
Role of Senescence-Resumed Proliferation in Keloid Pathogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
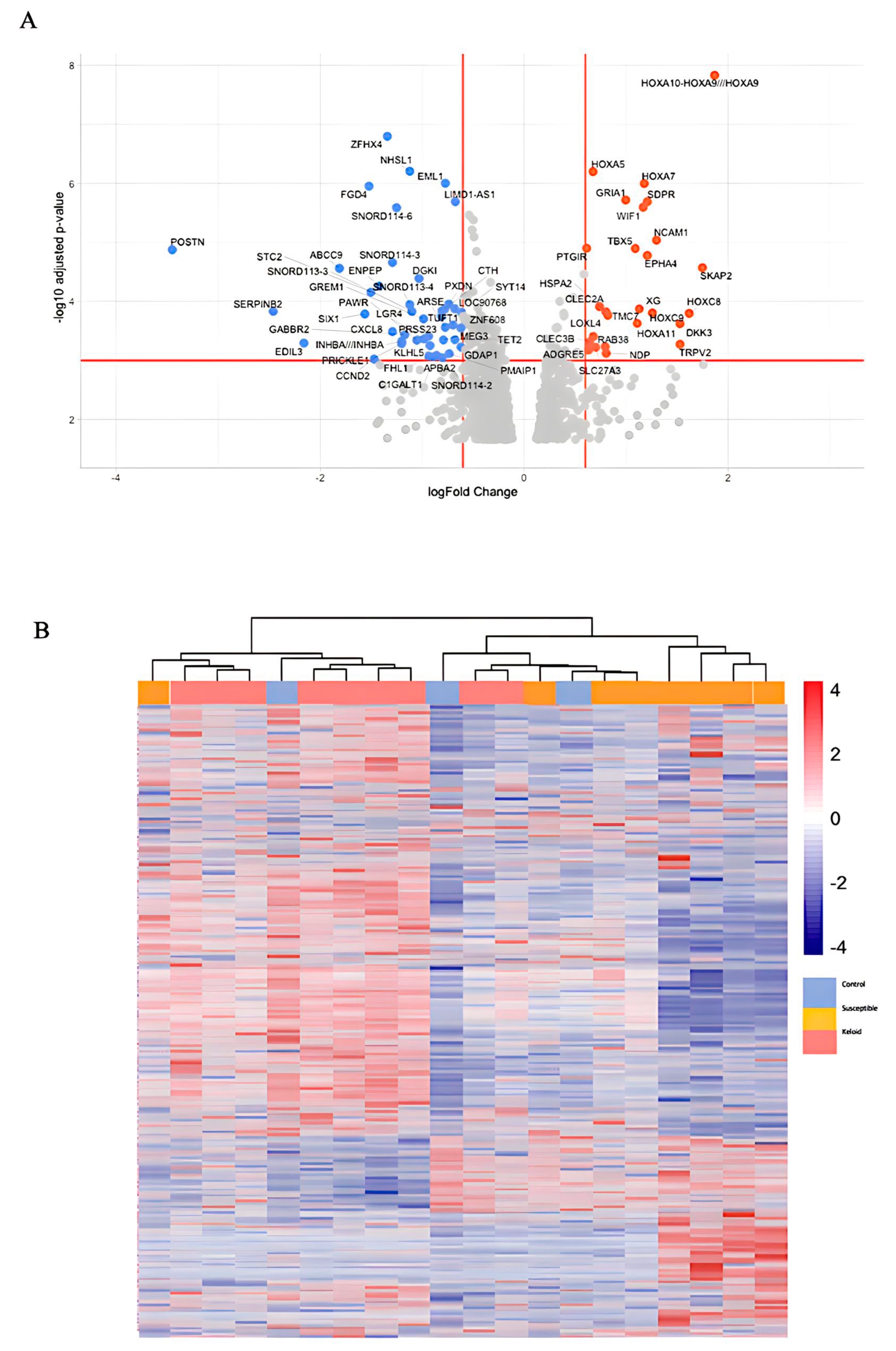
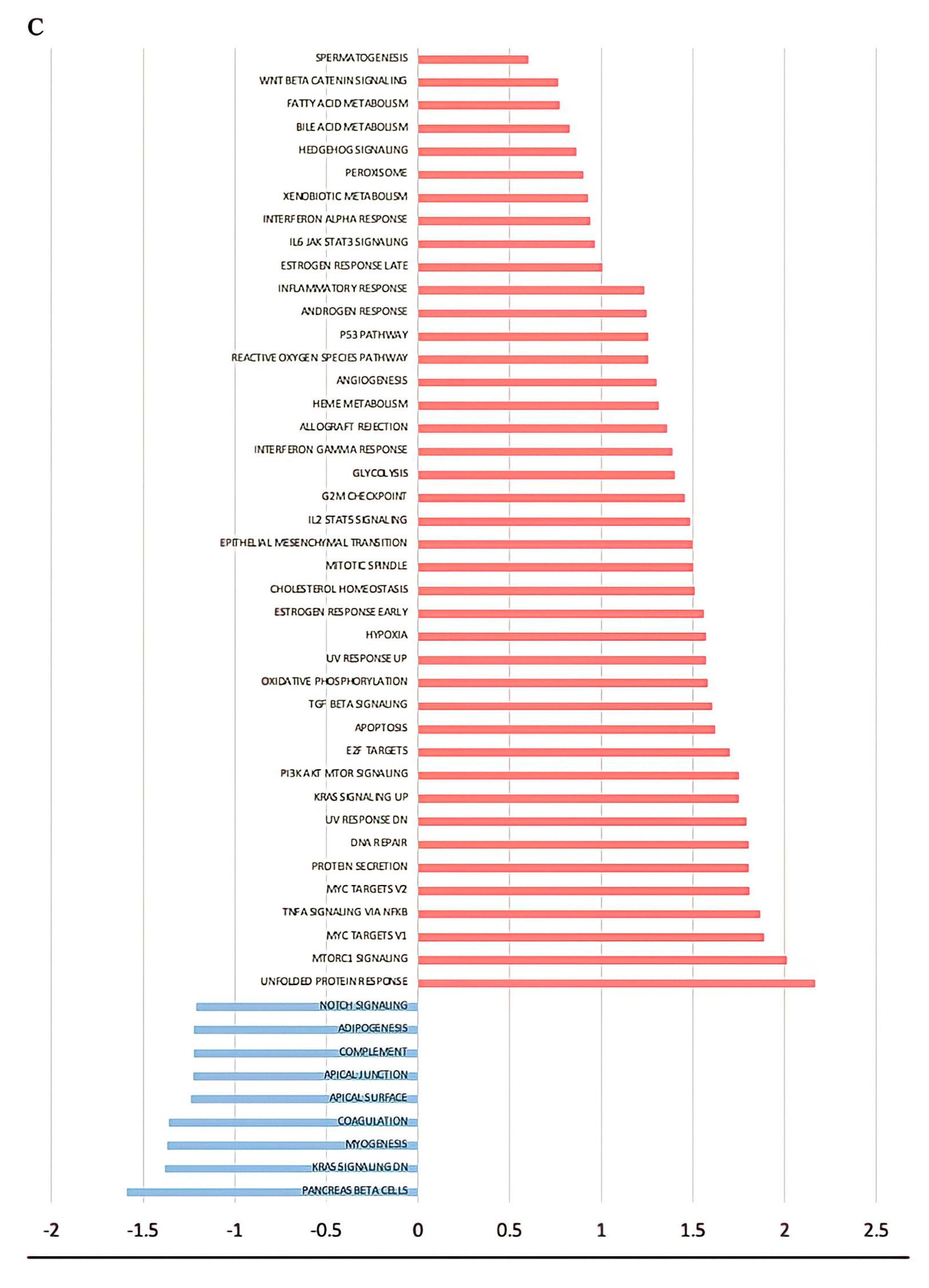
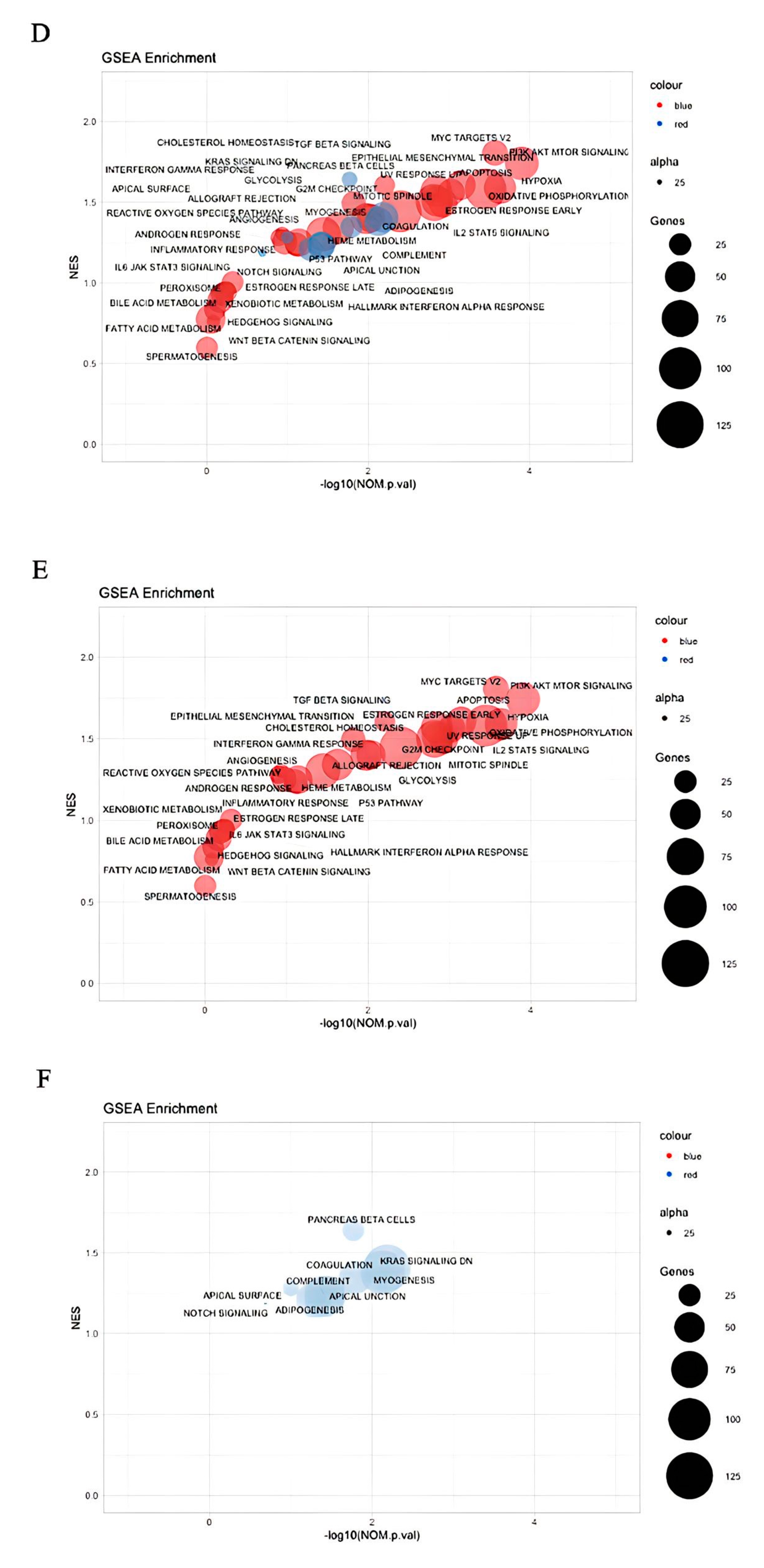
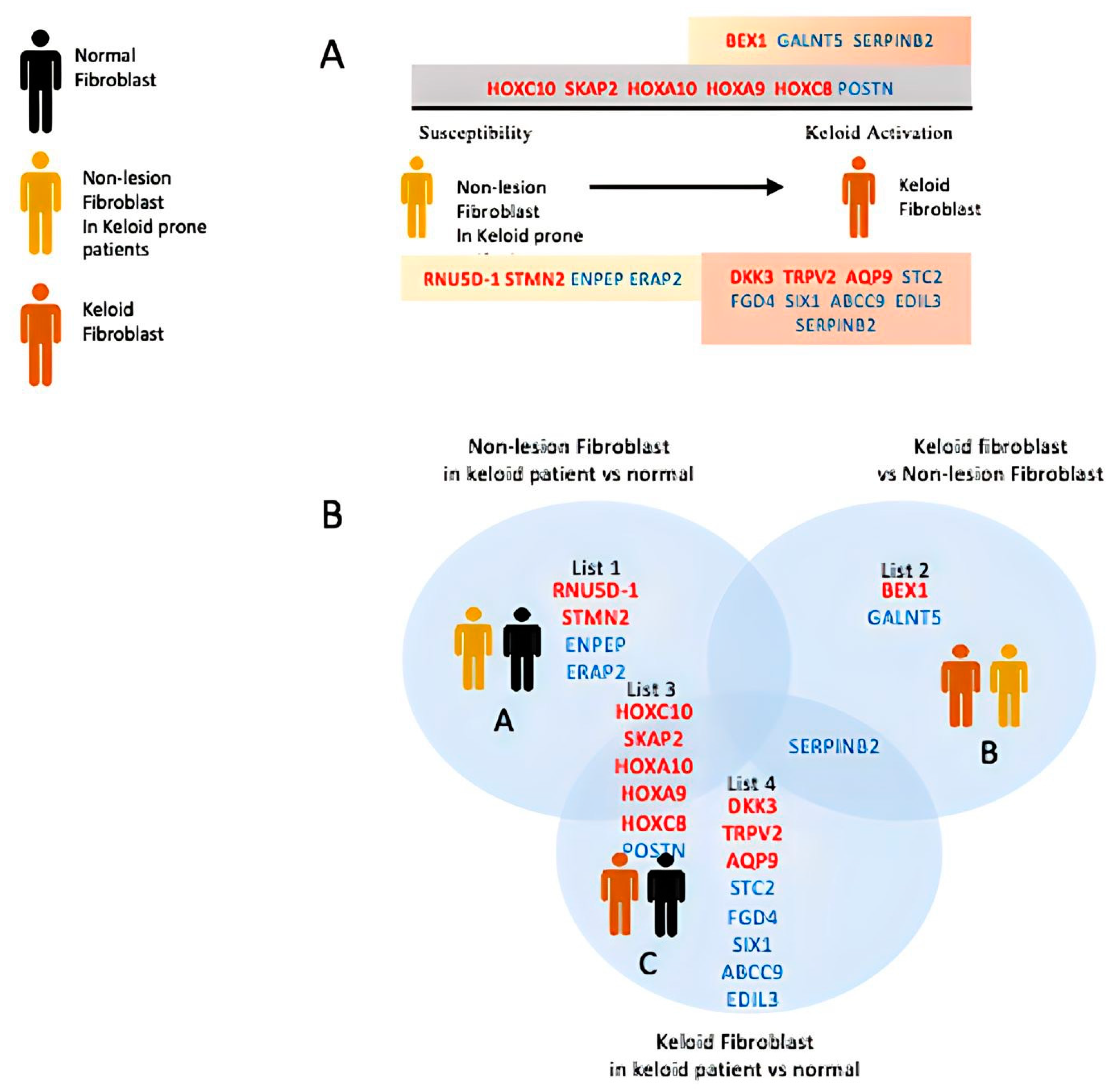
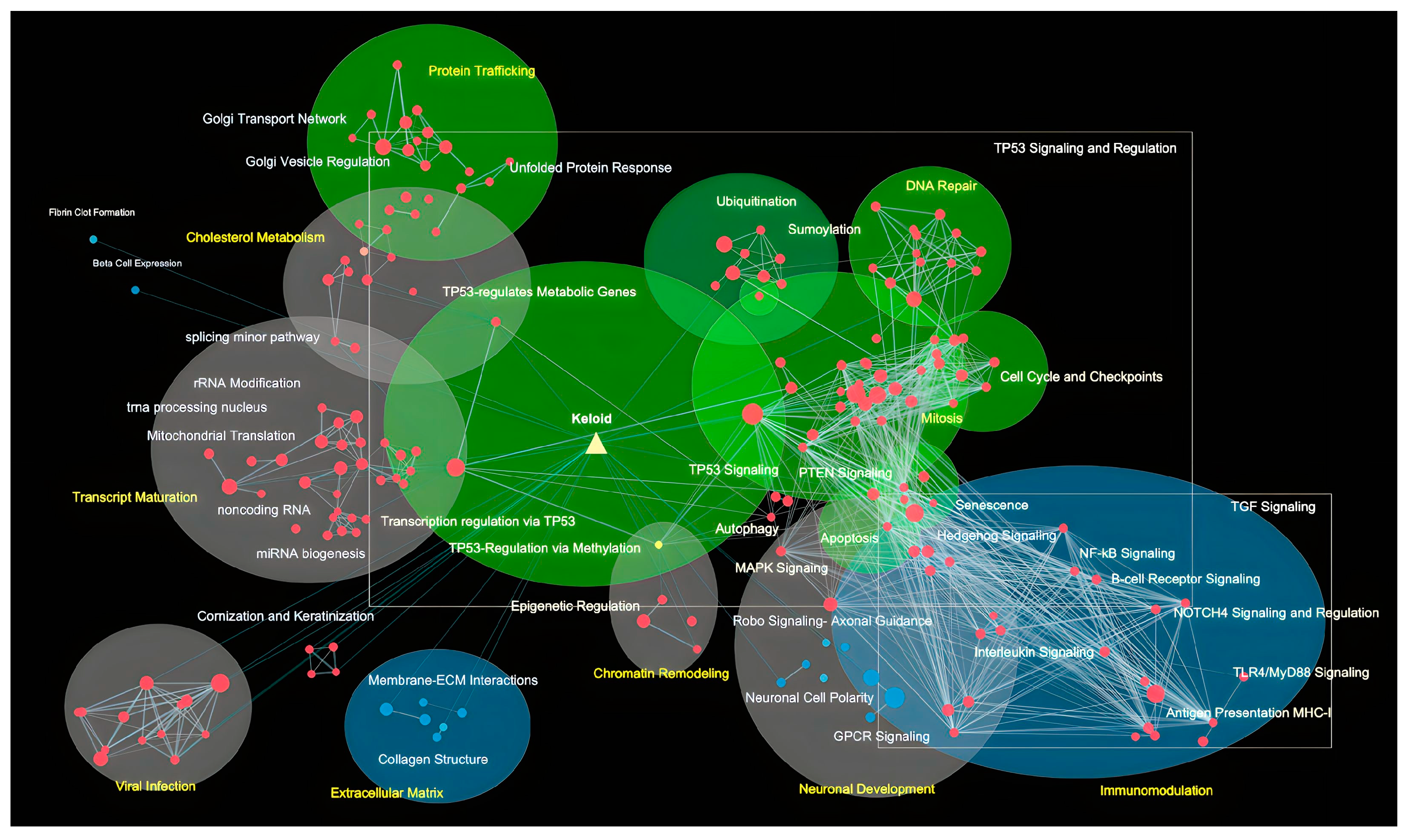
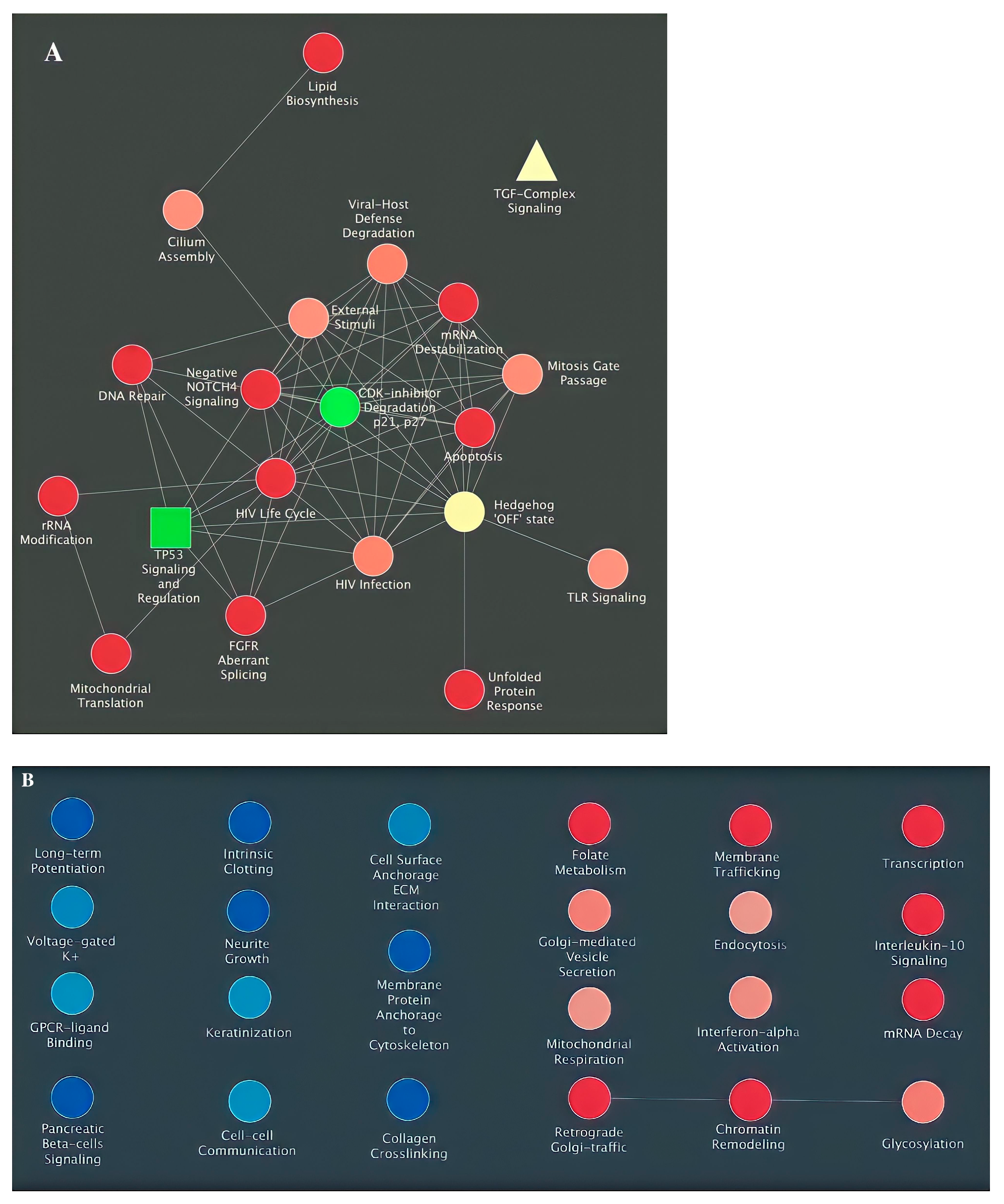
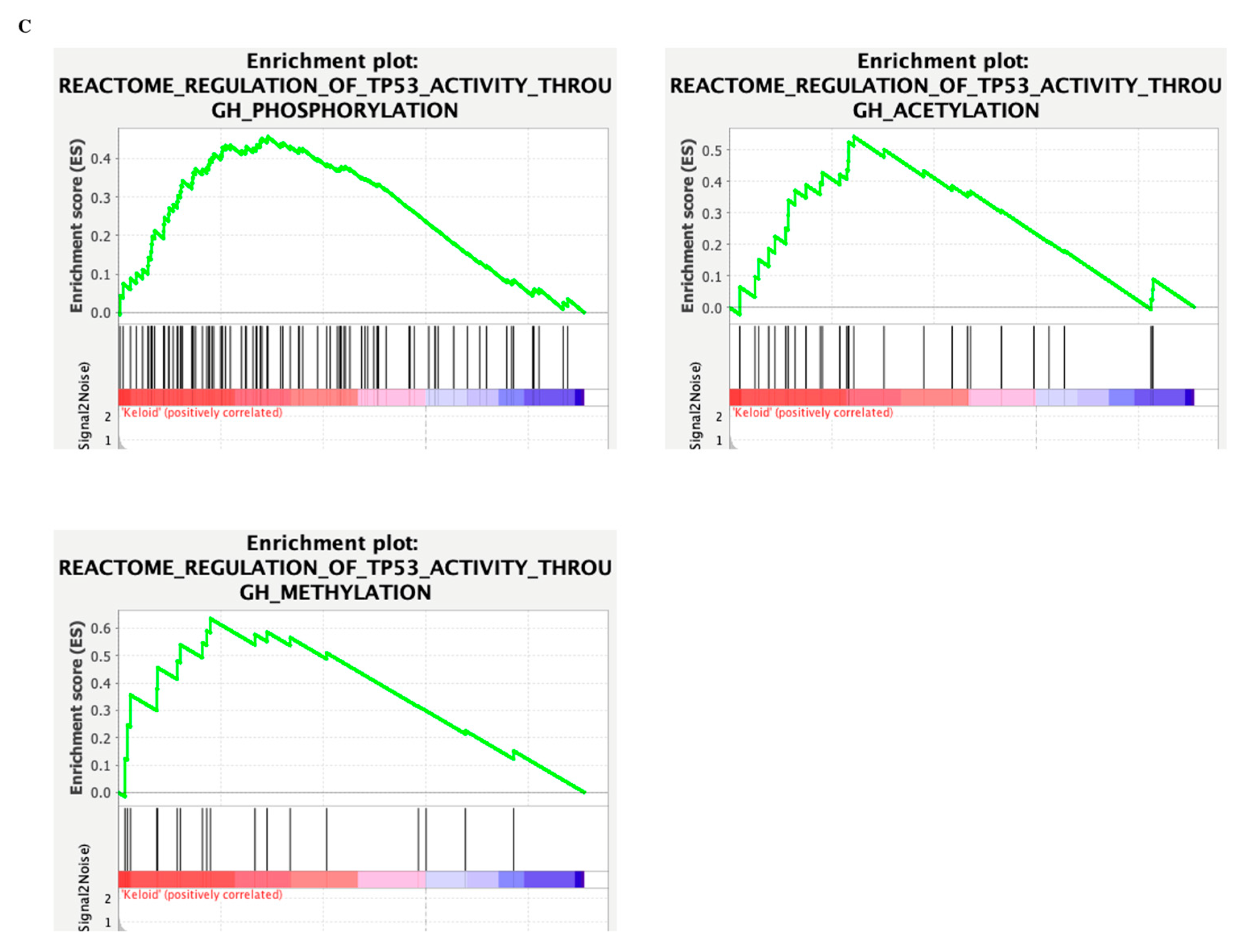
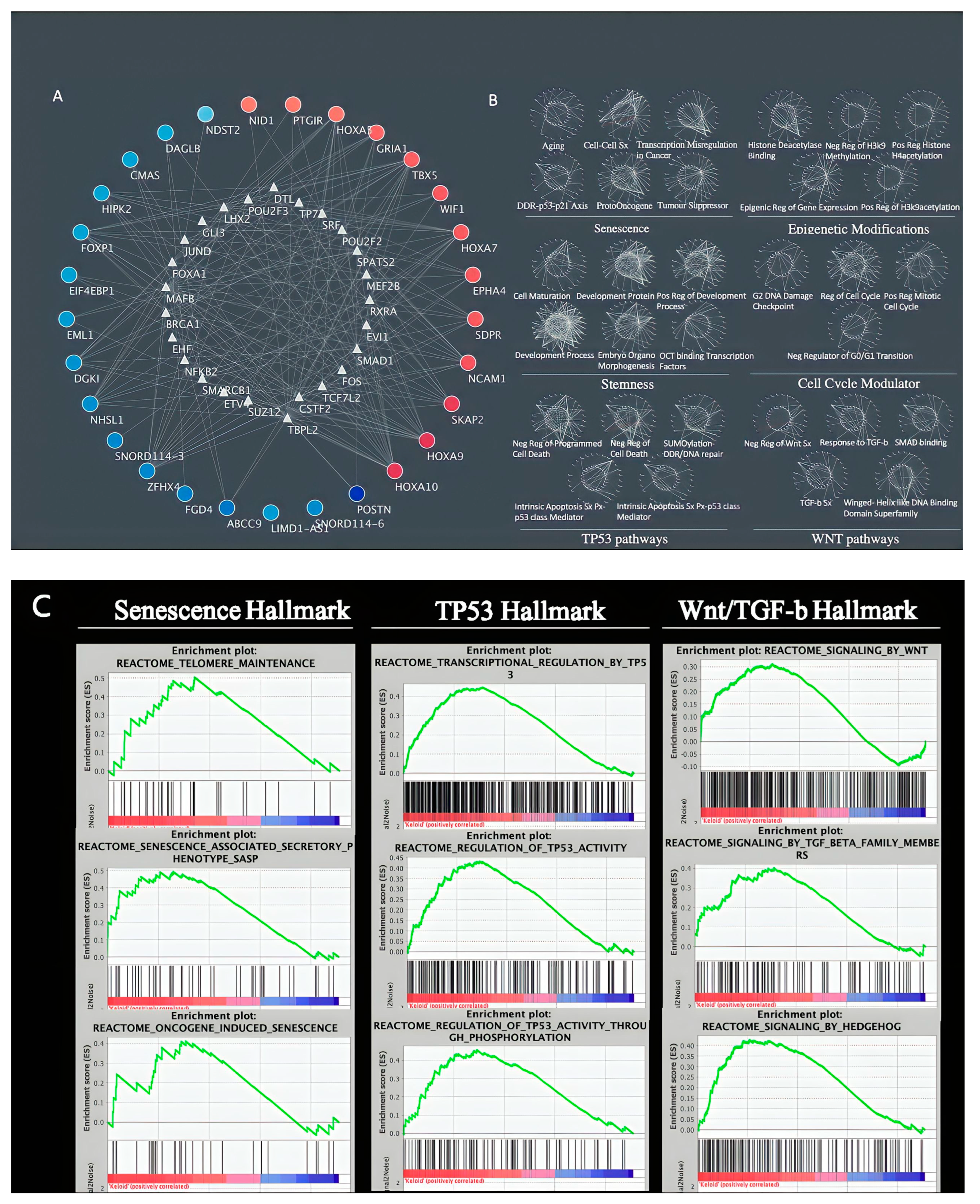
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Takeuchi, A.; Imatani, A.; Saito, M.; Jin, X.; Hatta, W.; Uno, K.; Koike, T.; Masamune, A. Wnt Signaling and Aging of the Gastrointestinal Tract. Int. J. Mol. Sci. 2022, 23, 12210. [Google Scholar] [CrossRef] [PubMed]
- Domen, A.; Deben, C.; Verswyvel, J.; Flieswasser, T.; Prenen, H.; Peeters, M.; Lardon, F.; Wouters, A. Cellular senescence in cancer: Clinical detection and prognostic implications. J. Exp. Clin. Cancer Res. 2022, 41, 360. [Google Scholar] [CrossRef] [PubMed]
- Piskorz, W.M.; Cechowska-Pasko, M. Senescence of Tumor Cells in Anticancer Therapy—Beneficial and Detrimental Effects. Int. J. Mol. Sci. 2022, 23, 11082. [Google Scholar] [CrossRef] [PubMed]
- Wen, G.-M.; Xu, X.-Y.; Xia, P. Metabolism in Cancer Stem Cells: Targets for Clinical Treatment. Cells 2022, 11, 3790. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; VanderKraats, N.D.; Shah, P.P.; Van Tuyn, J.; Rai, T.S.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.D.; Narita, M. Autophagy at the intersection of aging, senescence, and cancer. Mol. Oncol. 2022, 16, 3259–3275. [Google Scholar] [CrossRef]
- Crouch, J.; Shvedova, M.; Thanapaul, R.J.R.S.; Botchkarev, V.; Roh, D. Epigenetic Regulation of Cellular Senescence. Cells 2022, 11, 672. [Google Scholar] [CrossRef]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; van Deursen, J.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Hoare, M.; Das, T.; Alexander, G. Ageing, telomeres, senescence, and liver injury. J. Hepatol. 2010, 53, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Hua, F.; Hu, Z.W. The regulation of beta-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [PubMed]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Nicolas, M.A.-S. WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, L.; Li, L.; Qu, S.; Yu, B.; Sun, Y.; Wan, F.; Chen, X.; Liang, R.; Zhu, X. A positive feedback loop between Wnt/beta-catenin signaling and hTERT regulates the cancer stem cell-like traits in radioresistant nasopharyngeal carcinoma cells. J. Cell Biochem. 2020, 121, 4612–4622. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Saleh, T.; Carpenter, V.J.; Tyutyunyk-Massey, L.; Murray, G.; Leverson, J.; Souers, A.; Alotaibi, M.; Faber, A.; Reed, J.; Harada, H.; et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-X(L)-BAX interaction. Mol. Oncol. 2020, 14, 2504–2519. [Google Scholar]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.; Ling, Y.; Stout, M.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.; Giles, C.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sanomachi, T.; Suzuki, S.; Togashi, K.; Sugai, A.; Seino, S.; Sato, A.; Okada, M.; Kitanaka, C. Gemcitabine radiosensitization primes irradiated malignant meningioma cells for senolytic elimination by navitoclax. Neurooncol. Adv. 2021, 3, vdab148. [Google Scholar] [CrossRef]
- Ahmadinejad, F.; Bos, T.; Hu, B.; Britt, E.; Koblinski, J.; Souers, A.J.; Leverson, J.D.; Faber, A.C.; Gewirtz, D.A.; Harada, H. Senolytic-Mediated Elimination of Head and Neck Tumor Cells Induced Into Senescence by Cisplatin. Mol. Pharmacol. 2022, 101, 168–180. [Google Scholar] [CrossRef]
- Carpenter, V.; Saleh, T.; Lee, S.M.; Murray, G.; Reed, J.; Souers, A.; Faber, A.C.; Harada, H.; Gewirtz, D.A. Androgen-deprivation induced senescence in prostate cancer cells is permissive for the development of castration-resistance but susceptible to senolytic therapy. Biochem. Pharmacol. 2021, 193, 114765. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Cao, H.; Shen, D.; Li, S.; Yan, L.; Chen, C.; Xing, S.; Dou, F. Quercetin protects against atherosclerosis by regulating the expression of PCSK9, CD36, PPARgamma, LXRalpha and ABCA1. Int. J. Mol. Med. 2019, 44, 893–902. [Google Scholar] [CrossRef]
- Wong, S.; Chin, K.-Y.; Ima-Nirwana, S. Quercetin as an Agent for Protecting the Bone: A Review of the Current Evidence. Int. J. Mol. Sci. 2020, 21, 6448. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Barsoumian, H.; Yang, L.; Sezen, D.; Wasley, M.; Masrorpour, F.; Cortez, M.; Welsh, J. Senolytic Cocktail Dasatinib Plus Quercetin Enhances the Antitumor Effect of Senescence-Inducing Radiotherapy in a Preclinical Model of Melanoma. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, S57. [Google Scholar] [CrossRef]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic Cocktail Dasatinib+Quercetin (D+Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front. Oncol. 2018, 8, 459. [Google Scholar] [CrossRef]
- Tan, S.; Khumalo, N.P.; Bayat, A. Understanding Keloid Pathobiology From a Quasi-Neoplastic Perspective: Less of a Scar and More of a Chronic Inflammatory Disease With Cancer-Like Tendencies. Front. Immunol. 2019, 10, 1810. [Google Scholar] [CrossRef]
- De Felice, B.; Ciarmiello, L.; Mondola, P.; Damiano, S.; Seru, R.; Argenziano, C.; Nacca, M.; Santoriello, M.; Garbi, C. Differential p63 and p53 expression in human keloid fibroblasts and hypertrophic scar fibroblasts. DNA Cell Biol. 2007, 26, 541–547. [Google Scholar] [CrossRef]
- Saed, G.M.; Ladin, D.; Olson, J.; Han, X.; Hou, Z.; Fivenson, D. Analysis of p53 gene mutations in keloids using polymerase chain reaction-based single-strand conformational polymorphism and DNA sequencing. Arch. Dermatol. 1998, 134, 963–967. [Google Scholar] [CrossRef]
- Ladin, D.A.; Hou, Z.; Patel, D.; McPhail, M.; Olson, J.C.; Saed, G.M.; Fivenson, D.P. p53 and apoptosis alterations in keloids and keloid fibroblasts. Wound Repair Regen. 1998, 6, 28–37. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Yu, X.F.; Huang, J.Q.; Li, D.L. The mechanisms of beta-catenin on keloid fibroblast cells proliferation and apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 888–895. [Google Scholar]
- Cai, Y.; Zhu, S.; Yang, W.; Pan, M.; Wang, C.; Wu, W. Downregulation of beta-catenin blocks fibrosis via Wnt2 signaling in human keloid fibroblasts. Tumour Biol. 2017, 39, 1010428317707423. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Liang, Y.-C.; Wu, P.; Kulber, D.A.; Tanabe, K.; Chuong, C.-M.; Widelitz, R.; Tuan, T.-L. STAT3 signalling pathway is implicated in keloid pathogenesis by preliminary transcriptome and open chromatin analyses. Exp. Dermatol. 2019, 28, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Shang, Y.; Yuan, J.; Ding, S.; Luo, S.; Hao, L. Wnt/beta-Catenin Signaling Exacerbates Keloid Cell Proliferation by Regulating Telomerase. Cell Physiol. Biochem. 2016, 39, 2001–2013. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.; Chudakova, D.A.; Itinteang, T.; Chibnall, A.M.; Brasch, H.D.; Davis, P.F.; Tan, S.T. Expression of embryonic stem cell markers in keloid-associated lymphoid tissue. J. Clin. Pathol. 2016, 69, 643–646. [Google Scholar] [CrossRef]
- Varmeh, S.; Egia, A.; McGrouther, D.; Tahan, S.R.; Bayat, A.; Pandolfi, P.P. Cellular senescence as a possible mechanism for halting progression of keloid lesions. Genes Cancer 2011, 2, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.M.; Glaser, K.; McFarland, K.L.; Aronow, B.J.; Boyce, S.T.; Supp, D.M. Keloid-derived keratinocytes exhibit an abnormal gene expression profile consistent with a distinct causal role in keloid pathology. Wound Repair Regen. 2013, 21, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment Map: A Network-Based Method for Gene-Set Enrichment Visualization and Interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Janky, R.; Verfaillie, A.; Imrichova, H.; Van de Sande, B.; Standaert, L.; Christiaens, V.; Hulselmans, G.; Herten, K.; Sanchez, M.N.; Potier, D.; et al. iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLOS Comput. Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Raman, V.; Martensen, S.A.; Reisman, D.; Evron, E.; Odenwald, W.F.; Jaffee, E.; Marks, J.; Sukumar, S. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature 2000, 405, 974–978. [Google Scholar] [CrossRef]
- Svingen, T.; Tonissen, K. Hox transcription factors and their elusive mammalian gene targets. Heredity 2006, 97, 88–96. [Google Scholar] [CrossRef]
- Chang, C.-J.; Chen, Y.-L.; Hsieh, C.-H.; Liu, Y.-J.; Yu, S.-L.; Chen, J.J.; Wang, C.-C. HOXA5 and p53 cooperate to suppress lung cancer cell invasion and serve as good prognostic factors in non-small cell lung cancer. J. Cancer 2017, 8, 1071–1081. [Google Scholar] [CrossRef]
- Akdemir, K.C.; Jain, A.K.; Allton, K.; Aronow, B.; Xu, X.; Cooney, A.J.; Li, W.; Barton, M.C. Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 2013, 42, 205–223. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Zhang, C.; Sine, C.C.; van Deursen, E.-J.; Jeganathan, K.B.; Hamada, N.; Grasic, J.; Friedman, D.; Stutchman, J.T.; Can, I.; et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 2021, 374, eabb3420. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. ATM/miR-34a-5p axis regulates a p21-dependent senescence-apoptosis switch in non-small cell lung cancer: A Boolean model of G1/S checkpoint regulation. FEBS Lett. 2020, 594, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Limandjaja, G.C.; Niessen, F.B.; Scheper, R.J.; Gibbs, S. The Keloid Disorder: Heterogeneity, Histopathology, Mechanisms and Models. Front. Cell Dev. Biol. 2020, 8, 360. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Gao, J.; Ogawa, R.; Hyakusoku, H.; Ou, C. Biological differences between fibroblasts derived from peripheral and central areas of keloid tissues. Plast. Reconstr. Surg. 2007, 120, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Appleton, I.; Brown, N.J.; Willoughby, D.A. Apoptosis, necrosis, and proliferation: Possible implications in the etiology of keloids. Am. J. Pathol. 1996, 149, 1441–1447. [Google Scholar]
- Akasaka, Y.; Fujita, K.; Ishikawa, Y.; Asuwa, N.; Inuzuka, K.; Ishihara, M.; Ito, M.; Masuda, T.; Akishima, Y.; Zhang, L.; et al. Detection of apoptosis in keloids and a comparative study on apoptosis between keloids, hypertrophic scars, normal healed flat scars, and dermatofibroma. Wound Repair Regen. 2001, 9, 501–506. [Google Scholar] [CrossRef]
- Huang, C.; Akaishi, S.; Hyakusoku, H.; Ogawa, R. Are keloid and hypertrophic scar different forms of the same disorder? A fibroproliferative skin disorder hypothesis based on keloid findings. Int. Wound J. 2014, 11, 517–522. [Google Scholar] [CrossRef]
- Le, A.D.; Zhang, Q.; Wu, Y.; Messadi, D.V.; Akhondzadeh, A.; Nguyen, A.L.; Aghaloo, T.L.; Kelly, A.P.; Bertolami, C.N. Elevated vascular endothelial growth factor in keloids: Relevance to tissue fibrosis. Cells Tissues Organs 2004, 176, 87–94. [Google Scholar] [CrossRef]
- Touchi, R.; Ueda, K.; Kurokawa, N.; Tsuji, M. Central regions of keloids are severely ischaemic. J. Plast. Reconstr. Aesthe.t Surg. 2016, 69, e35–e41. [Google Scholar] [CrossRef]
- Louw, L.; Van Der Westhuizen, J.P.; De Wit, L.D.; Edwards, G. Keloids: Peripheral and central differences in cell morphology and fatty acid compositions of lipids. Adv. Exp. Med. Biol. 1997, 407, 515–520. [Google Scholar]
- Lim, I.J.; Phan, T.-T.; Bay, B.-H.; Qi, R.; Huynh, H.; Tan, W.T.-L.; Lee, S.-T.; Longaker, M.T. Fibroblasts cocultured with keloid keratinocytes: Normal fibroblasts secrete collagen in a keloidlike manner. Am. J. Physiol. Cell Physiol. 2002, 283, C212–C222. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, J.; Xu, B.; Long, X.; Qin, H.; Zhao, R.C.; Wang, X. Keloid-derived keratinocytes acquire a fibroblast-like appearance and an enhanced invasive capacity in a hypoxic microenvironment in vitro. Int. J. Mol. Med. 2015, 35, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Funayama, E.; Chodon, T.; Oyama, A.; Sugihara, T. Keratinocytes promote proliferation and inhibit apoptosis of the underlying fibroblasts: An important role in the pathogenesis of keloid. J. Invest. Dermatol. 2003, 121, 1326–1331. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-Y.; Wu, C.-W.; Lin, T.-Y. Role of Senescence-Resumed Proliferation in Keloid Pathogenesis. Future Pharmacol. 2023, 3, 198-212. https://doi.org/10.3390/futurepharmacol3010014
Wang C-Y, Wu C-W, Lin T-Y. Role of Senescence-Resumed Proliferation in Keloid Pathogenesis. Future Pharmacology. 2023; 3(1):198-212. https://doi.org/10.3390/futurepharmacol3010014
Chicago/Turabian StyleWang, Ching-Yun, Chieh-Wen Wu, and Ting-Yi Lin. 2023. "Role of Senescence-Resumed Proliferation in Keloid Pathogenesis" Future Pharmacology 3, no. 1: 198-212. https://doi.org/10.3390/futurepharmacol3010014
APA StyleWang, C.-Y., Wu, C.-W., & Lin, T.-Y. (2023). Role of Senescence-Resumed Proliferation in Keloid Pathogenesis. Future Pharmacology, 3(1), 198-212. https://doi.org/10.3390/futurepharmacol3010014