
IL-11 Expression in Systemic Sclerosis Is Dependent on Caspase-1 Activity but Does Not Increase Collagen Deposition



Abstract
1. Introduction
2. Materials and Methods
Statistical Analyses
3. Results
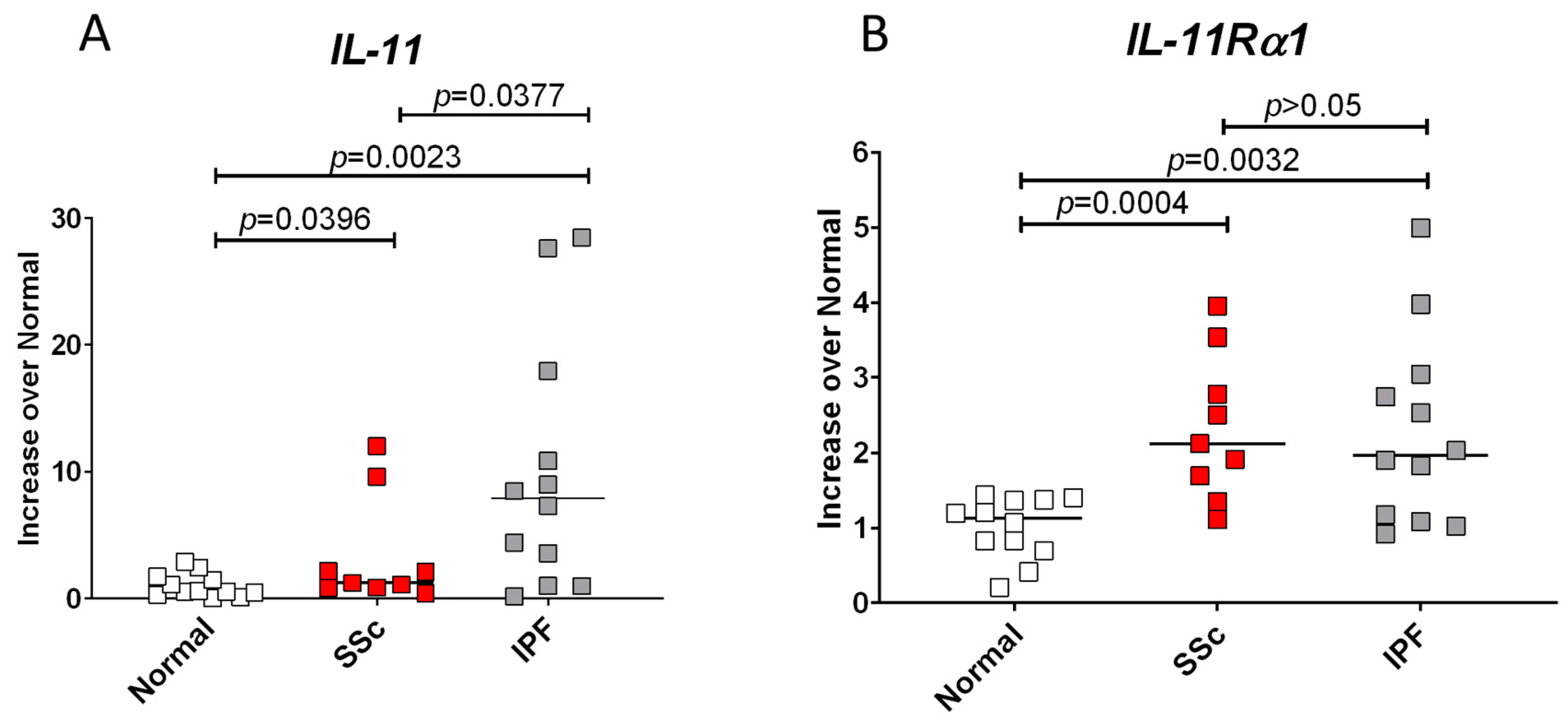
3.1. IL-11 and Its Receptor, IL-11Rα1, Have Increased Expression in SSc and IPF Fibroblasts
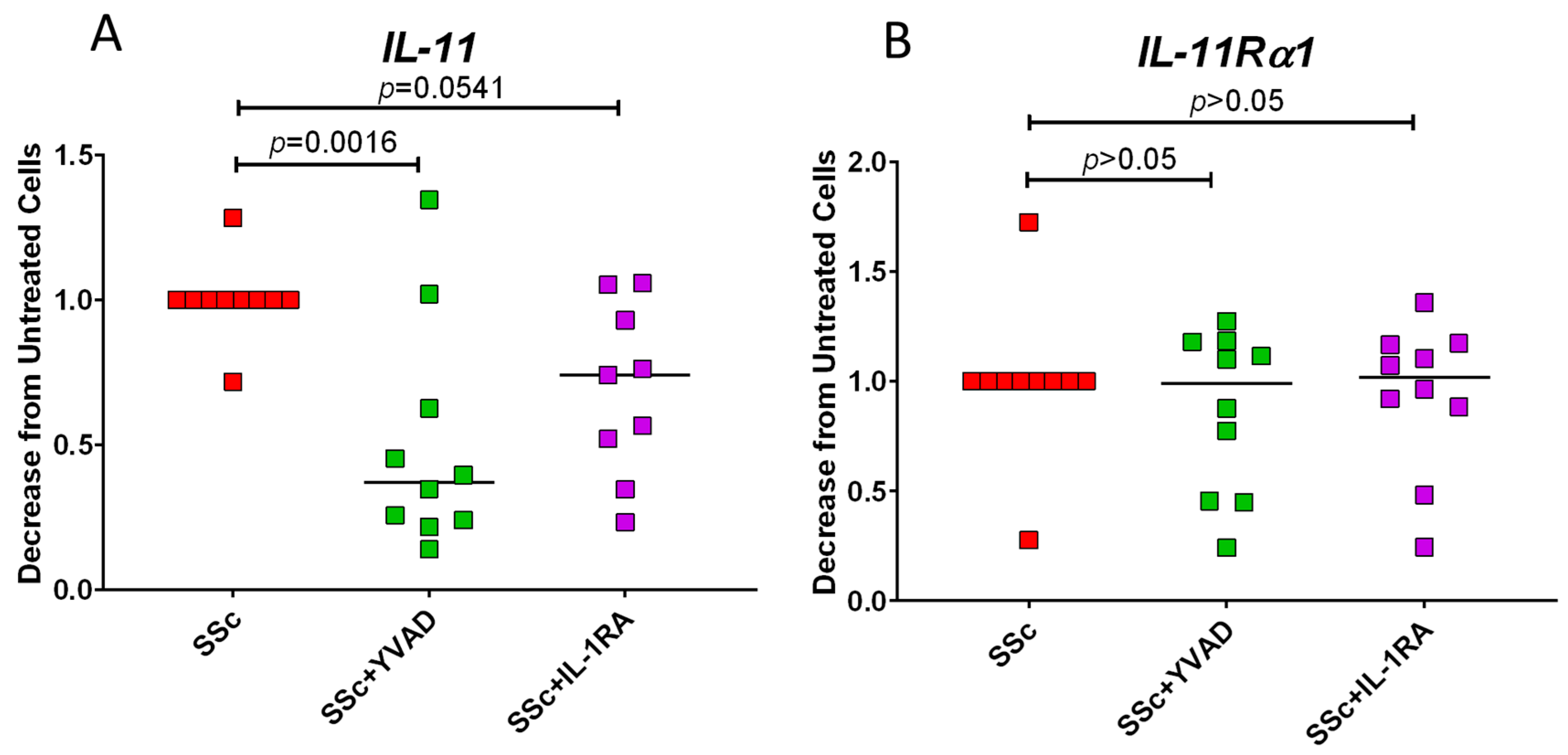
3.2. Inhibition of Caspase-1 Lowers IL-11 Expression in SSc Fibroblasts
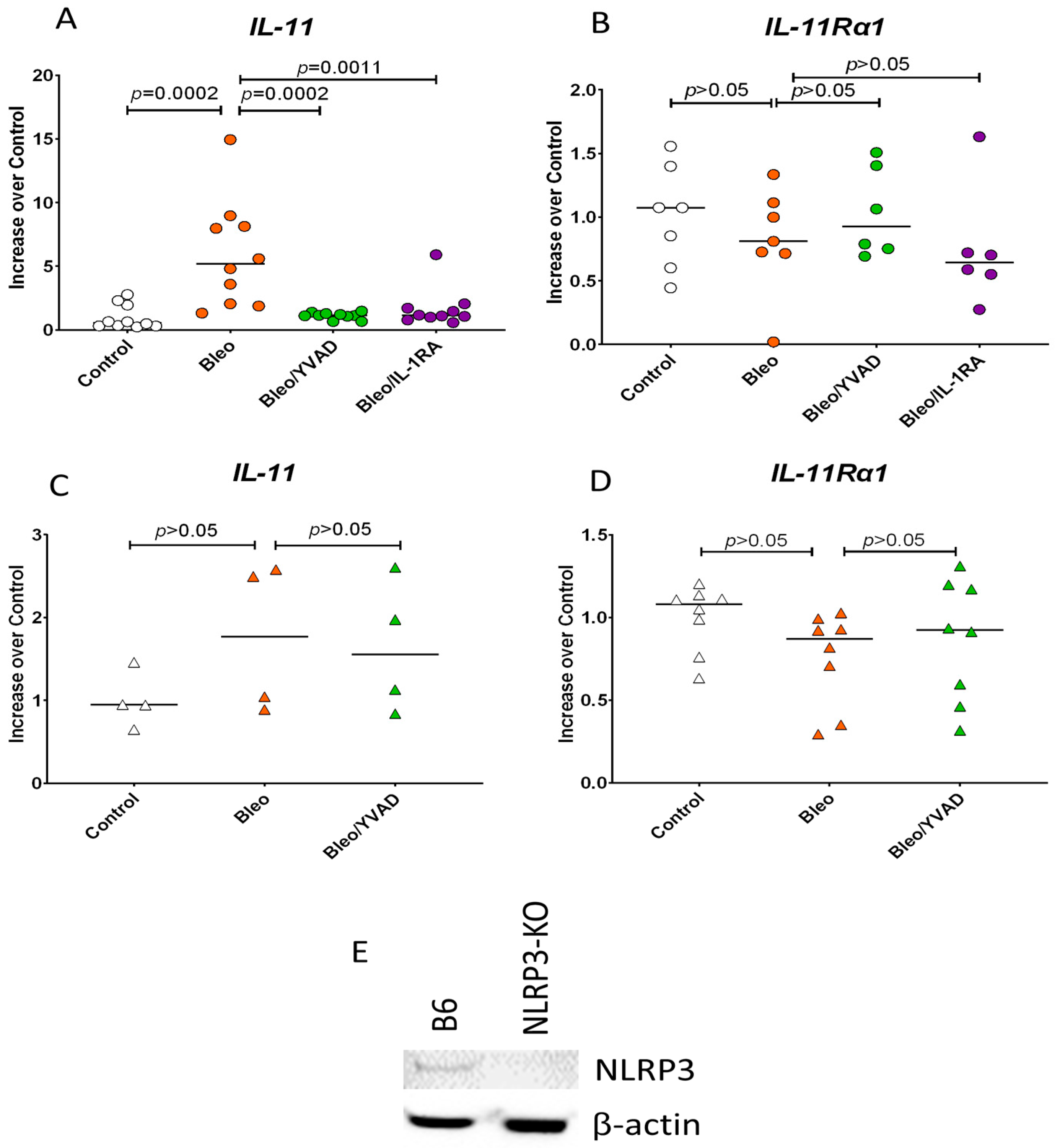
3.3. NLRP3 Inflammasome Activation of Caspase-1 Induces IL-11 Expression
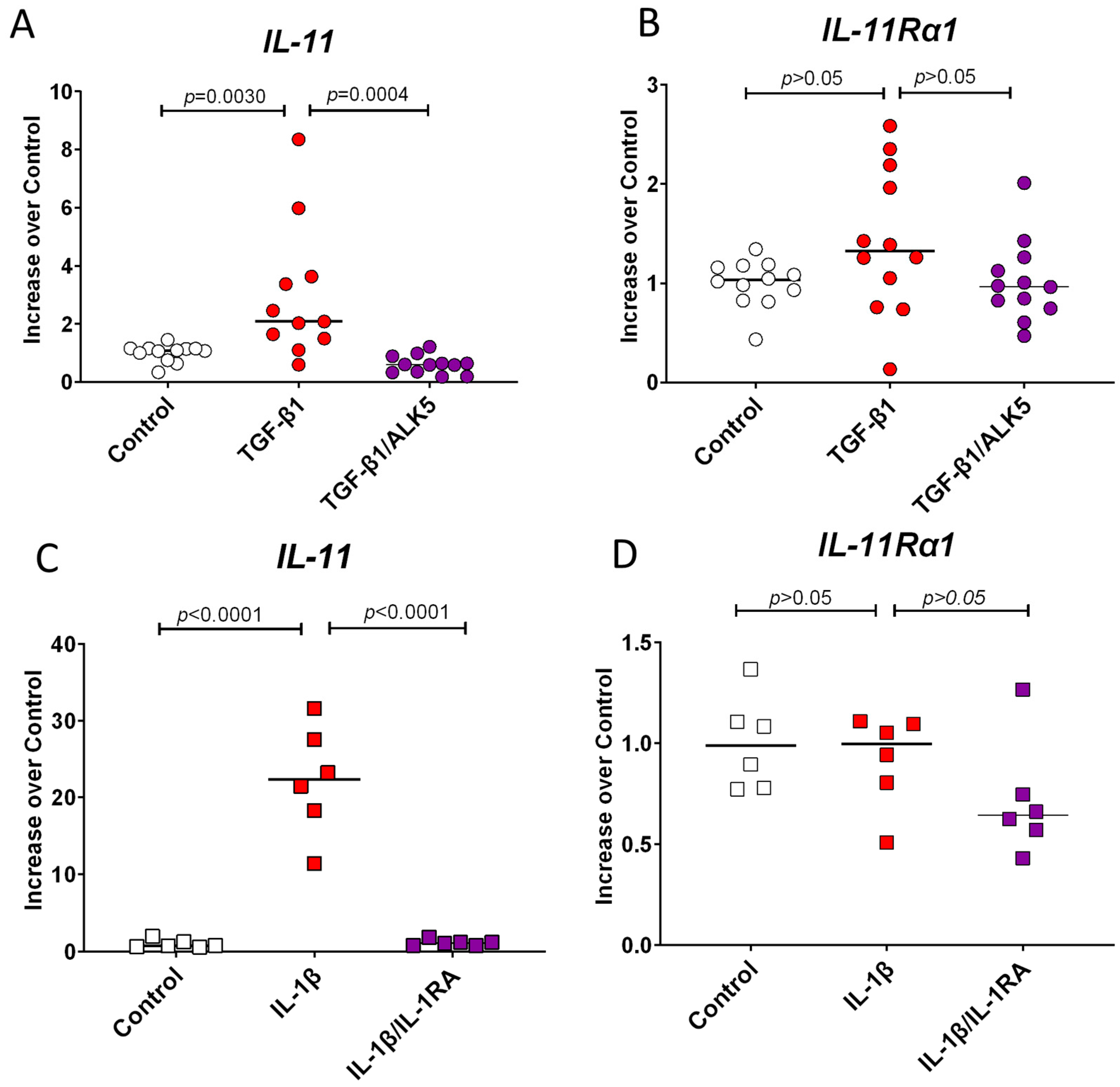
3.4. TGF-β1 and IL-1β Induce IL-11 Expression but Do Not Change the Expression of IL-11Ra1
3.5. TLR4 Signaling Induces IL-11 and IL-11Rα1 Expression
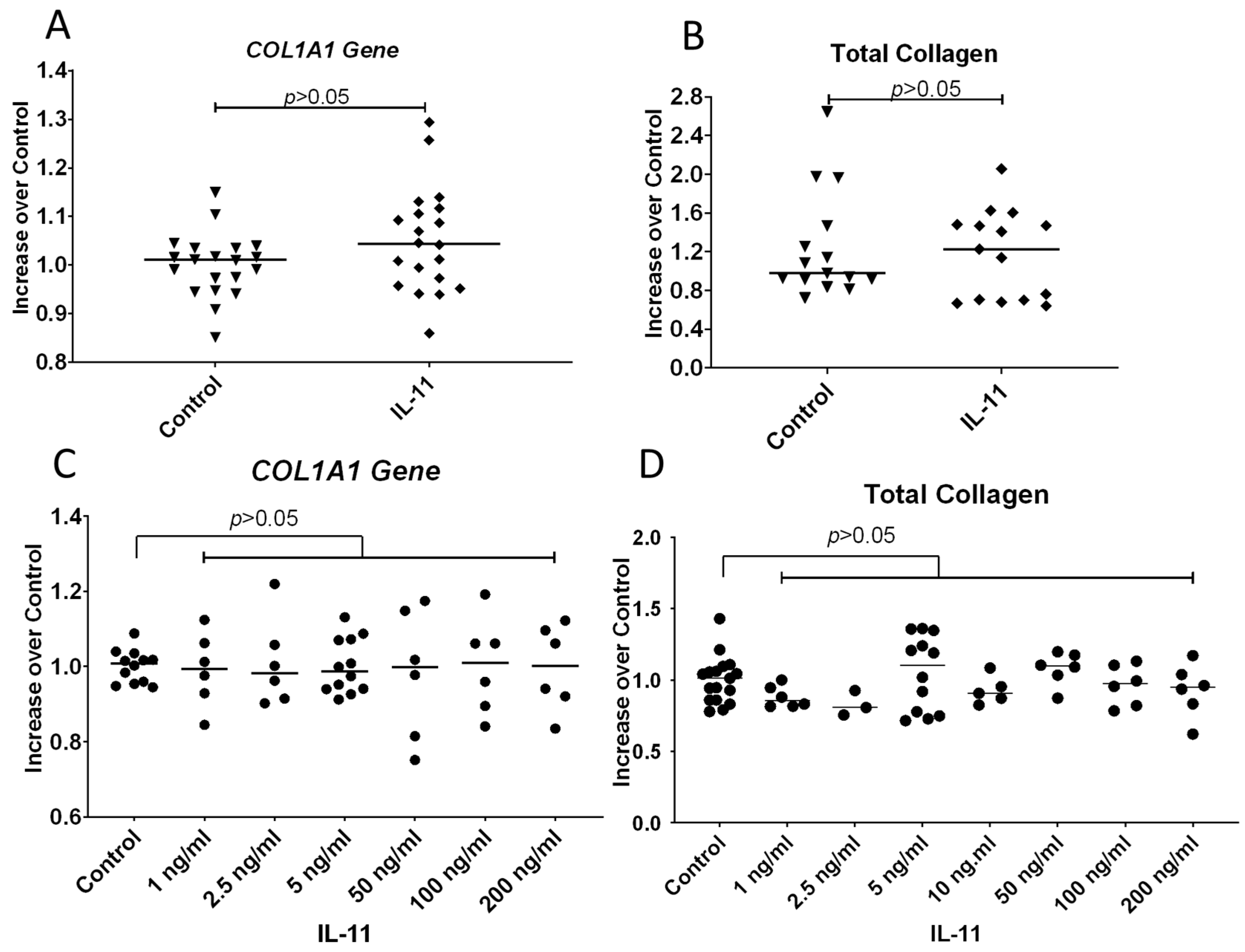
3.6. IL-11 Does Not Increase COL1A1 Gene Expression or Total Collagen Protein Secreted by Fibroblasts
3.7. ERK1/2 (p44/p42) Is Weakly Phosphorylated by Human Recombinant IL-11
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mayes, M.D.; Lacey, J.V., Jr.; Beebe-Dimmer, J.; Gillespie, B.W.; Cooper, B.; Laing, T.J.; Schottenfeld, D. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003, 48, 2246–2255. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, B.W.; Mathai, S.C.; Tedford, R.J.; Damico, R.L.; Corona-Villalobos, C.; Kolb, T.M.; Chaisson, N.F.; Harris, T.H.; Zimmerman, S.L.; Kamel, I.R.; et al. Right ventricular remodeling in idiopathic and scleroderma-associated pulmonary arterial hypertension: Two distinct phenotypes. Pulm. Circ. 2015, 5, 327–334. [Google Scholar] [CrossRef] [PubMed]
- McNearney, T.A.; Reveille, J.D.; Fischbach, M.; Friedman, A.W.; Lisse, J.R.; Goel, N.; Tan, F.K.; Zhou, X.; Ahn, C.; Feghali-Bostwick, C.A.; et al. Pulmonary involvement in systemic sclerosis: Associations with genetic, serologic, sociodemographic, and behavioral factors. Arthritis Rheum. 2007, 57, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Rosendahl, A.H.; Schönborn, K.; Krieg, T. Pathophysiology of systemic sclerosis (scleroderma). Kaohsiung J. Med. Sci. 2022, 38, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Liu, Z.; Zuo, H.; Yang, Z.; Qu, J. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Front. Pharmacol. 2021, 12, 797292. [Google Scholar] [CrossRef]
- Swarnakar, R.; Garje, Y.; Markandeywar, N.; Mehta, S. Exploring the common pathophysiological links between IPF, SSc-ILD and post-COVID fibrosis. Lung India 2022, 39, 279–285. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Steadman, T.; O’Reilly, S. Elevated interleukin-11 in systemic sclerosis and role in disease pathogenesis. J. Dermatol. 2023, 50, 1255–1261. [Google Scholar] [CrossRef]
- Ng, B.; Dong, J.; D’Agostino, G.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Ko, N.S.J.; Tan, J.; Chothani, S.P.; Huang, B.; et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2019, 11, eaaw1237. [Google Scholar] [CrossRef]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef]
- Ng, B.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Shekeran, S.G.; Goh, J.W.T.; Tan, J.; Kuthubudeen, F.; Lim, S.Y.; Xie, C.; et al. IL11 Activates Pancreatic Stellate Cells and Causes Pancreatic Inflammation, Fibrosis and Atrophy in a Mouse Model of Pancreatitis. Int. J. Mol. Sci. 2022, 23, 3549. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, A.A.; Singh, B.K.; Adami, E.; Viswanathan, S.; Dong, J.; D’Agostino, G.A.; Ng, B.; Lim, W.W.; Tan, J.; Paleja, B.S.; et al. Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology 2019, 157, 777–792.e714. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Roger, I.; Montero, P.; Artigues, E.; Escrivá, J.; Del Río, R.; Cortijo, J. Targeting IL-11 to reduce fibrocyte circulation and lung accumulation in animal models of pulmonary hypertension-associated lung fibrosis. Br. J. Pharmacol. 2024, 181, 2991–3009. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Zhao, G.; Chen, Q.; Li, Z.; Gao, M.; Ho, W.; Xu, X.; Zhang, X.Q. Inhaled siRNA nanoparticles targeting IL11 inhibit lung fibrosis and improve pulmonary function post-bleomycin challenge. Sci. Adv. 2022, 8, eabn7162. [Google Scholar] [CrossRef] [PubMed]
- Allanki, S.; Strilic, B.; Scheinberger, L.; Onderwater, Y.L.; Marks, A.; Günther, S.; Preussner, J.; Kikhi, K.; Looso, M.; Stainier, D.Y.R.; et al. Interleukin-11 signaling promotes cellular reprogramming and limits fibrotic scarring during tissue regeneration. Sci. Adv. 2021, 7, eabg6497. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Mosallanejad, K.; Zhang, Q.; O’Brien, S.; Clements, M.; Perper, S.; Wilson, S.; Chaulagain, S.; Wang, J.; Abdalla, M.; et al. IL11-mediated stromal cell activation may not be the master regulator of pro-fibrotic signaling downstream of TGFβ. Front. Immunol. 2024, 15, 1293883. [Google Scholar] [CrossRef]
- Widjaja, A.A.; Viswanathan, S.; Jinrui, D.; Singh, B.K.; Tan, J.; Wei Ting, J.G.; Lamb, D.; Shekeran, S.G.; George, B.L.; Schafer, S.; et al. Molecular Dissection of Pro-Fibrotic IL11 Signaling in Cardiac and Pulmonary Fibroblasts. Front. Mol. Biosci. 2021, 8, 740650. [Google Scholar] [CrossRef]
- Adami, E.; Viswanathan, S.; Widjaja, A.A.; Ng, B.; Chothani, S.; Zhihao, N.; Tan, J.; Lio, P.M.; George, B.L.; Altunoglu, U.; et al. IL11 is elevated in systemic sclerosis and IL11-dependent ERK signalling underlies TGFβ-mediated activation of dermal fibroblasts. Rheumatology 2021, 60, 5820–5826. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Yan, J.; Liu, J.; Chen, B.; Zhang, L.; Cheng, L. Efficacy and safety of recombinant human interleukin-11 in the treatment of acute leukaemia patients with chemotherapy-induced thrombocytopenia: A systematic review and meta-analysis. J. Eval. Clin. Pract. 2020, 26, 262–271. [Google Scholar] [CrossRef]
- Chen, M.; Li, L.; Xia, Q.; Chen, X.; Liao, Z.; Wang, C.; Shen, B.; Zhou, M.; Zhang, Q.; Zhang, Y.; et al. A real-world observation on thrombopoietic agents for patients with cancer treatment-induced thrombocytopenia in China: A multicenter, cross-sectional study. Cancer 2024, 130, 1524–1538. [Google Scholar] [CrossRef]
- Bhatia, M.; Davenport, V.; Cairo, M.S. The role of interleukin-11 to prevent chemotherapy-induced thrombocytopenia in patients with solid tumors, lymphoma, acute myeloid leukemia and bone marrow failure syndromes. Leuk. Lymphoma 2007, 48, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Chen, C.; Lou, L.H.; Mora, M. Blood thrombopoietin, IL-6 and IL-11 levels in patients with agnogenic myeloid metaplasia. Leukemia 1997, 11, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Owada, Y.; Izumi, Y.; Nonin, S.; Sugioka, K.; Nakatani, D.; Iwata, S.; Mizutani, K.; Nishimura, S.; Ito, A.; et al. Four cases of investigational therapy with interleukin-11 against acute myocardial infarction. Heart Vessel. 2016, 31, 1574–1578. [Google Scholar] [CrossRef] [PubMed]
- Obana, M.; Maeda, M.; Takeda, K.; Hayama, A.; Mohri, T.; Yamashita, T.; Nakaoka, Y.; Komuro, I.; Takeda, K.; Matsumiya, G.; et al. Therapeutic activation of signal transducer and activator of transcription 3 by interleukin-11 ameliorates cardiac fibrosis after myocardial infarction. Circulation 2010, 121, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Lu, B.; Cao, Q.; Wu, Z.; Xu, Z.; Li, W.; Yao, X.; Liu, F. IL-11 Attenuates Liver Ischemia/Reperfusion Injury (IRI) through STAT3 Signaling Pathway in Mice. PLoS ONE 2015, 10, e0126296. [Google Scholar] [CrossRef]
- Cook, S.A. Understanding interleukin 11 as a disease gene and therapeutic target. Biochem. J. 2023, 480, 1987–2008. [Google Scholar] [CrossRef]
- Sweeney, M.; O’Fee, K.; Villanueva-Hayes, C.; Rahman, E.; Lee, M.; Vanezis, K.; Andrew, I.; Lim, W.W.; Widjaja, A.; Barton, P.J.R.; et al. Cardiomyocyte-Restricted Expression of IL11 Causes Cardiac Fibrosis, Inflammation, and Dysfunction. Int. J. Mol. Sci. 2023, 24, 12989. [Google Scholar] [CrossRef]
- Lokau, J.; Nitz, R.; Agthe, M.; Monhasery, N.; Aparicio-Siegmund, S.; Schumacher, N.; Wolf, J.; Möller-Hackbarth, K.; Waetzig, G.H.; Grötzinger, J.; et al. Proteolytic Cleavage Governs Interleukin-11 Trans-signaling. Cell Rep. 2016, 14, 1761–1773. [Google Scholar] [CrossRef]
- Kolb, M.; Margetts, P.J.; Anthony, D.C.; Pitossi, F.; Gauldie, J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Investig. 2001, 107, 1529–1536. [Google Scholar] [CrossRef]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; van der Goot, F.G. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 2006, 126, 1135–1145. [Google Scholar] [CrossRef]
- Hussein, M.R.; Hassan, H.I.; Hofny, E.R.M.; Elkholy, M.; Fatehy, N.A.; Abd Elmoniem, A.E.A.; Ezz El-Din, A.M.; Afifi, O.A.; Rashed, H.G. Alterations in mononuclear inflammatory cells, CD4/CD8+ T cells, interleukin-1á, and tumor necrosis factor à in the bronchoalveolar lavage fluid, peripheral blood, and skin of patients with systemic sclerosis. J. Clin. Pathol. 2005, 58, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M.; Sassi-Gaha, S.; Rieger, J.L.; Boesteanu, A.C.; Feghali-Bostwick, C.A.; Katsikis, P.D. The inflammasome activating caspase-1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum. 2011, 63, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.D.; Fielding, C.; Phillips, A.; Fraser, D. Interleukin-1 beta regulates proximal tubular cell transforming growth factor beta-1 signalling. Nephrol. Dial. Transplant. 2009, 24, 2655–2665. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Li, B.; Subbarao Malireddi, R.K.; Lamkanfi, M.; Geiger, T.L.; Kanneganti, T.D. Chronic TLR Stimulation Controls NLRP3 Inflammasome Activation through IL-10 Mediated Regulation of NLRP3 Expression and Caspase-8 Activation. Sci. Rep. 2015, 5, 14488. [Google Scholar] [CrossRef] [PubMed]
- Mezzasoma, L.; Schmidt-Weber, C.B.; Fallarino, F. In Vitro Study of TLR4-NLRP3-Inflammasome Activation in Innate Immune Response. Methods Mol. Biol. 2023, 2700, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Huo, M.; Guo, W.; Ding, L. Benidipine Hydrochloride Inhibits NLRP3 Inflammasome Activation by Inhibiting LPS-Induced NF-κB Signaling in THP-1 Macrophages. J. Inflamm. Res. 2024, 17, 6307–6316. [Google Scholar] [CrossRef]
- Fernández Fabrellas, E.; Peris Sánchez, R.; Sabater Abad, C.; Juan Samper, G. Prognosis and Follow-Up of Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 6, 51. [Google Scholar] [CrossRef]
- Volkmann, E.R.; Fischer, A. Update on Morbidity and Mortality in Systemic Sclerosis-Related Interstitial Lung Disease. J. Scleroderma Relat. Disord. 2021, 6, 11–20. [Google Scholar] [CrossRef]
- Martínez-Godínez, M.A.; Cruz-Domínguez, M.P.; Jara, L.J.; Domínguez-López, A.; Jarillo-Luna, R.A.; Vera-Lastra, O.; Montes-Cortes, D.H.; Campos-Rodríguez, R.; López-Sánchez, D.M.; Mejía-Barradas, C.M.; et al. Expression of NLRP3 inflammasome, cytokines and vascular mediators in the skin of systemic sclerosis patients. Isr. Med. Assoc. J. IMAJ 2015, 17, 5–10. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Gene | Forward Primer | Reverse Primer |
|---|---|---|---|
| Human | β-actin | 5′–TTGCCGACAGGATGCAGAA–3′ | 5′–GCCGATCCACACGGAGTACTT–3′ |
| IL-11 | 5′–GGGGACATGAACTGTGTTTGC–3′ | 5′–GGGCGACAGCTGTATCTGG–3′ | |
| IL-11Rα1 | 5′–CTGGGCTAGGGCATGAACTG–3′ | 5′–CTGGGACTCCAAGTGCAAGA–3′ | |
| Mouse | β-actin | 5′–ACGGCCAGGTCATCACTATTG–3′ | 5′–CAAGAAGGAAGGCTGGAAAAGA–3′ |
| IL-11 | 5′–TTGGGATCTTTGCAGCCTTCCT–3′ | 5′–CATGCCGGAGGTAGGACATC–3′ | |
| IL-11Rα1 | 5′–CAACTCAGTGGAGCGGGAG–3′ | 5′–AGCTGCTGCTCATCTTGATAAT–3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McFalls, C.M.; Connolly, L.M.; Fustakgi, A.G.; Artlett, C.M. IL-11 Expression in Systemic Sclerosis Is Dependent on Caspase-1 Activity but Does Not Increase Collagen Deposition. Rheumato 2024, 4, 163-175. https://doi.org/10.3390/rheumato4040013
McFalls CM, Connolly LM, Fustakgi AG, Artlett CM. IL-11 Expression in Systemic Sclerosis Is Dependent on Caspase-1 Activity but Does Not Increase Collagen Deposition. Rheumato. 2024; 4(4):163-175. https://doi.org/10.3390/rheumato4040013
Chicago/Turabian StyleMcFalls, Caya M., Lianne M. Connolly, Alfred G. Fustakgi, and Carol M. Artlett. 2024. "IL-11 Expression in Systemic Sclerosis Is Dependent on Caspase-1 Activity but Does Not Increase Collagen Deposition" Rheumato 4, no. 4: 163-175. https://doi.org/10.3390/rheumato4040013
APA StyleMcFalls, C. M., Connolly, L. M., Fustakgi, A. G., & Artlett, C. M. (2024). IL-11 Expression in Systemic Sclerosis Is Dependent on Caspase-1 Activity but Does Not Increase Collagen Deposition. Rheumato, 4(4), 163-175. https://doi.org/10.3390/rheumato4040013