Abstract
Next-Generation Sequencing (NGS) is used as a diagnostic strategy for identifying pathogenic genetic variants in children and adults. However, the analysis is complex, requiring specialized bioinformaticians, and it can take weeks to finalize one study. This has been a limiting factor for the application of NGS in the screening of populations for rare genetic diseases. In this work, we show two case studies, where we applied an AI-driven bioinformatics framework in a diagnostic and a preventive scenario, respectively. The AI analysis was accurate and substantially faster than using conventional bioinformatics tools. Our results support the concept that AI-driven bioinformatics is a scalable solution for rendering accurate results and enabling a more widely available genetic screening for rare diseases.
1. Introduction
Whole Exome Sequencing (WES) using Next-Generation Sequencing (NGS) is a clinically accepted diagnostic technology for the identification of pathogenic genetic variants in children and adults [1]. Finding gene-function-disruptive variants (SNPs and INDELs) in sequences is fundamental in determining the cause of the genetic disease and for genetic counselling consultations. Additionally, the application of this method at the pre-conception stage can also enable parents to make informed decisions regarding the possible birth of children with a particular genetic disease. Databases such as ClinVar and OMIM have been accumulating information on an ever-increasing number of new pathogenic variants [2]. In these databases, gene–disease associations have also been growing over time, leading to more than eight thousand having been already reported [3]. Public and private healthcare facilities are beginning to use these data as a front-line tool over conventional techniques to diagnose pediatric rare genetic diseases [1,4]. However, the analysis of WES using bioinformatics is complex and requires specialist skills and training, hence it can take several weeks from sample to diagnosis [5]. The relative complexity associated with the high labor intensity is a substantial bottleneck in the field, leading to a heavy cost in human resources. This has been a limiting factor for the screening and prevention of rare diseases in the general population. Artificial Intelligence (AI) is considered to be a solution for automating complex analysis and decision-making [6]. In this work, we present two case studies where we applied an AI-driven bioinformatics framework in a diagnostic and a preventive scenario, respectively.
2. Methodology
2.1. Clinical Samples and Sequencing
Saliva samples were collected in DNA/RNA saliva collection tubes (GeneFix™, Isohelix) using the commercial ExoMart and SureMart kits from MolMart Ltd., Manchester, United Kingdom. Relevant clinical data were submitted by the referring clinician into the MolMart online form for kit activations (https://molmartgenomics.com, accessed on 27 October 2022). WES was performed by NGS using the Illumina platform. The exome library was prepared with Agilent’s SureSelect V6+UTR-post kit.
2.2. Bioinformatics
Variant Calling Files (VCF) were generated from FASTQ files using a standard bioinformatics pipeline [7,8]. BWA (Burrows–Wheeler Alignment Tool) software version 0.7.12 and reference human genome version hg38 were used for read mapping and alignment. Variant calling and variant annotation of genetic modifications was made using GATK (Genome Analysis Toolkit) software version 3.4.0 and SnpEff version 4.1, respectively. The MolMart Artificial Intelligence Analyst (MAIA) was used for pathogenic gene variant candidate identification and ranking on the clinical observations of the Variant Calling Files (VCF). Clinical observation matching and pathogenic scoring were performed by MAIA, considering both experimental evidence on databases and sequence predictions.
3. Results
We applied an AI-driven bioinformatics framework to analyze two case studies, one a diagnostic (Case Study 1) and the other a preventive scenario (Case Study 2).
3.1. Case Study 1
An 8-month-old infant was referred for genetic testing with hypotonia, delayed development, hepatosplenomegaly and strabismus. We applied AI-driven bioinformatics on the sequenced exome containing about 114,000 gene variants, taking into account the clinical phenotype (Figure 1). From all gene variants, the AI took ~5 s to identify a total of 757 putative pathogenic variants, where only 15 had high-scoring matches on disease database annotations that related to the clinical observations. Furthermore, the top-ranked variant (Figure 1) was the one chosen by independent molecular geneticists as causative of the phenotype by manually checking in the OMIM and ClinVar databases.
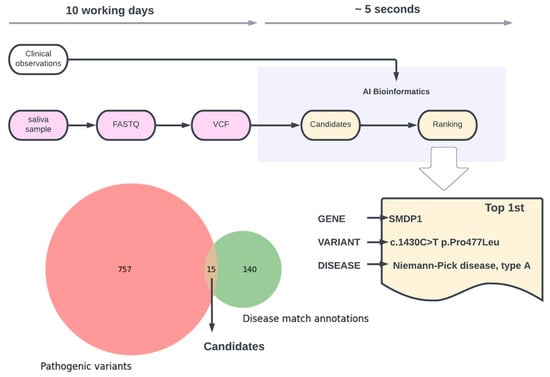
Figure 1.
Overview of the bioinformatics analysis pipeline and final outcome on case study 1.
3.2. Case Study 2
In this case, we screened a healthy couple at the pre-conception stage for their potential risk of having a child affected by a genetic disease. We applied AI-driven bioinformatics on the male and female exomes containing about 112,000 and 113,000 gene variants, respectively (Figure 2). The AI took ~12 s to identify six putative pathogenic gene variants that can be transmitted from both males and females. From these, only one raised some concern based on strong gene–disease association evidence, with an estimated probability of 23% of having a child with mannose binding deficiency.
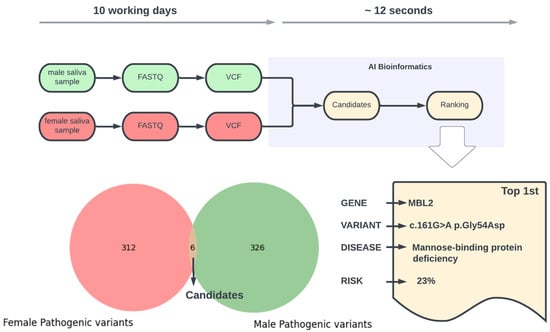
Figure 2.
Overview of the bioinformatics analysis pipeline and final outcome on case study 2.
4. Conclusions
The case studies shown here demonstrate that AI-driven bioinformatics analysis is substantially faster than conventional bioinformatics tools and platforms. Furthermore, our results support the concept that AI-driven bioinformatics is an accurate and scalable solution which can make population-wide genetic screening for rare diseases possible.
Author Contributions
Conceptualization, R.P., M.M. and T.K.; methodology, R.P., M.M. and T.K.; formal analysis, R.P., A.C., Y.Z., Y.S. and T.R.; investigation, R.P., A.C., Y.Z., Y.S., T.R. and M.G.M.; data curation, R.P., A.C., Y.Z., Y.S., T.R. and M.G.M.; writing—original draft preparation, R.P.; writing—review and editing, M.M.; supervision, R.P., M.G.M. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee).
Informed Consent Statement
All patients have given their consent to use their clinical and genomic data for research and clinical purposes.
Data Availability Statement
Medical and genomic data are not publicly available due to privacy restrictions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Frésard, L.; Montgomery, S.B. Diagnosing Rare Diseases after the Exome. Mol. Case Stud. 2018, 4, a003392. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Oliveira, J.; Sousa, M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J. Clin. Med. 2020, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Fridman, H.; Yntema, H.G.; Mägi, R.; Andreson, R.; Metspalu, A.; Mezzavila, M.; Tyler-Smith, C.; Xue, Y.; Carmi, S.; Levy-Lahad, E.; et al. The Landscape of Autosomal-Recessive Pathogenic Variants in European Populations Reveals Phenotype-Specific Effects. Am. J. Hum. Genet. 2021, 108, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Thareja, G.; Al-Sarraj, Y.; Belkadi, A.; Almotawa, M.; Ismail, S.; Al-Muftah, W.; Badji, R.; Mbarek, H.; Darwish, D.; Fadl, T.; et al. Whole Genome Sequencing in the Middle Eastern Qatari Population Identifies Genetic Associations with 45 Clinically Relevant Traits. Nat. Commun. 2021, 12, 1250. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Kumar, C.; Zeng, W.F.; Strauss, M.T. Artificial Intelligence for Proteomics and Biomarker Discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, M.; Kramer, M.; Parla, J.; Goes, F.S.; Potash, J.B.; McCombie, W.R.; Zandi, P.P. Validation and Assessment of Variant Calling Pipelines for Next-Generation Sequencing. Hum. Genom. 2014, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Coldren, C.; Karunamurthy, A.; Kip, N.S.; Klee, E.W.; Lincoln, S.E.; Leon, A.; Pullambhatla, M.; Temple-Smolkin, R.L.; Voelkerding, K.V.; et al. Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines: A Joint Recommendation of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn. 2018, 20, 4–27. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).