Many useful chemicals have been industrially produced using genetic recombination technology in microorganisms and animal cells. However, in many cases, this takes a long period of research and development through trial and error; furthermore, a high cost is required to raise the target substance’s productivity to a sufficient level. Additionally, many of these chemicals cannot be produced, even though there is a business need for their applications.
Synthetic biology is expected to overcome these problems and dramatically expand the range of bio-producible substances, quickly achieving high productivity and producing chemicals previously thought unproducible by living organisms. Synthetic biology is based on one of the fundamental tenets of biology—to learn by making. However, we will focus on synthetic biology, which is specialized for industrial use and called “synthetic bioengineering” [1].
The approach to producing useful chemicals through synthetic bioengineering differs from the approach based on conventional molecular biology. In the approach of molecular biology, for example, when a microorganism that produces a novel secondary metabolite is discovered through intensive screening, a gene cluster that produces the compound is found, the function of each gene in the cluster is clarified, and then genetic modification is performed in the microorganism to increase productivity. In other words, development proceeds in the flow of DDBT: “discover ⇒ design ⇒ built (recombine) ⇒ test” (Figure 1), which is an approach that relies on human wisdom and experiences based on the molecular biological approach. On the other hand, the purpose of the synthetic bioengineering approach is to design an artificial gene cluster by assembling molecular parts for a target substance using a database and an artificial metabolic design algorithm; the artificial gene cluster is then synthesized as DNA modules and incorporated into microorganisms, and its productivity is measured. Then, we can find ways to improve it by using machine learning and other methods based on the obtained results. The results are then used in machine learning and other methods to find improvement strategies. In other words, the DBTL cycle (design ⇒ build ⇒ test ⇒ learn) is used to create the target system (Figure 1), which is a computer-assisted design. This approach has become possible due to the rapid research-based development of various genome and enzyme databases. In actual bioproduction strategy, the molecular and synthetic biotechnology approaches are complementary; creating a platform that integrates them is imperative.
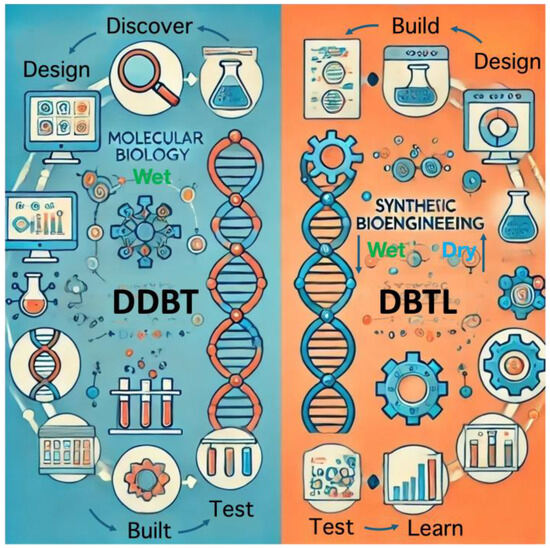
Figure 1.
Concept of discover–design–build–test (DDBT) and design–build–test–learn (DBTL). Wet means based on high-throughput experiments; dry means based on in silico modeling and simulation. The artificial intelligence (AI) generator partially generated the illustration based on the concepts.
To rationally design the metabolic functions of manufacturing microorganisms in the context of synthetic biotechnology, a knowledge/information base that serves as a design basis (working hypothesis) and metabolic pathway mining using this knowledge/information base are necessary. In general, metabolic information consists of metabolites, enzyme reactions, and gene information, and databases such as KEGG [2], BRENDA [3], and BioCyc [4] can be used to compile the knowledge and data. In particular, the KEGG database provides an information repository that incorporates and visualizes diverse data as a unique PATHWAY database; this database is a standard in metabolic pathway analysis. These databases also provide a web interface, which is helpful for bottom-up metabolic design. This can be effectively utilized for top-down metabolic pathway design by employing data mining on the accumulated data.
Various design tools have been developed for metabolic pathway design based on metabolic databases, which can be categorized into two main categories based on their distinct approaches. One approach is to design the metabolic pathways necessary for producing the target substance using only the information on enzyme reactions and metabolites in the metabolism-related databases. The primary objective of this method is to identify enzyme genes from the heterologous hosts required for the target metabolic pathway and reconstruct the metabolic pathway using model microorganisms. In response to the recent increase in data accumulation, new algorithmic design tools such as FMM [5], MRSD [6], and DESHARKY [7] have been developed; each of these employs various databases, data quality and quantities, and metabolic pathway reconstruction methodologies. However, these design tools rely solely on information from existing databases, making the prediction of entirely novel metabolic pathways problematic.
Design tools based on other approaches have been developed to design novel metabolic pathways. Through delineating the characteristics of enzyme reactions without specifying their genomic origin, BNICE [8] has developed an algorithm to predict new enzyme reactions. PathPred [9], a metabolic pathway prediction utility that uses KEGG-specific enzyme reaction information, and EC-BLAST, which specializes in enzyme reaction comparisons, have been designed with similar approaches. This technique is becoming indispensable for designing de novo metabolic pathways in model microorganisms.
In addition to the described metabolic pathway design, more precise metabolic modeling methodologies have been devised through quantitatively treating metabolic models. Palsson et al. [10] have developed a metabolic model (Genome Scale Model) based on existing metabolic pathway data to develop a basis for the steady-state analysis of metabolic fluxes; they are currently conducting metabolic modeling to enhance the production of target substances [11]. Additionally, OptKnock, a gene design tool [12], has been developed to generate a knockout model of a gene based on the Genome Scale Model; this tool is anticipated to be utilized in the future for metabolic pathway design.
In addition to in silico prediction methods using databases and metabolic models, estimating the rate-limiting points of metabolic flux and key genetic control points is essential. This can be achieved by understanding the cell’s actual physiological conditions for the rational design of metabolic pathways in manufacturing microorganisms. A comprehensive analysis of intracellular gene expression and metabolites, such as transcriptome and metabolome analysis, is effective for such a data-driven design [13]. Changes in gene expression levels can be determined with relative ease using microarray analysis, which is currently available for a wide range of hosts. Several methods have been devised for metabolome analysis using mass spectrometry to quantify intracellular metabolic intermediates’ accumulation and conversion (metabolic flux). With the emergence of next-generation sequencers (NGS), it is now possible for individual researchers to decode the genome sequences of microorganisms. The variety of microorganisms that can be used as manufacturing hosts has expanded significantly. In addition, it is now possible to isolate differences in genome-level nucleotide sequences between high-producing strains obtained through evolutionary engineering techniques and reference strains. Since these omics analysis methods are based on measured data, they are utilized as effective and practicable instruments for rational design determination.
Genetic recombination techniques are essential for implementing in silico and omics-based metabolic designs in microorganism hosts. Metabolic engineering typically requires the expression or disruption of genes at multiple loci. Through combining operons, bacteria such as Escherichia coli can express multiple genes with a single promoter; eukaryotes such as yeast must, in principle, prepare promoters for each gene individually. Eukaryotes are being used to develop genetic recombination techniques and vectors that enable multiple gene expressions to implement such a design. In addition, while site-directed recombinases (Flp from the yeast Saccharomyces cerevisiae, Cre from the bacteriophage P1, and R from the yeast Zygosaccharomyces rouxii) are commonly used to disrupt genes [14], genome-editing technologies such as CRISPR-Cas9 have recently been developed to disrupt multiple genes simultaneously. In addition to simple robust gene expression and disruption, more complex and sophisticated gene expression regulation systems, such as siRNA [15] and gene switches [16], have begun to be utilized, enabling more complex and sophisticated gene expression regulation designs.
Dynamic and nonlinear interactions comprise the transcription, translation, post-translational modifications, and metabolic networks in the expression of each gene. By characterizing the gene network as a mathematical model consisting of of these aforementioned interactions, it is possible to simulate the behavior of cells and predict their behavior when the interactions are altered. In addition, sensitivity analysis can be used to predict which interactions can be modified to increase manufacturing productivity.
Mathematical models for characterizing gene networks can be classified into three principal types: static, stochastic, and dynamic models. The Boolean network [17] exemplifies the static model, which characterizes the relationship between genes using a logical function based on the expression profile of a single gene disruption strain or exhaustively measured time series. A Bayesian network [18] is a stochastic model that describes the regulatory relationship between genes via conditional probability. Although both Boolean and Bayesian networks can manage time series data, differential equation models that explicitly characterize the changes in the expression levels of each gene are suitable for a more natural and detailed analysis of the behavior of dynamic networks. Models of generalized nonlinear differential equations incorporate the Generalized Mass Action (GMA) rule [19], a generalization of the mass action rule, and the S-system, a conceptual model based on GMA [20].
Regarding the analysis of gene networks, a generalized mathematical model is typically developed to describe the relationships between genes, and the model’s internal parameters are estimated using experimental values such as disrupted strains and expression profiles sampled over time. Nevertheless, it is challenging to estimate the size of the network uniquely. This is particularly challenging when there is a large discrepancy between the degree of freedom of the differential equation model and the number of experimental values acquired and when the computational burden increases exponentially with the size of the network. The computational burden increases exponentially with network size. On the other hand, information on the network’s structure can be obtained with or without dynamic parameter values based on what is known about gene networks, studies on the typology of gene network structures, and predictions derived from the static and stochastic methods previously described. Through the utilization of this information on the network structure, we are devising methods to estimate gene networks more efficiently than conventional methods that simultaneously estimate network structure and dynamic parameters from inception.
The recent rapid development of experimental techniques has increased the amount of measured data. The advancements in simulation and network estimation techniques and increased computing power have made dynamic and nonlinear gene network analysis feasible. The regulation and design of gene expression will increasingly incorporate these methods. Recently, the missing link via the correlation between the changes in the genome (genotype) and the fluctuation in the omics (phenotype) has been discovered in a robust yeast strain during its fermentation in the presence of inhibitory chemical compounds [21]. Deep learning approaches coupled with the capability of artificial intelligence (AI) can examine and narrow the gap in the intricate relationships between genotypes and phenotypes by analyzing large-scale genetic and phenotypic data [22,23]. Genotypes refer to the genetic makeup of an organism, typically represented by its DNA sequence; phenotypes encompass the observable traits and characteristics of that organism, such as its physical traits, stress susceptibility, or behavior. These technologies are expected to significantly impact the advancement of synthetic biology, enabling the creation of novel material production systems and fostering the emergence of associated entrepreneurial ventures.
SynBio should assume a prominent position as an esteemed scientific publication; it is currently positioned to prioritize accepting submissions that align with the themes discussed in this article. Synthetic biology is an area where researchers are active worldwide and where close collaboration between wet- and dry-based researchers is required. We hope that this issue of “Insights in synthetic bioengineering and aspects to create a new biotechnology” will bring together biotechnology researchers from across the globe and encourage more young researchers to take an active role in this field, leading the world in the research, development, and application of biotechnology.
Conflicts of Interest
The author declares no conflict of interest.
References
- Kondo, A.; Ishii, J.; Hara, K.Y.; Hasunuma, T.; Matsuda, F. Development of microbial cell factories for bio-refinery through synthetic bioengineering. J. Biotechnol. 2013, 163, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36 (Suppl. 1), D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, I.; Chang, A.; Placzek, S.; Söhngen, C.; Rother, M.; Lang, M.; Munaretto, C.; Ulas, S.; Stelzer, M.; Grote, A.; et al. BRENDA in 2013: Integrated reactions, kinetic data, enzyme function data, improved disease classification: New options and contents in BRENDA. Nucleic Acids Res. 2012, 41, D764–D772. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Ouzounis, C.A.; Moore-Kochlacs, C.; Goldovsky, L.; Kaipa, P.; Ahrén, D.; Tsoka, S.; Darzentas, N.; Kunin, V.; López-Bigas, N. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res. 2005, 33, 6083–6089. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Chang, W.C.; Chiu, C.M.; Huang, C.C.; Huang, H.D. FMM: A web server for metabolic pathway reconstruction and comparative analysis. Nucleic Acids Res. 2009, 37 (Suppl. 2), W129–W134. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Zheng, H.; Liu, Z.; Li, G.; Li, J.; Hong, J.; Zhao, K. MRSD: A web server for metabolic route search and design. Bioinformatics 2011, 27, 1581–1582. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodrigo, G.; Carrera, J.; Prather, K.J.; Jaramillo, A. DESHARKY: Automatic design of metabolic pathways for optimal cell growth. Bioinformatics 2008, 24, 2554–2556. [Google Scholar] [CrossRef] [PubMed]
- Hatzimanikatis, V.; Li, C.; Ionita, J.A.; Henry, C.S.; Jankowski, M.D.; Broadbelt, L.J. Exploring the diversity of complex metabolic networks. Bioinformatics 2005, 21, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Shigemizu, D.; Hattori, M.; Tokimatsu, T.; Kotera, M.; Goto, S.; Kanehisa, M. PathPred: An enzyme-catalyzed metabolic pathway prediction server. Nucleic Acids Res. 2010, 38 (Suppl. 2), W138–W143. [Google Scholar] [CrossRef] [PubMed]
- Palsson, B.O. Systems Biology: Properties of Reconstructed Networks; Cambridge University Press: New York, NY, USA, 2006. [Google Scholar]
- Bordbar, A.; Monk, J.M.; King, Z.A.; Palsson, B.O. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 2014, 15, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. Optknock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 2003, 84, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Hasunuma, T.; Sung, K.M.; Sanda, T.; Yoshimura, K.; Matsuda, F.; Kondo, A. Efficient fermentation of xylose to ethanol at high formic acid concentrations by metabolically engineered Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2011, 90, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Kilby, N.J.; Snaith, M.R.; Murray, J.A. Site-specific recombinases: Tools for genome engineering. Trends Genet. 1993, 9, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Na, D.; Yoo, S.M.; Chung, H.; Park, H.; Park, J.H.; Lee, S.Y. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol. 2013, 31, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Nozaki, K.; Umeno, D.; Ishii, J.; Kondo, A. Robust and flexible platform for directed evolution of yeast genetic switches. Nat. Commun. 2021, 12, 1846. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, S. Homeostasis and differentiation in random genetic control networks. Nature 1969, 224, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Friedman, N. The Bayesian structural EM algorithm. In Proceedings of the Fourteenth Conference on Uncertainty in Artificial Intelligence, Madison, WI, USA, 24–26 July 1998; pp. 129–138. [Google Scholar]
- Kacser, H.; Burns, J.A. The control of flux. Symp. Soc. Exp. Biol. 1973, 27, 65. [Google Scholar] [CrossRef] [PubMed]
- Savageau, M.A. Biochemical System Analysis: A Study of Function and Design in Molecular Biology; Addison-Wesley: Boston, MA, USA, 1976. [Google Scholar]
- Kahar, P.; Itomi, A.; Tsuboi, H.; Ishizaki, M.; Yasuda, M.; Kihira, C.; Otsuka, H.; binti Azmi, N.; Matsumoto, H.; Ogino, C.; et al. The flocculant Saccharomyces cerevisiae strain gains robustness via alteration of the cell wall hydrophobicity. Metab. Eng. 2022, 72, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- Goshisht, M.K. Machine Learning and Deep Learning in Synthetic Biology: Key Architectures, Applications, and Challenges. ACS Omega 2024, 9, 9921–9945. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).