. Educ. Program 2022, 2022, 569–578. [Google Scholar] [CrossRef]
- Symington, E.; Rangarajan, S.; Lester, W.; Madan, B.; Pierce, G.F.; Raheja, P.; Millar, C.; Osmond, D.; Li, M.; Robinson, T.M. Valoctocogene roxaparvovec gene therapy provides durable haemostatic control for up to 7 years for haemophilia A. Haemophilia 2024, 30, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Pipe, S.W.; Leebeek, F.W.G.; Recht, M.; Key, N.S.; Castaman, G.; Miesbach, W.; Lattimore, S.; Peerlinck, K.; Van der Valk, P.; Coppens, M.; et al. Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. N. Engl. J. Med. 2023, 388, 706–718. [Google Scholar] [CrossRef]
- Rodríguez-Merchan, E.C. Surgical approaches to hemophilic arthropathy. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 2019, 30 (Suppl. S1), S11–S13. [Google Scholar] [CrossRef]
- Siegel, M.E.; Siegel, H.J.; Luck, J.V. Radiosynovectomy’s clinical applications and cost effectiveness: A review. Semin. Nucl. Med. 1997, 27, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Merchán, E.C. The role of orthopaedic surgery in haemophilia: Current rationale, indications and results. EFORT Open Rev. 2019, 4, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Pulles, A.E.; Mastbergen, S.C.; Schutgens, R.E.G.; Lafeber, F.P.J.G.; Van Vulpen, L.F.D. Pathophysiology of hemophilic arthropathy and potential targets for therapy. Pharmacol. Res. 2017, 115, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Buckner, T.W.; Sidonio, R.; Witkop, M.; Guelcher, C.; Cutter, S.; Iyer, N.N.; Cooper, D.L. Correlations between patient-reported outcomes and self-reported characteristics in adults with hemophilia B and caregivers of children with hemophilia B: Analysis of the B-HERO-S study. Patient Relat. Outcome Meas. 2019, 10, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Plug, I.; Peters, M.; Mauser-Bunschoten, E.P.; de Goede-Bolder, A.; Heijnen, L.; Smit, C.; Willemse, J.; Rosendaal, F.R.; van der Bom, J.G. Social participation of patients with hemophilia in the Netherlands. Blood 2008, 111, 1811–1815. [Google Scholar] [CrossRef]
- Buckner, T.W.; Witkop, M.; Guelcher, C.; Sidonio, R.; Kessler, C.M.; Clark, D.B.; Owens, W.; Frick, N.; Iyer, N.N.; Cooper, D.L. Impact of hemophilia B on quality of life in affected men, women, and caregivers—Assessment of patient-reported outcomes in the B-HERO-S study. Eur. J. Haematol. 2018, 100, 592–602. [Google Scholar] [CrossRef]
- Mazepa, M.A.; Monahan, P.E.; Baker, J.R.; Riske, B.K.; Soucie, J.M.; US Hemophilia Treatment Center Network. Men with severe hemophilia in the United States: Birth cohort analysis of a large national database. Blood 2016, 127, 3073–3081. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).