Abstract
mRNAs have some regulatory codes which can define the fate of an individual mRNA in translation. We have developed a flexible online database JetGene (https://jetgene.bioset.org/; accessed on 23 October 2020) that contains cDNA, CDS, 5′-UTR, 3′-UTR sequences from Bacteria, Fungi, Metazoa, Plants, Protists and Vertebrates with the aim of searching regulatory codes in mRNA and studying their correlation with the translational efficiency. It has a friendly interface and puts together a set of tools which are necessary for designing experiments. JetGene allows doing a benchmark analysis of sequences, namely: (1) to estimate the variation of length, nucleotide composition, frequency of codon usage, to analyze GC-content, CpG-islands, to study nucleotides surrounding the start codon and much more; (2) to identify and define the statistically significant representation of potential regulatory contexts at mRNA with different translation efficiency. A user can make a bioinformatics analysis for full-length transcripts or for a fragment of transcripts, or for coding/non-coding regions. Every step of the work is accompanied by a graphical interpretation of the results. Moreover, the beta-version of JetGene (https://beta.bioset.org, under construction; accessed on 23 October 2020) allows users to compare two datasets of mRNA and to apply omics data for searching and predicting the regulatory determinants of translation.
1. Introduction
Translation is a fundamental process and an important starting point in gene expression regulation for the cells of all living organisms because, in this process, the encoding potential of mRNA is exposed via the protein molecule. In the current view, translational control, in general, is decisive in the continuity of cell events and, for example, in the response of plant cells to various environmental factors and different metabolites [1]. The special attention of researchers is focused on the discrepancy between mRNAs levels and translation effectivity in eukaryotic cells, in particular in plant cells [2,3]. The experimental data of the various elegant studies show that when decoding their genomes, organisms are able to widely use the regulation and decoding rules of higher orders along with the canonical translational rules, thereby suggesting the presence of specific regulatory codes characteristic of mRNA translation.
As we know, cDNA includes the following parts: 5′ untranslated region (5′-UTR), coding region (CDS), and 3′ untranslated region (3′-UTR). These regions modulate translation at “control points”: initiation, elongation, and translation termination. According to the current opinion, numerous regulatory codes could be hidden in nucleotide contexts of such cDNA regions. Each element separately or some of them in combination can determine the fate of an individual mRNA in the translational process [4]. An in silico analysis of the cDNA parts, which have mentioned above, CDS, 5′-UTR and 3′-UTR, is applied for prediction of these regulatory codes.
For the purpose of such regulatory codes, discovery in mRNA, and their correlation with the efficiency of translation, we have created an online database JetGene (https://jetgene.bioset.org/; accessed on 23 October 2020). In addition, JetGene allows estimation of the variation of nucleotide composition, codon usage, frequency to study nucleotides surrounding the start codon, and much more.
2. Experiments
2.1. The Motivation for the Development of JetGene
Our goal for the creation of JetGene is to provide users that have minimal experience in programming and in a bioinformatics analysis with a simple and useable toolkit for analyzing and planning an experiment. So, in JetGene, we have put together a wide set of options that are useful for any researcher. JetGene allows to make a comparative analysis of sequences, such as (1) to estimate the variation of length, nucleotide composition, frequency of codon usage, to analyze GC-content, CpG-islands, to study nucleotides surrounding the start codon and much more; (2) to identify and define the statistically significant representation of potential regulatory contexts at mRNA with different translation efficiency. JetGene contains cDNA, CDS, 5′-UTR, 3′-UTR sequences for six groups of living organisms: Bacteria, Fungi, Metazoa, Plants, Protists, and Vertebrates. It should be noted that the analysis could be performed both on full-length transcripts and on truncated transcripts, and on coding/non-coding regions.
In addition, the beta-version of JetGene (https://beta.bioset.org, under construction; accessed on 23 October 2020) allows users to compare two mRNA datasets (Figure 1) and to apply omics data for searching the regulatory determinants of translation.
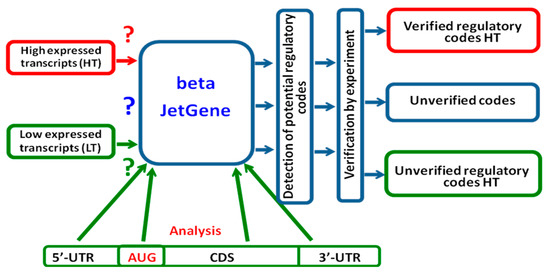
Figure 1.
Schematic comparison of two individual user datasets with the aim of detecting and verifying regulatory codes in high expressed/low expressed transcripts.
2.2. “System of Nested Datasets” Algorithm
Another important advantage of JetGene is a “System of nested datasets” algorithm, which we have implemented in our work (Figure 2). Its essence is that at the first stage of work, a researcher selects a certain criterion as a primary one, for example, (1) cDNA with the specified length “CDNA length” and creates the main dataset. At subsequent stages, a researcher can use the remaining parameters as additional ones, for example, (2) add parameter “5′-UTR length”. It will allow to choose sequences with the specified 5′-UTR length and to create a second-order dataset. Then a researcher can add the next parameter, for example, motive search “Motifs”. As a result of such step, JetGene will select sequences containing this motif from the second-order dataset.
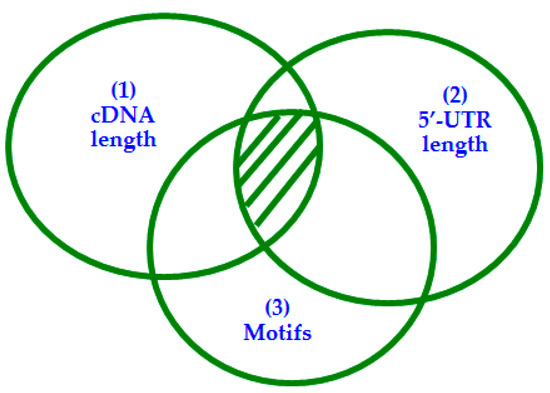
Figure 2.
“System of nested datasets” algorithm. Three overlapping circles represent an opportunity to choose sequences by criteria “cDNA length”, “5′-UTR length”, “Motifs”. (1) “cDNA length” is selected as a primary criterion, (2) “5′-UTR length” and (3) “Motifs”—as additional ones. The resulting dataset is located at intersection of all circles and is shaded.
So, a user has the ability to create a series of subsequent datasets, each of which is based on the previous ones without extracting intermediate results from JetGene. A researcher can define the criteria hierarchy (main and auxiliary). As a result, a user has to obtain different variants of biological texts that satisfy nontrivial parameter combinations. The number of such combinations is unlimited. Besides, graphical representation of analysis results is realized in JetGene. All of this greatly simplifies in silico analysis.
3. Results
3.1. Database Overview
Transcriptomic data of six key groups of living organisms: Bacteria (44048 species), Fungi (782 species), Metazoa (68 species), Plants (45 species), Protists (195 species), and Vertebrates (139 species) were downloaded from Ensembl (https://www.ensembl.org/index.html; accessed on 23 October 2020) [5] on 28 June 2017 and updated regularly (once a week). Description of each transcriptome includes information about assembly. Gene Ontology Annotation (GO) [6] is given for many transcriptomes. The main interface of JetGene contains four major sections: cDNA data, CDS data, 5′-UTR data, and 3′-UTR data (Figure 3) for most eukaryotic organisms. It should be noted that we obtain information about 5′-UTR and 3′-UTR as subtraction CDS from cDNA. In addition to the major ones, JetGene has one auxiliary GO-section. It is presented only when information about GO annotations is provided by the Ensembl server, and this section is unrelated to the major ones.
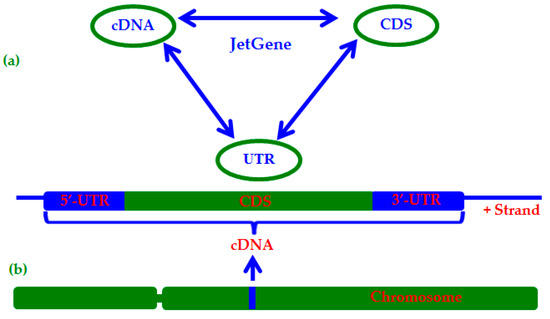
Figure 3.
(a) General structure of JetGene. Arrows indicate that a user could start the analysis with any section (CDNA, for example) and continue it at any other section (CDS or UTR, for example) without extracting intermediate results from JetGene; (b) Schematic gene representation on a chromosome (introns are removed).
JetGene is implemented in a modular form. Modules could be applied both individually and in combination for conducting extended and continuous research. The web interface of the database consists of 10 main modules inherent to any of the four major sections (“CDS”, “cDNA”, “5′-UTR”, “3′-UTR”) and three modules inherent to the section “CDS data”. The list of modules available for every section is shown in Figure 4.
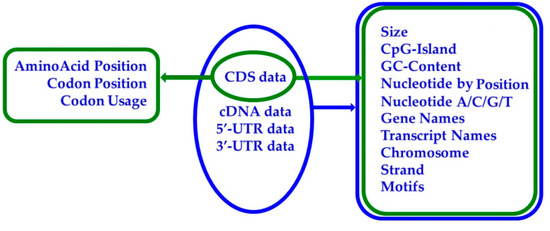
Figure 4.
A toolkit of four major sections of JetGene. Tools that are accessible for every of the available data types (CDS, CDNA, 5′-UTR, 3′-UTR) are shown schematically.
It is important to note that a user can extract the obtained sequences in a FASTA-format at any step of the work. Moreover, JetGene gives a visual representation for comparison of the performed analysis of the narrow user dataset with an initial transcriptome dataset for researched organisms. Besides, there is a possibility to upload a user dataset and analyze it (this option is available after free registration). In this case, all toolkits will be available except “chromosome”, “motifs”, and “strand”, besides that, a sequence does not markup on CDS, CDNA, 5′-UTR, 3′-UTR.
Here we give a list of modules specific for every section (major and auxiliary).
Modules specific to “CDS data” only:
- Amino Acid Position
- Codon Position
- Codon Usage
Modules specific to “CDS data”, “cDNA data”, “5′-UTR data”, “3′-UTR data”:
- CDS/cDNA/5′-UTR/3′-UTR Length
- CpG-Island in CDS/cDNA/5′-UTR/3′-UTR
- GC-Content in CDS/cDNA/5′-UTR/3′-UTR
- Nucleotide by Position in CDS/cDNA/5′-UTR/3′-UTR
- Nucleotide A/C/G/T in CDS/cDNA/5′-UTR/3′-UTR
- Gene Names
- Transcript Names
- Chromosome
- Strand
- Motifs
Module specific to GO:
- Gene Ontology Annotations
Further, we provide a brief description of modules specific for each of the four major sections “CDS data”, “cDNA data”, “5′-UTR data”, “3′-UTR data” in detail.
3.2. Modules Specific to “CDS Data” Only
3.2.1. Amino Acid Position
This module makes it possible to display an amino acid that is located on the sequence in positions 1–10, both from the C-terminus and N-terminus. It can be helpful for the analysis and design of signal peptides [7] and for applying the N-end rule. According to this rule, the second N-terminal amino acid of a protein determines its half-life [8].
3.2.2. Codon Position
This utility is similar to the previous one. It defines which nucleotide triplets are located in the position 1–10 forming 5′-end or 3′-end of CDS. With this application, users can study the N-terminal region of the protein or of the signal peptide at a nucleotide level.
3.2.3. Codon Usage
The current tool shows triplets encoding amino acids in CDS and also their numerical and percentage composition (we take the sum of all triplets encoding present amino acid as 100%, but we do not take the sum of all triplets in CDS). This tool allows to study full-length CDSs and truncated sequences of CDSs (an option “Sequence region to calculate data (%)”). Such utility will be helpful for works similar to [9], in which authors analyzed the codon usage of adenoviral proteins and evaluate their adaptation to the host codons.
3.3. Modules Specific to “CDS Data”, “cDNA Data”, “5′-UTR Data”, “3′-UTR Data”
3.3.1. CDS/CDNA/5′-UTR/3′-UTR Length
This module displays all lengths of CDS/CDNA/5’-UTR/3’-UTR sequences in the transcriptome of the studied organism. It gives the possibility to choose sequences of a certain length (scale division is 500 nucleic acids) or to set a length range at option “Values interval to calculate data”. Such utility can be useful when choosing sequences, with a maximum length for gene cloning into a certain vector.
3.3.2. CpG-Island in CDS/CDNA/5′-UTR/3′-UTR
This application analyzes CpG-islands and calculates the percent of CpG dinucleotides in CpG-islands in CDS/cDNA/5′-UTR/3′-UTR. The tool allows choosing all sequences with a certain percent interval of CpG dinucleotides in CpG-islands. Moreover, it works both with full-length and truncated sequences (an option “Sequence region to calculate data (%)”).
3.3.3. GC-Content in CDS
The current tool is similar to the “GpC-island in CDS/CDNA/5′-UTR/3′-UTR”, but it takes into account all G and C nucleotides in transcripts. Users have the ability to pick up all transcripts that have certain GC content (scale division is 1%). This utility can be applied in research similar to [10], in which authors analyzed codon usage in CDSs of H. manillensis and also distribution of GC dinucleotides content in CDSs.
3.3.4. Nucleotide by Position in CDS/CDNA/5’-UTR/3’-UTR
This application shows what nucleotide is located in the position 1–10 form 5′-end or from 3′-end of CDS/CDNA/5′-UTR/3′-UTR. It can be useful in works similar to [11], in which authors analyzed the immediate upstream region of the 5′-UTR from the AUG start codon in different genes of A. thaliana and showed that a region from positions −1 to −5 is most important for translational efficiency.
3.3.5. Nucleotide A/C/G/T in CDS/CDNA/5′-UTR/3′-UTR
This utility can calculate the percentage of A/C/G/T in CDS/CDNA/5′-UTR/3′-UTR. It analyzes both the full-length and truncated sequences (an option “Sequence region to calculate data (%)”). Moreover, users can pick up all sequences with a certain percentage (option “Values interval to calculate data”) and form a dataset of sequences with a certain nucleotide composition. Such a tool could be useful in works like [4] in which scientists revealed the influence of 5′-UTR mono- and di-nucleotide composition on ribosome loading in A. thaliana.
3.3.6. Gene Names
This module enables to select sequences by a list of names or select sequences that have the common part of their names. Apart from that, this module allows to upload the user datasets by standard gene names if information about an organism is represented in JetGene.
3.3.7. Transcript Names
The current application is similar to “Gene names”, but users can find unique transcript(s) or all transcripts related to a certain gene or transcripts with a common part of their names. The utility enables easy identification of all isoforms of a certain gene and finding some difference between them.
3.3.8. Chromosome
The current tool shows sequence distribution on chromosomes and on mitochondrial DNA. It can be useful in cases when a researcher is interested in sequences that are located on a certain chromosome or when the user compares two datasets obtained for two different chromosomes.
3.3.9. Strain
The current utility allows to distinguish transcripts located on the forward strand from transcripts located on the reverse strand and then to divide the dataset into two different parts based on this parameter. For bacteria, such simple manipulation makes it possible to find genes that are assigned incorrectly to the one operon. Moreover, this tool can be useful in research like [12], in which authors showed little asymmetry between forward/reverse strands on open reading frame number and between lengths of genes in C. acetobutylicum.
3.3.10. Motifs
This module finds out sequences that contain a certain motif. It can search several motifs simultaneously (by means of an operator AND) or one of the listed motifs (by means of an operator OR). Users can perform an analysis on full-length sequences and on truncated transcripts. The results are visually presented as a bar graph that displays motif occurrence frequency.
4. Discussion
4.1. Comparison JetGene with Other Online Databases
We have created JetGene that is accessible via the web interface and very simple to use. It is developed not only for experienced bioinformatics but for experimentalists who have minimal experience in bioinformatics analysis and in programming. Let us compare JetGene with other online databases.
Currently, biological texts of sequences are stored in different web servers. Most frequently, such recourses contain CDSs and protein sequences corresponding to them, as for example, in GenBank [13] and in KEGG [14,15]. Furthermore, they contain metabolic pathways maps, software package Blast [16,17] for searching homologous sequences, a list of publications, links to external Internet resources which provide a comprehensive description of the studied gene or protein, and much more. Notwithstanding the diversity of represented information, when the user works with such databases, the search is only possible at a trivial level: find a sequence with a given function or detect a homologous sequence.
Then, we should describe web resources that allow conducting a complex analysis of sequences. These include Ensembl (https://www.ensembl.org/; accessed on 23 October 2020) [5], which served as the basis for JetGene. It should be mentioned that JetGene contains information about all organisms and about all nucleotide sequences represented in Ensembl. Nowadays, Ensembl is one of the most important Internet resources which stores information about gene annotation, genetics, comparative genomics, and epigenomics for a huge number of living organisms. Possibilities of using Ensembl range from a quick overview of information to whole-genome in silico analysis. Meanwhile, Ensembl supports access via BioMart [18], Perl and REST APIs [19,20] or via FTP for providing access to information the user is interested in. However, BioMart does not use the whole information that is stored in Ensembl. For example, BioMart does not use information about many organisms represented in Ensembl. Besides, using API и FTP requires programming skills that not all users have.
However, BioMart provides an opportunity to work separately with CDS, CDNA, 5′-UTR, 3′-UTR, and with protein sequences. Biomart toolkit is larger than JetGene toolkit. In particular, BioMart allows to set chromosome coordinates, to obtain information about intron-exon structure, to do a search by phenotype, to find orthologous in other organisms, and much more. Herewith, the intersection between the BioMart toolkit and the JetGene toolkit is insignificant. Particularly both BioMart and JetGene give the opportunity to display CDS, CDNA, 5′-UTR, 3′-UTR sequences, to find a gene by ID or some genes by GO (gene ontology annotation), to choose chromosome for an analysis. Nevertheless, such essential information as sequence length, GC-content, sequence location at the forward/reverse strand is displayed in the resulting file. So, a user should select sequences manually from the resulting file by the parameters mentioned above, and this increases the time of analysis.
In addition, some information, for instance, percentage of nucleotide A/C/G/T or what nucleotide is located in the position 1–10, the distribution of triplets within the dataset, is not provided by BioMart. The possibility to work with truncated sequences implemented by BioMart is not so clear as by JetGene. Apart from that, the graphical representation of the analyzed results by the selected parameter is omitted.
Moreover, there are a number of limitations when the user tries to make several iterations of the analysis or when the user is trying to do transfers between CDNA/CDS/UTR. For example, it is more difficult to begin with a 5′-UTR analysis, than to transfer it to the analysis of cDNA (cDNA which contains researched 5′-UTR) without additional supporting actions.
UCSC Genome Browser (https://genome.ucsc.edu/; accessed on 15 October 2020) [21,22] is another information resource that allows making a comprehensive search and analysis of sequences. It contains information about more than 100 species, and for some of them, it has several variants of transcriptome assembly. At the same time, UCSC Genome Browser covers fewer kingdoms than JetGene. Moreover, any kingdom includes fewer organisms than JetGene. For instance, it does not contain any information about Plants; besides that, information about Fungi is only provided for S. cerevisiae.
UCSC Table Browser is a flexible and powerful graphical interface designed for manipulating and querying the UCSC Genome Browser. Table Browser like JetGene allows to select sequences by several user criteria, to form sequences dataset with the help of some tools and extract the obtained dataset in FASTA-format. Nevertheless, UCSC settings are less clear than JetGene settings. In order to be able to form a correct request or to apply multiple query criteria, to download user data, and to use the information of this internet resource, the user should study the structure of the input/output data, the description of filters, and to have some bioinformatics knowledge. When researchers solve similar tasks regularly, it is justified. However, learning settings and options regularly takes considerable time when tasks change rapidly or when the selection of sequences is based on different criteria. It should be noted that graphical interpretation of results is not realized in UCSC Table Browser.
At the same time, data from UCSC Table Browser can be exported directly to the open web-based platform Galaxy (http://usegalaxy.org; accessed on 15 October 2020) [23], but it takes additional time. Some options of Galaxy are the same as for the JetGene tools (for example, CDS, CDNA, 5′-UTR, 3′-UTR analysis, GC-content analysis, an ability to choose sequences by length, an opportunity to study both full-length and truncated sequences, a possibility to extract sequences in FASTA-format) and graphical visualization of results is implemented in it. Nevertheless, Galaxy options are not so clearly defined as JetGene tools. Additionally, it should be noted that both Galaxy and JetGene can do transfers between CDNA/CDS/5′-UTR/3′-UTR. However, Galaxy makes such transfers in a less trivial form, and they take a longer period of time.
4.2. Usage of JetGene
As an example of JetGene usage (it was called FlowGene initially), we can cite the article [24]. In this work, the authors studied the influence of the 5′-UTR nucleotide context on the gene expression in plants and used JetGene for a bioinformatics analysis. They applied the “System of nested datasets” algorithm. Researchers selected the (1) 5′-UTR of not less than two thousand base pairs (minimum size of CpG-island) as a primary parameter and created the main dataset. Then they selected additional criteria for creating subsequent datasets: (2) GC-content higher than 50% (one of the characteristics of CpG-islands); (3) nucleotides surrounding the start codon at positions +4 and −3 according to Kozak sequence [25]; (4) the absence of alternative start and stop codons within sequences. Then they searched for six-nucleotide motifs, which are contained not less than 50% in all sequences from the result dataset. Subsequently, these motifs were incorporated into the design of the synthetic sequence.
5. Conclusions
Fluctuations in nucleotide composition are revealed in genomes of all organisms, and they define gene expression efficiency for any species [26,27,28,29]. Knowledge about the fine mechanisms of translation is very important for understanding what makes organisms switch genes. Applying information about nucleotide context variations helps to develop antiviral vaccines [30], selecting host expression system for an experiment [31], predict genes based on genomic sequences [32,33], design degenerate primers [34], and much more. Research on fluctuations in nucleotide composition occupies a central position in such important areas as molecular evolution [35] and biotechnology [36]. The availability of genome-wide sequences allows a unique opportunity to identify regularities in the distribution of various properties [37] both across the whole genome and for parts of separate transcripts. Thus, for example, it was identified a dependence between nucleotide composition and efficiency of protein translation [38]. It was established that dependence exists between nucleotide composition and level of gene expression [39,40]. Moreover, it was also shown to change in nucleotide composition depending on the localization of the sequence [41] and much more.
In such studies, success is highly dependent on the ability to form sequences datasets of biological texts based on a wide range of criteria. The greater the number of parameters involved in the analysis, the higher the potential for creating and manipulating sequence datasets. So, the potential for searching and identifying characteristics that influence the biological properties of sequences will be greater. In accordance with all the above requirements, we have created an online database JetGene which allows to carry out such analysis quickly and efficiently. It is important to note that currently, it gives a comprehensive understanding of the structure–function potential of the biological texts encoded in mRNAs. JetGene is only developed for the analysis of nucleotide sequences and aimed at experimentalists who have minimal experience in bioinformatics analysis. The uniqueness of our database is that any user is able to scan huge amounts of information within the shortest time or can create different datasets of nucleotide sequences de novo, which satisfy the goals of the experiment. In this way, a researcher can apply a wide set of options based on different user criteria for conducting a comprehensive analysis, then form a nucleotide sequences dataset and extract it in FASTA-format from JetGene. In addition, a graphical representation of results accompanies every phase of the study. Such cute details have greatly facilitated the work of any user.
Supplementary Materials
The poster presentation is available online at https://www.mdpi.com/article/10.3390/IECPS2020-08624/s1.
Author Contributions
O.M. developed JetGene; N.S., A.T. and I.D. tested JetGene; N.S. and I.G.-P. wrote the paper; I.G.-P. supervised the project. All authors have read and agreed to the published version of the manuscript.
Funding
The authors gratefully acknowledge the financial support from the Russian Science Foundation (project no. 18-14-00026; IVG-P).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CDS | Coding DNA Sequence |
| cDNA | Complementary DNA |
| GO | Gene Ontology Annotation |
| HT | High Expressed Transcripts |
| LT | Low Expressed Transcripts |
| UTR | Untranslated Region |
References
- Guerra, D.; Crosatti, C.; Khoshro, H.; Mastrangelo, A.M.; Mica, E.; Mazzucotelli, E. Post-transcriptional and post-translational regulations of drought and heat response in plants: A spider’s web of mechanisms. Front. Plant Sci. 2015, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Bärenfaller, K.; Grossmann, J.; Grobei, M.A.; Hull, R.; Hirsch-Hoffmann, M.; Yalovsky, S.; Zimmermann, P.; Grossniklaus, U.; Gruissem, W.; Baginsky, S. Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 2008, 320, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Bärenfaller, K.; Massonnet, C.; Walsh, S.; Baginsky, S.; Buhlmann, P.; Hennig, L.; Hirsch-Hoffmann, M.; Howell, K.A.; Kahlau, S.; Radziejwoski, A.; et al. Systems-based analysis of Arabidopsis leaf growth reveals adaptation to water deficit. Mol. Syst. Biol. 2012, 8, 606. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Bailey-Serres, J. mRNA sequence features that contribute to translational regulation in Arabidopsis. Nucleic Acids Res. 2005, 33, 955–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.; Achuthan, P.; Akann, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.; Azov, A.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Douglass, E.; Dunn, N.; Good, B.; Harris, N.L.; Lewis, S.E.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; et al. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Jeiranikhameneh, M.; Moshiri, F.; Keyhan Falasafi, S.; Zomorodipour, A. Designing Signal Peptides for Efficient Periplasmic Expression of Human Growth Hormone in Escherichia coli. J. Microbiol. Biotechnol. 2017, 27, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Tasaki, T.; Sriram, S.M.; Park, K.S.; Kwon, Y.T. The N-end rule pathway. Annu. Rev. Biochem. 2012, 81, 261–289. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, E.; Martí-Solano, M.; Fillat, C. Codon optimization of the adenoviral fiber negatively impacts structural protein expression and viral fitness. Sci. Rep. 2016, 6, 27546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, D.L.; Ma, L.B.; Khan, M.S.; Zhang, X.X.; Xu, S.Q.; Xie, J.Y. Analysis of codon usage patterns in Hirudinaria manillensis reveals a preference for GC-ending codons caused by dominant selection constraints. BMC Genom. 2018, 19, 542. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, G.; Jeon, E.; Sohn, E.J.; Lee, Y.; Kang, H.; Lee, D.W.; Kim, D.H.; Hwang, I. The immediate upstream region of the 5′-UTR from the AUG start codon has a pronounced effect on the translational efficiency in Arabidopsis thaliana. Nucleic Acids Res. 2014, 42, 485–498. [Google Scholar] [CrossRef]
- Zhao, H.L.; Xia, Z.K.; Hua, Z.G.; Wei, W. Selectional versus mutational mechanism underlying genomic features of bacterial strand asymmetry: A case study in Clostridium acetobutylicum. Genet. Mol. Res. 2015, 14, 1911–1925. [Google Scholar] [CrossRef]
- Benson, D.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.; Ostell, J.; Sayers, E. GenBank. Nucleic Acids Res. 2017, 45, D37–D42. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boratyn, G.; Camacho, C.; Cooper, P.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.; Matten, W.; McGinnis, S.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef] [Green Version]
- Kinsella, R.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef] [PubMed]
- Ruffier, M.; Kähäri, A.; Komorowska, M.; Keenan, S.; Laird, M.; Longden, I.; Proctor, G.; Searle, S.; Staines, D.; Taylor, K.; et al. Ensembl core software resources: Storage and programmatic access for DNA sequence and genome annotation. Database 2017, 2017, bax020. [Google Scholar] [CrossRef]
- Yates, A.; Beal, K.; Keenan, S.; McLaren, W.; Pignatelli, M.; Ritchie, G.R.; Ruffier, M.; Taylor, K.; Vullo, A.; Flicek, P. The Ensembl REST API: Ensembl Data for Any Language. Bioinformatics 2015, 31, 143–145. [Google Scholar] [CrossRef] [Green Version]
- Hung, J.H.; Weng, Z. Visualizing Genomic Annotations with the UCSC Genome Browser. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot093062. [Google Scholar] [CrossRef]
- Haeussler, M.; Zweig, A.; Tyner, C.; Speir, M.; Rosenbloom, K.; Raney, B.; Lee, C.; Lee, B.; Hinrichs, A.; Gonzalez, J.; et al. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 2019, 47, D853–D858. [Google Scholar] [CrossRef] [Green Version]
- Goecks, J.; Nekrutenko, A.; Taylor, J.; Team, G. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyurin, A.; Kabardaeva, K.; Gra, O.; Mustafaev, O.; Sadovskaya, N.; Pavlenko, O.; Goldenkova-Pavlova, I. Efficient Expression of a heterologous gene in plants depends on the nucleotide composition of mRNA’s 5’-region. Russ. J. Plant. Physiol. 2016, 63, 511–522. [Google Scholar] [CrossRef]
- Kozak, M. Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol. Cell. Biol. 1989, 9, 5134–5142. [Google Scholar] [CrossRef]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Quax, T.; Claassens, N.; Söll, D.; van der Oost, J. Codon Bias as a Means to Fine-Tune Gene Expression. Mol. Cell. 2015, 59, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Gao, H.; Liu, J.; Tian, P.; Nan, Z. Comprehensive analysis of correlations among codon usage bias, gene expression, and substitution rate in Arachis duranensis and Arachis ipaënsis orthologs. Sci. Rep. 2017, 7, 14853. [Google Scholar] [CrossRef] [Green Version]
- Lingemann, M.; Liu, X.; Surman, S.; Liang, B.; Herbert, R.; Hackenberg, A.; Buchholz, U.; Collins, P.; Munir, S. Attenuated Human Parainfluenza Virus Type 1 Expressing Ebola Virus Glycoprotein GP Administered Intranasally Is Immunogenic in African Green Monkeys. J. Virol. 2017, 91, e02469-16. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhao, W.M.; Wang, H.; Zhou, Y.B.; Luan, Y.; Qi, M.; Cheng, Y.Z.; Tang, W.; Liu, J.; Yu, H.; et al. Codon usage bias in Chlamydia trachomatis and the effect of codon modification in the MOMP gene on immune responses to vaccination. Biochem. Cell Biol. 2007, 85, 218–226. [Google Scholar] [CrossRef]
- Picardi, E.; Pesole, G. Computational methods for ab initio and comparative gene finding. Methods Mol. Biol. 2010, 609, 269–284. [Google Scholar] [CrossRef]
- König, S.; Romoth, L.; Gerischer, L.; Stanke, M. Simultaneous gene finding in multiple genomes. Bioinformatics 2016, 32, 3388–3395. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Gu, W.; Ma, J.; Sun, X.; Lu, Z. Analysis of synonymous codon usage in H5N1 virus and other influenza A viruses. Biosystems 2005, 81, 77–86. [Google Scholar] [CrossRef]
- Kim, N.; Lim, S.; Chae, H.; Park, Y. Complete mitochondrial genome of the Amur hedgehog Erinaceus amurensis (Erinaceidae) and higher phylogeny of the family Erinaceidae. Genet. Mol. Res. 2017, 16. [Google Scholar] [CrossRef]
- Kinkema, M.; Geijskes, J.; Delucca, P.; Palupe, A.; Shand, K.; Coleman, H.; Brinin, A.; Williams, B.; Sainz, M.; Dale, J. Improved molecular tools for sugar cane biotechnology. Plant Mol. Biol. 2014, 84, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Chaney, J.; Clark, P. Roles for Synonymous Codon Usage in Protein Biogenesis. Annu. Rev. Biophys. 2015, 44, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Tuller, T.; Carmi, A.; Vestsigian, K.; Navon, S.; Dorfan, Y.; Zaborske, J.; Pan, T.; Dahan, O.; Furman, I.; Pilpel, Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 2010, 141, 344–354. [Google Scholar] [CrossRef] [Green Version]
- Whittle, C.; Extavour, C. Expression-Linked Patterns of Codon Usage, Amino Acid Frequency, and Protein Length in the Basally Branching Arthropod Parasteatoda tepidariorum. Genome Biol. Evol. 2016, 8, 2722–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Yan, Y.; Yue, Q.; Liu, X.; Chu, X.; Wu, N.; Fan, Y. Predicting synonymous codon usage and optimizing the heterologous gene for expression in E. coli. Sci. Rep. 2017, 7, 9926. [Google Scholar] [CrossRef]
- Diament, A.; Pinter, R.; Tuller, T. Three-dimensional eukaryotic genomic organization is strongly correlated with codon usage expression and function. Nat. Commun. 2014, 5, 5876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).