
The Use of Personalized Medicine in Pancreatic Ductal Adenocarcinoma (PDAC): New Therapeutic Opportunities



Abstract
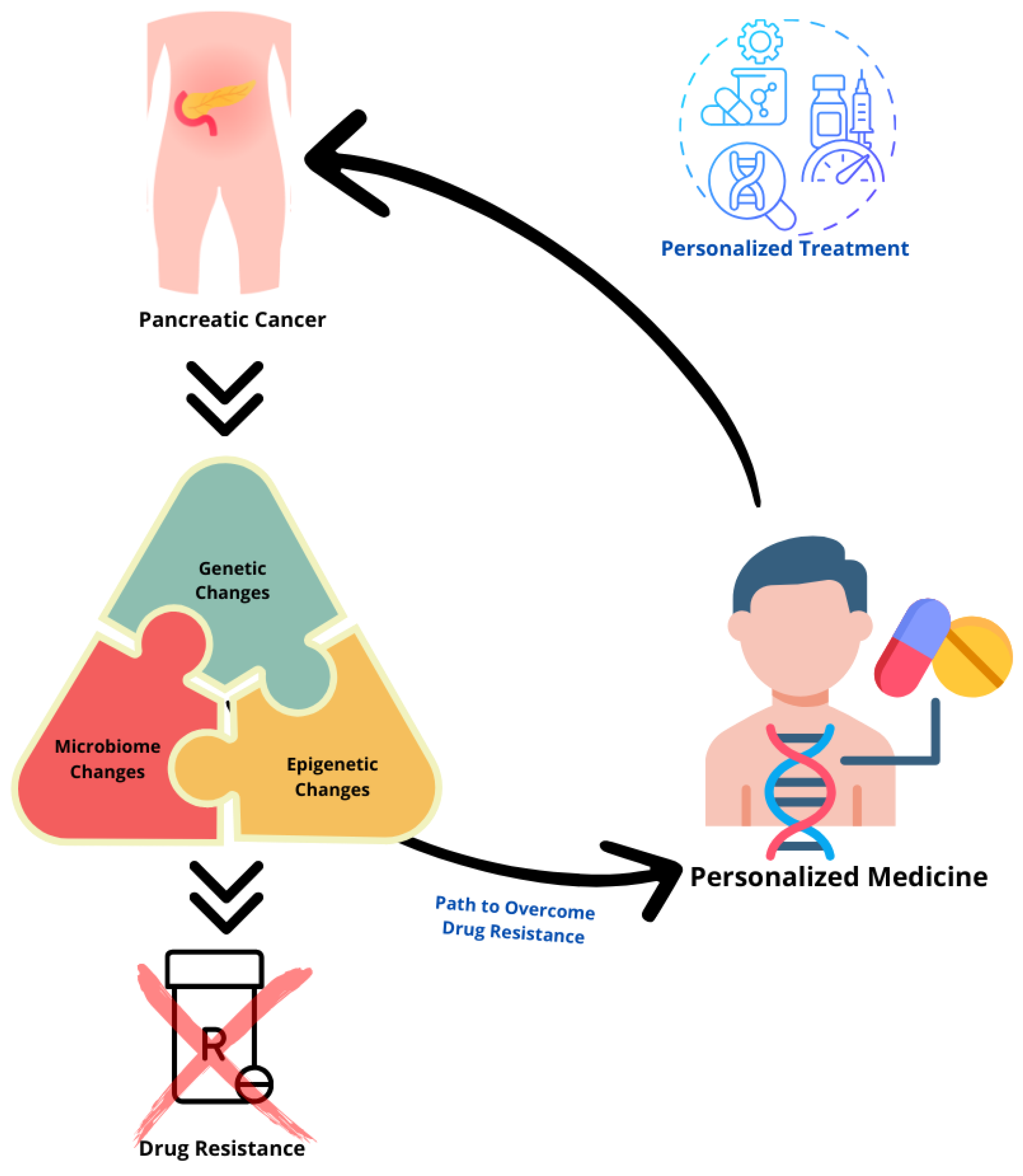
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Pancreatic Ductal Adenocarcinoma (PDAC): A Global Health Concern
2.1. Dilemma in Diagnosis of PDAC
2.2. Dilemma in Chemotherapy of PDAC
3. Standard Chemotherapy of PDAC
3.1. Antimetabolite Drug 5-FU in PDAC
3.1.1. 5-FU Mechanism of Action
3.1.2. Chemoresistance to and Cytotoxicity of 5-FU
3.2. Gemcitabine in PDAC
4. Pathogenesis of PDAC
4.1. Genetics and PDAC
4.2. Epigenetics and PDAC
4.3. Microbiome and PDAC
5. Personalized Approaches That Influence Standard Therapies Response in PDAC
5.1. Genetic Polymorphism of DPYD
5.2. Chronomodulated Treatment with 5-FU
5.3. cNEK6 Induces GEM Resistance
6. Precision Medicine: Engineering the Microbiome to Improve Drug Outcomes
Personalized Alteration of the Gut Microbiota: Future Therapy Opportunities
- (1)
- Inhibition of metabolic activities performed by gut bacterial enzymes: Microbial enzymes that catalyze undesirable reactions within the gastrointestinal tract present potential targets for therapeutic intervention. The development of selective and non-lethal enzyme inhibitors aimed at these enzymes could offer a promising approach to modulating gut health [110].
- (2)
- Removal of specific bacterial species or strains: An additional method for microbiome modification entails the targeted depletion of specific strains exhibiting detrimental activities, particularly those that metabolize pharmacological agents into toxic metabolites. This strategy seeks to improve the safety and efficacy of therapeutic applications [102,111].
- (3)
- Introduction or engrafting of engineered strains into the gut: In addition to efforts aimed at eliminating specific strains, there is a growing focus on the introduction of engineered strains into the host as live bacterial therapeutics. This approach bears similarities to the application of probiotics, which are designed to provide beneficial functions [102,112].
- (4)
- Direct genetic modification of bacterial cells present within the gastrointestinal tract: Recent advancements in the modification of gut microbiota indicate a paradigm shift from the introduction of engineered strains to the direct genetic modification of bacterial populations that naturally colonize the gastrointestinal tract. This progressive approach, known as in vivo or in situ engineering, involves the execution of genetic modification techniques within the host organism, as opposed to traditional methodologies conducted within a laboratory environment [102,113].
7. New Therapeutic Strategies: Future Directions
7.1. Probiotic-Based Regimens Combined with Chemotherapy Drugs
7.2. Gene Therapy and Oncolytic Virotherapy for PDAC
7.3. Epigenetics Engineering: Modifying miRNAs with GEM and 5-FU for Developing miRNA-Based Therapeutics
7.4. Bacteria and Aptamer-Drug Conjugates (ApDCs)
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ApDC | aptamer-drug conjugates |
| CH2THF | 5,10-methylenetetrahydrofolate |
| DHFU | dihydrofluorouracil |
| DPD | dihydropyrimidine dehydrogenase |
| dTTP | deoxythymidine triphosphate |
| DUT | dUTPase |
| dUMP | deoxyuridine monophosphate |
| dUTP | deoxyuridine triphosphate |
| FBAL | α-2-fluoro-β-alanine |
| FdUDP | fluorodeoxyuridine diphosphate |
| FdUMP | fluorodeoxyuridine monophosphate |
| FUMP | 5-fluoro-uridine-monophosphate |
| FUPA | α-2-fluoro-β-ureido propionic acid |
| FUR | fluorouridine |
| FUTP | fluorodeoxyuridine triphosphate |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GEM | gemcitabine |
| IPMN | intraductal papillary mucinous neoplasm |
| LTR | long terminal repeat sequences |
| miRNAs | microRNAs |
| MR | mendelian randomization |
| OPRT | orotate phosphorylase |
| PanIN | pancreatic intraepithelial neoplasm |
| PDAC | pancreatic ductal adenocarcinoma |
| RNR | ribonucleotide reductase |
| RRV | retroviral replicating vector |
| TK | thymidine kinase |
| TME | tumor microenvironment |
| Toca 511 | vocimagene amiretrorepvec |
| TP | thymidine phosphorylase |
| TS | thymidylate synthase |
| UK | uridine kinase |
| UP | uridine phosphorylase |
| yCD | cytosine deaminase |
| 5-FU | 5-fluorouracil |
| 5-FC | 5-fluorocytosine |
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Klatte, D.C.F.; Boekestijn, B.; Onnekink, A.M.; Dekker, F.W.; van der Geest, L.G.; Wasser, M.N.J.M.; Feshtali, S.; Mieog, J.S.D.; Luelmo, S.A.C.; Morreau, H.; et al. Surveillance for Pancreatic Cancer in High-Risk Individuals Leads to Improved Outcomes: A Propensity Score-Matched Analysis. Gastroenterology 2023, 164, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Grant, T.J.; Hua, K.; Singh, A. Molecular Pathogenesis of Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275. [Google Scholar] [PubMed]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Kommalapati, A.; Tella, S.H.; Goyal, G.; Ma, W.W.; Mahipal, A. Contemporary Management of Localized Resectable Pancreatic Cancer. Cancers 2018, 10, 24. [Google Scholar] [CrossRef]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402.e1. [Google Scholar] [CrossRef]
- Elhussein, A.; Baymuradov, U.; NYGC ALS Consortium; Elhadad, N.; Natarajan, K.; Gürsoy, G. A framework for sharing of clinical and genetic data for precision medicine applications. Nat. Med. 2024, 30, 3578–3589. [Google Scholar] [CrossRef]
- Kim, R.; Kim, S.; Oh, B.B.; Yu, W.S.; Kim, C.W.; Hur, H.; Son, S.Y.; Yang, M.J.; Cho, D.S.; Ha, T.; et al. Clinical application of whole-genome sequencing of solid tumors for precision oncology. Exp. Mol. Med. 2024, 56, 1856–1868. [Google Scholar] [CrossRef]
- Quaresma, M.; Coleman, M.P.; Rachet, B. 40-year trends in an index of survival for all cancers combined and survival adjusted for age and sex for each cancer in England and Wales, 1971–2011: A population-based study. Lancet 2015, 385, 1206–1218. [Google Scholar] [CrossRef]
- Arnold, M.; Rutherford, M.J.; Bardot, A.; Ferlay, J.; Andersson, T.M.; Myklebust, T.Å.; Tervonen, H.; Thursfield, V.; Ransom, D.; Shack, L.; et al. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995–2014 (ICBP SURVMARK-2): A population-based study. Lancet Oncol. 2019, 20, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- George, B. Precision Medicine and Pancreatic Cancer. Surg. Oncol. Clin. N. Am. 2021, 30, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, Y.; Hou, Y.; Wang, Q.; Long, D.; Liu, X.; Tian, X.; Yang, Y. Single cell RNA-seq reveals the CCL5/SDC1 receptor-ligand interaction between T cells and tumor cells in pancreatic cancer. Cancer Lett. 2022, 545, 215834. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Ren, H. Precision treatment of pancreatic ductal adenocarcinoma. Cancer Lett. 2024, 585, 216636. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Javed, M.A.; Beyer, G.; Le, N.; Vinci, A.; Wong, H.; Palmer, D.; Morgan, R.D.; Lamarca, A.; Hubner, R.A.; Valle, J.W.; et al. Impact of intensified chemotherapy in metastatic pancreatic ductal adenocarcinoma (PDAC) in clinical routine in Europe. Pancreatology 2019, 19, 97–104. [Google Scholar] [CrossRef]
- Raptis, D.A.; Fessas, C.; Belasyse-Smith, P.; Kurzawinski, T.R. Clinical presentation and waiting time targets do not affect prognosis in patients with pancreatic cancer. Surgeon 2010, 8, 239–246. [Google Scholar] [CrossRef]
- Collisson, E.A.; Trejo, C.L.; Silva, J.M.; Gu, S.; Korkola, J.E.; Heiser, L.M.; Charles, R.P.; Rabinovich, B.A.; Hann, B.; Dankort, D.; et al. A central role for RAF→MEK→ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012, 2, 685–693. [Google Scholar] [CrossRef]
- Gong, J.; Tuli, R.; Shinde, A.; Hendifar, A.E. Meta-analyses of treatment standards for pancreatic cancer. Mol. Clin. Oncol. 2016, 4, 315–325. [Google Scholar] [CrossRef]
- Aprile, G.; Negri, F.V.; Giuliani, F.; De Carlo, E.; Melisi, D.; Simionato, F.; Silvestris, N.; Brunetti, O.; Leone, F.; Marino, D.; et al. Second-line chemotherapy for advanced pancreatic cancer: Which is the best option? Crit. Rev. Oncol. Hematol. 2017, 115, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Gibiino, G.; Sbrancia, M.; Coluccio, C.; Cazzato, M.; Carloni, L.; Cucchetti, A.; Ercolani, G.; Sambri, V.; Fabbri, C. Microbiota in the Natural History of Pancreatic Cancer: From Predisposition to Therapy. Cancers 2022, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, M.; Hou, S.; Tian, B. Role of the microbiome in systemic therapy for pancreatic ductal adenocarcinoma (Review). Int. J. Oncol. 2021, 59, 101. [Google Scholar] [CrossRef]
- Rutman, R.J.; Cantarow, A.; Paschkis, K.E. Studies in 2-acetylaminofluorene carcinogenesis. III. The utilization of uracil-2-C14 by preneoplastic rat liver and rat hepatoma. Cancer Res. 1954, 14, 119–123. [Google Scholar]
- Sethy, C.; Kundu, C.N. 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer: Implication of DNA repair inhibition. Biomed. Pharmacother. 2021, 137, 111285. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Hagenkort, A.; Paulin, C.B.J.; Desroses, M.; Sarno, A.; Wiita, E.; Mortusewicz, O.; Koolmeister, T.; Loseva, O.; Jemth, A.S.; Almlöf, I.; et al. dUTPase inhibition augments replication defects of 5-Fluorouracil. Oncotarget 2017, 8, 23713–23726. [Google Scholar] [CrossRef]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef]
- Khorana, A.A.; McKernin, S.E.; Berlin, J.; Hong, T.S.; Maitra, A.; Moravek, C.; Mumber, M.; Schulick, R.; Zeh, H.J.; Katz, M.H.G. Potentially Curable Pancreatic Adenocarcinoma: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2019, 37, 2082–2088. [Google Scholar] [CrossRef]
- Pinedo, H.M.; Peters, G.F. Fluorouracil: Biochemistry and pharmacology. J. Clin. Oncol. 1988, 6, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Donadio, M.D.S.; Carraro, D.M.; Torrezan, G.T.; de Mello, C.A.L. Dihydropyrimidine dehydrogenase (DPD) polymorphisms knocking on the door. Ecancermedicalscience 2022, 16, 1344. [Google Scholar] [CrossRef] [PubMed]
- Forouzesh, D.C.; Moran, G.R. Mammalian dihydropyrimidine dehydrogenase. Arch. Biochem. Biophys. 2021, 714, 109066. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Siolas, D.; Morrissey, C.; Oberstein, P.E. The Achilles’ Heel of Pancreatic Cancer: Targeting pancreatic cancer’s unique immunologic characteristics and metabolic dependencies in clinical trials. J. Pancreatol. 2020, 3, 121–131. [Google Scholar] [CrossRef]
- Saif, M.W.; Lee, Y.; Kim, R. Harnessing gemcitabine metabolism: A step towards personalized medicine for pancreatic cancer. Ther. Adv. Med. Oncol. 2012, 4, 341–346. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: A randomized controlled trial. JAMA 2007, 297, 267–277. [Google Scholar] [CrossRef]
- Liu, Z.D.; Shi, Y.H.; Xu, Q.C.; Zhao, G.Y.; Zhu, Y.Q.; Li, F.X.; Ma, M.J.; Ye, J.Y.; Huang, X.T.; Wang, X.Y.; et al. CSNK2A1 confers gemcitabine resistance to pancreatic ductal adenocarcinoma via inducing autophagy. Cancer Lett. 2024, 585, 216640. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Mendes, I.; Vale, N. Overcoming Microbiome-Acquired Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. Biomedicines 2024, 12, 227. [Google Scholar] [CrossRef] [PubMed]
- Basturk, O.; Hong, S.M.; Wood, L.D.; Adsay, N.V.; Albores-Saavedra, J.; Biankin, A.V.; Brosens, L.A.; Fukushima, N.; Goggins, M.; Hruban, R.H.; et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. Am. J. Surg. Pathol. 2015, 39, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Cameron, J.L.; Campbell, K.A.; Arnold, M.A.; Chang, D.C.; Coleman, J.; Hodgin, M.B.; Sauter, P.K.; Hruban, R.H.; Riall, T.S.; et al. 1423 pancreaticoduodenectomies for pancreatic cancer: A single-institution experience. J. Gastrointest. Surg. 2006, 10, 1199–1210; discussion 1210–1211. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, M.; Noë, M.; Masica, D.L.; Hosoda, W.; Chianchiano, P.; Fischer, C.G.; Lionheart, G.; Brosens, L.A.A.; Pea, A.; Yu, J.; et al. IPMNs with co-occurring invasive cancers: Neighbours but not always relatives. Gut 2018, 67, 1652–1662. [Google Scholar] [CrossRef]
- Zamboni, G.; Scarpa, A.; Bogina, G.; Iacono, C.; Bassi, C.; Talamini, G.; Sessa, F.; Capella, C.; Solcia, E.; Rickaert, F.; et al. Mucinous cystic tumors of the pancreas: Clinicopathological features, prognosis, and relationship to other mucinous cystic tumors. Am. J. Surg. Pathol. 1999, 23, 410–422. [Google Scholar] [CrossRef]
- Laffan, T.A.; Horton, K.M.; Klein, A.P.; Berlanstein, B.; Siegelman, S.S.; Kawamoto, S.; Johnson, P.T.; Fishman, E.K.; Hruban, R.H. Prevalence of unsuspected pancreatic cysts on MDCT. AJR Am. J. Roentgenol. 2008, 191, 802–807. [Google Scholar] [CrossRef]
- Grimont, A.; Leach, S.D.; Chandwani, R. Uncertain Beginnings: Acinar and Ductal Cell Plasticity in the Development of Pancreatic Cancer. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 369–382. [Google Scholar] [CrossRef]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef]
- Scarpa, A.; Capelli, P.; Mukai, K.; Zamboni, G.; Oda, T.; Iacono, C.; Hirohashi, S. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am. J. Pathol. 1993, 142, 1534–1543. [Google Scholar]
- Caldas, C.; Hahn, S.A.; da Costa, L.T.; Redston, M.S.; Schutte, M.; Seymour, A.B.; Weinstein, C.L.; Hruban, R.H.; Yeo, C.J.; Kern, S.E. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat. Genet. 1994, 8, 27–32. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar]
- Vincent, A.; Omura, N.; Hong, S.M.; Jaffe, A.; Eshleman, J.; Goggins, M. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clin. Cancer Res. 2011, 17, 4341–4354. [Google Scholar] [CrossRef]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef]
- Fukushima, N.; Sato, N.; Ueki, T.; Rosty, C.; Walter, K.M.; Wilentz, R.E.; Yeo, C.J.; Hruban, R.H.; Goggins, M. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am. J. Pathol. 2002, 160, 1573–1581. [Google Scholar] [CrossRef]
- Sato, N.; Fukushima, N.; Hruban, R.H.; Goggins, M. CpG island methylation profile of pancreatic intraepithelial neoplasia. Mod. Pathol. 2008, 21, 238–244. [Google Scholar] [CrossRef]
- Sato, N.; Ueki, T.; Fukushima, N.; Iacobuzio-Donahue, C.A.; Yeo, C.J.; Cameron, J.L.; Hruban, R.H.; Goggins, M. Aberrant methylation of CpG islands in intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology 2002, 123, 365–372. [Google Scholar] [CrossRef]
- Fujikura, K.; Alruwaii, Z.I.; Haffner, M.C.; Trujillo, M.A.; Roberts, N.J.; Hong, S.M.; Macgregor-Das, A.; Goggins, M.G.; Roy, S.; Meeker, A.K.; et al. Downregulation of 5-hydroxymethylcytosine is an early event in pancreatic tumorigenesis. J. Pathol. 2021, 254, 279–288. [Google Scholar] [CrossRef]
- Akshintala, V.S.; Talukdar, R.; Singh, V.K.; Goggins, M. The Gut Microbiome in Pancreatic Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 290–295. [Google Scholar] [CrossRef]
- Herremans, K.M.; Riner, A.N.; Cameron, M.E.; McKinley, K.L.; Triplett, E.W.; Hughes, S.J.; Trevino, J.G. The oral microbiome, pancreatic cancer and human diversity in the age of precision medicine. Microbiome 2022, 10, 93. [Google Scholar] [CrossRef]
- McAllister, F.; Khan, M.A.W.; Helmink, B.; Wargo, J.A. The Tumor Microbiome in Pancreatic Cancer: Bacteria and Beyond. Cancer Cell 2019, 36, 577–579. [Google Scholar] [CrossRef]
- Yin, H.; Pu, N.; Chen, Q.; Zhang, J.; Zhao, G.; Xu, X.; Wang, D.; Kuang, T.; Jin, D.; Lou, W.; et al. Gut-derived lipopolysaccharide remodels tumoral microenvironment and synergizes with PD-L1 checkpoint blockade via TLR4/MyD88/AKT/NF-κB pathway in pancreatic cancer. Cell Death Dis. 2021, 12, 1033. [Google Scholar] [CrossRef]
- Jusakul, A.; Khuntikeo, N.; Haigh, W.G.; Kuver, R.; Ioannou, G.N.; Loilome, W.; Namwat, N.; Bhudhisawasdi, V.; Pugkhem, A.; Pairojkul, C.; et al. Identification of biliary bile acids in patients with benign biliary diseases, hepatocellular carcinoma and cholangiocarcinoma. Asian Pac. J. Cancer Prev. 2012, 13, 77–82. [Google Scholar]
- Shukla, V.K.; Tiwari, S.C.; Roy, S.K. Biliary bile acids in cholelithiasis and carcinoma of the gall bladder. Eur. J. Cancer Prev. 1993, 2, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Wang, H.; Shi, Y.; Dong, Y.; Zhang, Y.; Wang, J. Impact of bile acids on the growth of human cholangiocarcinoma via FXR. J. Hematol. Oncol. 2011, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, X.; Li, G.; Yi, J.; Wang, J. Expressions of farnesoid X receptor and myeloid cell leukemia sequence 1 protein are associated with poor prognosis in patients with gallbladder cancer. Chin. Med. J. 2014, 127, 2637–2642. [Google Scholar] [PubMed]
- Su, H.; Ma, C.; Liu, J.; Li, N.; Gao, M.; Huang, A.; Wang, X.; Huang, W.; Huang, X. Downregulation of nuclear receptor FXR is associated with multiple malignant clinicopathological characteristics in human hepatocellular carcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1245–G1253. [Google Scholar] [CrossRef]
- Knudsen, C.; Neyrinck, A.M.; Lanthier, N.; Delzenne, N.M. Microbiota and nonalcoholic fatty liver disease: Promising prospects for clinical interventions? Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 393–400. [Google Scholar] [CrossRef]
- Badawy, A.A. Tryptophan metabolism and disposition in cancer biology and immunotherapy. Biosci. Rep. 2022, 42, BSR20221682. [Google Scholar] [CrossRef]
- Alvandi, E.; Wong, W.K.M.; Joglekar, M.V.; Spring, K.J.; Hardikar, A.A. Short-chain fatty acid concentrations in the incidence and risk-stratification of colorectal cancer: A systematic review and meta-analysis. BMC Med. 2022, 20, 323. [Google Scholar] [CrossRef]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef]
- Daniel, N.; Farinella, R.; Chatziioannou, A.C.; Jenab, M.; Mayén, A.L.; Rizzato, C.; Belluomini, F.; Canzian, F.; Tavanti, A.; Keski-Rahkonen, P.; et al. Genetically predicted gut bacteria, circulating bacteria-associated metabolites and pancreatic ductal adenocarcinoma: A Mendelian randomisation study. Sci. Rep. 2024, 14, 25144. [Google Scholar] [CrossRef]
- Froehlich, T.K.; Amstutz, U.; Aebi, S.; Joerger, M.; Largiadèr, C.R. Clinical importance of risk variants in the dihydropyrimidine dehydrogenase gene for the prediction of early-onset fluoropyrimidine toxicity. Int. J. Cancer 2015, 136, 730–739. [Google Scholar] [CrossRef]
- Meulendijks, D.; Henricks, L.M.; Sonke, G.S.; Deenen, M.J.; Froehlich, T.K.; Amstutz, U.; Largiadèr, C.R.; Jennings, B.A.; Marinaki, A.M.; Sanderson, J.D.; et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015, 16, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Amstutz, U.; Henricks, L.M.; Offer, S.M.; Barbarino, J.; Schellens, J.H.M.; Swen, J.J.; Klein, T.E.; McLeod, H.L.; Caudle, K.E.; Diasio, R.B.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin. Pharmacol. Ther. 2018, 103, 210–216. [Google Scholar] [CrossRef]
- Henricks, L.M.; Lunenburg, C.A.; Meulendijks, D.; Gelderblom, H.; Cats, A.; Swen, J.J.; Schellens, J.H.; Guchelaar, H.J. Translating DPYD genotype into DPD phenotype: Using the DPYD gene activity score. Pharmacogenomics 2015, 16, 1277–1286. [Google Scholar] [CrossRef]
- Henricks, L.M.; Lunenburg, C.A.T.C.; de Man, F.M.; Meulendijks, D.; Frederix, G.W.J.; Kienhuis, E.; Creemers, G.J.; Baars, A.; Dezentjé, V.O.; Imholz, A.L.T.; et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: A prospective safety analysis. Lancet Oncol. 2018, 19, 1459–1467. [Google Scholar] [CrossRef]
- Mormont, M.C.; Levi, F. Cancer chronotherapy: Principles, applications, and perspectives. Cancer 2003, 97, 155–169. [Google Scholar] [CrossRef]
- Jacobs, B.A.; Deenen, M.J.; Pluim, D.; van Hasselt, J.G.; Krähenbühl, M.D.; van Geel, R.M.; de Vries, N.; Rosing, H.; Meulendijks, D.; Burylo, A.M.; et al. Pronounced between-subject and circadian variability in thymidylate synthase and dihydropyrimidine dehydrogenase enzyme activity in human volunteers. Br. J. Clin. Pharmacol. 2016, 82, 706–716. [Google Scholar] [CrossRef]
- Raida, M.; Kliche, K.O.; Schwabe, W.; Häusler, P.; Clement, J.H.; Behnke, D.; Höffken, K. Circadian variation of dihydropyrimidine dehydrogenase mRNA expression in leukocytes and serum cortisol levels in patients with advanced gastrointestinal carcinomas compared to healthy controls. J. Cancer Res. Clin. Oncol. 2002, 128, 96–102. [Google Scholar] [CrossRef]
- Jiang, H.; Lu, J.; Ji, J. Circadian rhythm of dihydrouracil/uracil ratios in biological fluids: A potential biomarker for dihydropyrimidine dehydrogenase levels. Br. J. Pharmacol. 2004, 141, 616–623. [Google Scholar] [CrossRef]
- Harris, B.E.; Song, R.; Soong, S.J.; Diasio, R.B. Relationship between dihydropyrimidine dehydrogenase activity and plasma 5-fluorouracil levels with evidence for circadian variation of enzyme activity and plasma drug levels in cancer patients receiving 5-fluorouracil by protracted continuous infusion. Cancer Res. 1990, 50, 197–201. [Google Scholar]
- Metzger, G.; Massari, C.; Etienne, M.C.; Comisso, M.; Brienza, S.; Touitou, Y.; Milano, G.; Bastian, G.; Misset, J.L.; Lévi, F. Spontaneous or imposed circadian changes in plasma concentrations of 5-fluorouracil coadministered with folinic acid and oxaliplatin: Relationship with mucosal toxicity in patients with cancer. Clin. Pharmacol. Ther. 1994, 56, 190–201. [Google Scholar] [CrossRef]
- Lévi, F.; Giacchetti, S.; Adam, R.; Zidani, R.; Metzger, G.; Misset, J.L. Chronomodulation of chemotherapy against metastatic colorectal cancer. International Organization for Cancer Chronotherapy. Eur. J. Cancer 1995, 31A, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Petit, E.; Milano, G.; Lévi, F.; Thyss, A.; Bailleul, F.; Schneider, M. Circadian rhythm-varying plasma concentration of 5-fluorouracil during a five-day continuous venous infusion at a constant rate in cancer patients. Cancer Res. 1988, 48, 1676–1679. [Google Scholar] [PubMed]
- Lévi, F.; Okyar, A.; Dulong, S.; Innominato, P.F.; Clairambault, J. Circadian timing in cancer treatments. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 377–421. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.L.; Sun, J.; Guo, L.; Li, S.; Wu, M.W.; Qiu, F.; Jiang, W.Q.; Lévi, F.; Xian, L.J. Circadian rhythm in dihydropyrimidine dehydrogenase activity and reduced glutathione content in peripheral blood of nasopharyngeal carcinoma patients. Chronobiol. Int. 2005, 22, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.J.W.; Innominato, P.F.; Lévi, F.; Ballesta, A. Optimizing circadian drug infusion schedules towards personalized cancer chronotherapy. PLoS Comput. Biol. 2020, 16, e1007218. [Google Scholar] [CrossRef]
- Lévi, F.; Karaboué, A.; Etienne-Grimaldi, M.C.; Paintaud, G.; Focan, C.; Innominato, P.; Bouchahda, M.; Milano, G.; Chatelut, E. Pharmacokinetics of Irinotecan, Oxaliplatin and 5-Fluorouracil During Hepatic Artery Chronomodulated Infusion: A Translational European OPTILIV Study. Clin. Pharmacokinet. 2017, 56, 165–177. [Google Scholar] [CrossRef]
- Li, G.; She, F.F.; Liao, C.Y.; Wang, Z.W.; Wang, Y.T.; Wu, Y.D.; Huang, X.X.; Xie, C.K.; Lin, H.Y.; Zhu, S.C.; et al. cNEK6 induces gemcitabine resistance by promoting glycolysis in pancreatic ductal adenocarcinoma via the SNRPA/PPA2c/mTORC1 axis. Cell Death Dis. 2024, 15, 742. [Google Scholar] [CrossRef]
- Yu, T.; Wang, Y.; Fan, Y.; Fang, N.; Wang, T.; Xu, T.; Shu, Y. CircRNAs in cancer metabolism: A review. J. Hematol. Oncol. 2019, 12, 90. [Google Scholar] [CrossRef]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87.e7. [Google Scholar] [CrossRef]
- Ghanbari Movahed, Z.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharmacother. 2019, 112, 108690. [Google Scholar] [CrossRef]
- Nakao, K.; Minato, N.; Uemoto, S. (Eds.) Innovative Medicine: Basic Research and Development [Internet]; Springer: Tokyo, Japan, 2015. [Google Scholar]
- Lam, K.N.; Alexander, M.; Turnbaugh, P.J. Precision Medicine Goes Microscopic: Engineering the Microbiome to Improve Drug Outcomes. Cell Host Microbe 2019, 26, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Henrissat, B.; Gordon, J.I. Viewing the human microbiome through three-dimensional glasses: Integrating structural and functional studies to better define the properties of myriad carbohydrate-active enzymes. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Spanogiannopoulos, P.; Bess, E.N.; Carmody, R.N.; Turnbaugh, P.J. The microbial pharmacists within us: A metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 2016, 14, 273–287. [Google Scholar] [CrossRef]
- Klatt, N.R.; Cheu, R.; Birse, K.; Zevin, A.S.; Perner, M.; Noël-Romas, L.; Grobler, A.; Westmacott, G.; Xie, I.Y.; Butler, J.; et al. Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science 2017, 356, 938–945. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108, 4554–4561. [Google Scholar] [CrossRef]
- Mimee, M.; Citorik, R.J.; Lu, T.K. Microbiome therapeutics—Advances and challenges. Adv. Drug Deliv. Rev. 2016, 105, 44–54. [Google Scholar] [CrossRef]
- Wallace, B.D.; Redinbo, M.R. The human microbiome is a source of therapeutic drug targets. Curr. Opin. Chem. Biol. 2013, 17, 379–384. [Google Scholar] [CrossRef]
- Duerkop, B.A.; Huo, W.; Bhardwaj, P.; Palmer, K.L.; Hooper, L.V. Molecular Basis for Lytic Bacteriophage Resistance in Enterococci. mBio 2016, 7, e01304–e01316. [Google Scholar] [CrossRef]
- Isabella, V.M.; Ha, B.N.; Castillo, M.J.; Lubkowicz, D.J.; Rowe, S.E.; Millet, Y.A.; Anderson, C.L.; Li, N.; Fisher, A.B.; West, K.A.; et al. Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol. 2018, 36, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Ronda, C.; Chen, S.P.; Cabral, V.; Yaung, S.J.; Wang, H.H. Metagenomic engineering of the mammalian gut microbiome in situ. Nat. Methods 2019, 16, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.; Chieng, W.W.; Huang, S.W.; Hsu, L.J.; Jan, M.S. The synergistic tumor growth-inhibitory effect of probiotic Lactobacillus on transgenic mouse model of pancreatic cancer treated with gemcitabine. Sci. Rep. 2020, 10, 20319. [Google Scholar] [CrossRef]
- Ginn, S.L.; Alexander, I.E.; Edelstein, M.L.; Abedi, M.R.; Wixon, J. Gene therapy clinical trials worldwide to 2012—An update. J. Gene Med. 2013, 15, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.K.; Gottschalk, S.; Leen, A.M.; Vera, J.F. Is cancer gene therapy an empty suit? Lancet Oncol. 2013, 14, e447–e456. [Google Scholar] [CrossRef]
- Singh, H.M.; Ungerechts, G.; Tsimberidou, A.M. Gene and cell therapy for pancreatic cancer. Expert Opin. Biol. Ther. 2015, 15, 505–516. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Hecht, J.R.; Bedford, R.; Abbruzzese, J.L.; Lahoti, S.; Reid, T.R.; Soetikno, R.M.; Kirn, D.H.; Freeman, S.M. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin. Cancer Res. 2003, 9, 555–561. [Google Scholar]
- Hecht, J.R.; Farrell, J.J.; Senzer, N.; Nemunaitis, J.; Rosemurgy, A.; Chung, T.; Hanna, N.; Chang, K.J.; Javle, M.; Posner, M.; et al. EUS or percutaneously guided intratumoral TNFerade biologic with 5-fluorouracil and radiotherapy for first-line treatment of locally advanced pancreatic cancer: A phase I/II study. Gastrointest. Endosc. 2012, 75, 332–338. [Google Scholar] [CrossRef]
- Herman, J.M.; Wild, A.T.; Wang, H.; Tran, P.T.; Chang, K.J.; Taylor, G.E.; Donehower, R.C.; Pawlik, T.M.; Ziegler, M.A.; Cai, H.; et al. Randomized phase III multi-institutional study of TNFerade biologic with fluorouracil and radiotherapy for locally advanced pancreatic cancer: Final results. J. Clin. Oncol. 2013, 31, 886–894. [Google Scholar] [CrossRef]
- Nakao, A.; Kasuya, H.; Sahin, T.T.; Nomura, N.; Kanzaki, A.; Misawa, M.; Shirota, T.; Yamada, S.; Fujii, T.; Sugimoto, H.; et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 2011, 18, 167–175. [Google Scholar] [CrossRef]
- Noonan, A.M.; Farren, M.R.; Geyer, S.M.; Huang, Y.; Tahiri, S.; Ahn, D.; Mikhail, S.; Ciombor, K.K.; Pant, S.; Aparo, S.; et al. Randomized Phase 2 Trial of the Oncolytic Virus Pelareorep (Reolysin) in Upfront Treatment of Metastatic Pancreatic Adenocarcinoma. Mol. Ther. 2016, 24, 1150–1158. [Google Scholar] [CrossRef]
- Wang, W.J.; Tai, C.K.; Kasahara, N.; Chen, T.C. Highly efficient and tumor-restricted gene transfer to malignant gliomas by replication-competent retroviral vectors. Hum. Gene Ther. 2003, 14, 117–127. [Google Scholar] [CrossRef]
- Tai, C.K.; Wang, W.J.; Chen, T.C.; Kasahara, N. Single-shot, multicycle suicide gene therapy by replication-competent retrovirus vectors achieves long-term survival benefit in experimental glioma. Mol. Ther. 2005, 12, 842–851. [Google Scholar] [CrossRef]
- Perez, O.D.; Logg, C.R.; Hiraoka, K.; Diago, O.; Burnett, R.; Inagaki, A.; Jolson, D.; Amundson, K.; Buckley, T.; Lohse, D.; et al. Design and selection of Toca 511 for clinical use: Modified retroviral replicating vector with improved stability and gene expression. Mol. Ther. 2012, 20, 1689–1698. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Landolfi, J.; Hogan, D.J.; Bloomfield, S.; Carter, B.; Chen, C.C.; Elder, J.B.; Kalkanis, S.N.; Kesari, S.; Lai, A.; et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci. Transl. Med. 2016, 8, 341ra75. [Google Scholar] [CrossRef]
- Inoko, K.; Hiraoka, K.; Inagaki, A.; Takahashi, M.; Kushibiki, T.; Hontani, K.; Takano, H.; Sato, S.; Takeuchi, S.; Nakamura, T.; et al. Therapeutic activity of retroviral replicating vector-mediated prodrug activator gene therapy for pancreatic cancer. Cancer Gene Ther. 2018, 25, 184–195. [Google Scholar] [CrossRef]
- Yuen, J.G.; Hwang, G.R.; Fesler, A.; Intriago, E.; Pal, A.; Ojha, A.; Ju, J. Development of gemcitabine-modified miRNA mimics as cancer therapeutics for pancreatic ductal adenocarcinoma. Mol. Ther. Oncol. 2024, 32, 200769. [Google Scholar] [CrossRef]
- Xiao, Y.; Pan, T.; Da, W.; Liu, Y.; Chen, S.; Chen, D.; Liu, K.; Zheng, Y.; Xie, D.; Gao, Y.; et al. Aptamer-drug conjugates-loaded bacteria for pancreatic cancer synergistic therapy. Signal Transduct. Target. Ther. 2024, 9, 272. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes, I.; Vale, N. The Use of Personalized Medicine in Pancreatic Ductal Adenocarcinoma (PDAC): New Therapeutic Opportunities. Future Pharmacol. 2024, 4, 934-954. https://doi.org/10.3390/futurepharmacol4040049
Mendes I, Vale N. The Use of Personalized Medicine in Pancreatic Ductal Adenocarcinoma (PDAC): New Therapeutic Opportunities. Future Pharmacology. 2024; 4(4):934-954. https://doi.org/10.3390/futurepharmacol4040049
Chicago/Turabian StyleMendes, Inês, and Nuno Vale. 2024. "The Use of Personalized Medicine in Pancreatic Ductal Adenocarcinoma (PDAC): New Therapeutic Opportunities" Future Pharmacology 4, no. 4: 934-954. https://doi.org/10.3390/futurepharmacol4040049
APA StyleMendes, I., & Vale, N. (2024). The Use of Personalized Medicine in Pancreatic Ductal Adenocarcinoma (PDAC): New Therapeutic Opportunities. Future Pharmacology, 4(4), 934-954. https://doi.org/10.3390/futurepharmacol4040049