
In Silico ADME Profiling of Salubrinal and Its Analogues


Abstract
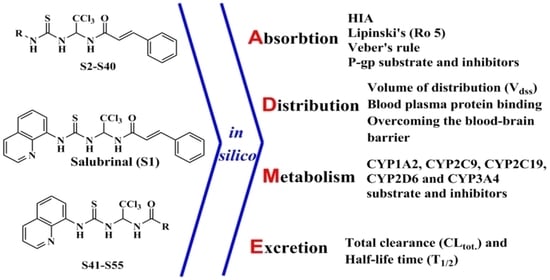
:
1. Introduction
2. Materials and Methods
2.1. Absorption
2.1.1. Calculation of the Lipophilicity Coefficient (LogP)
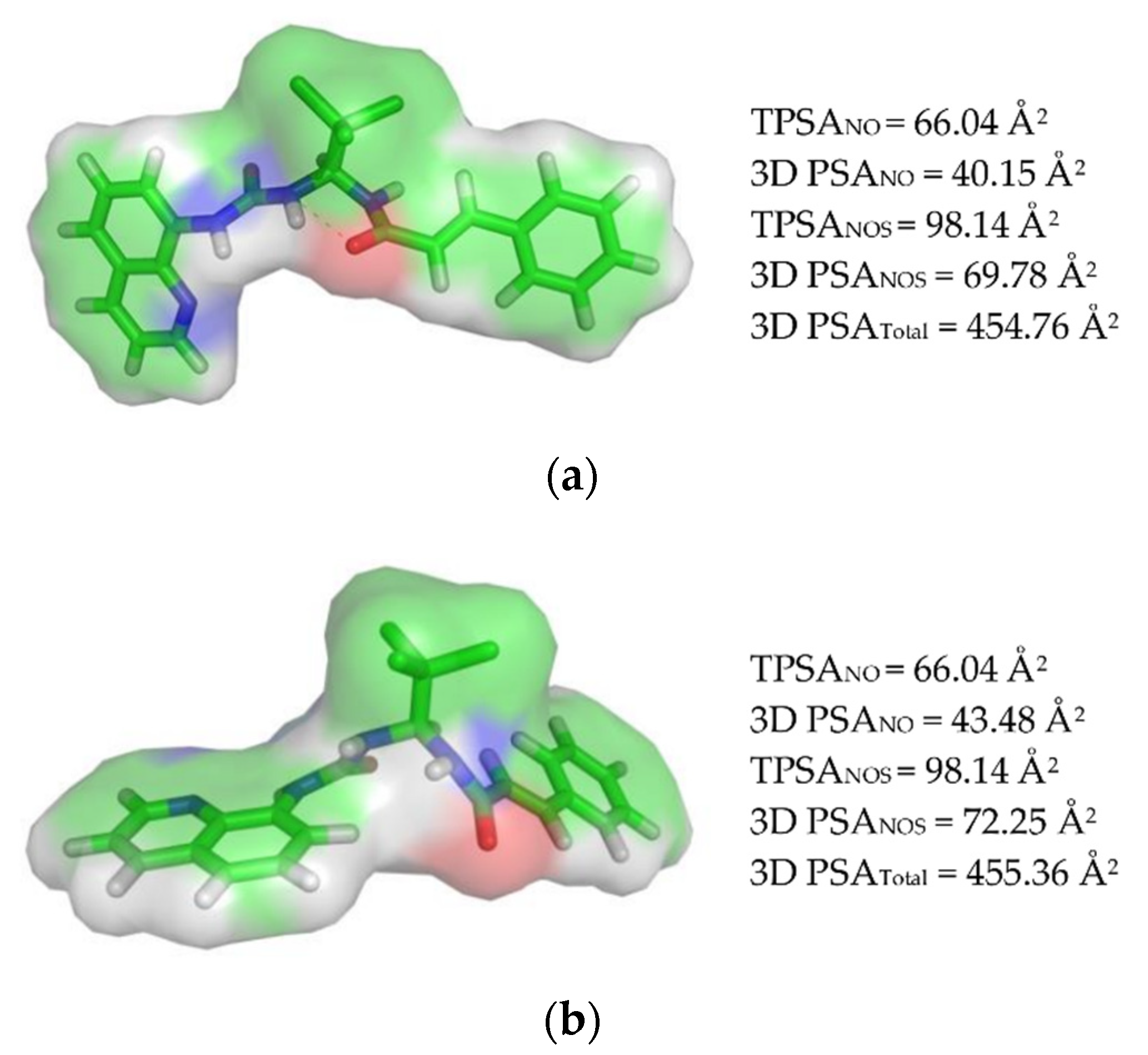
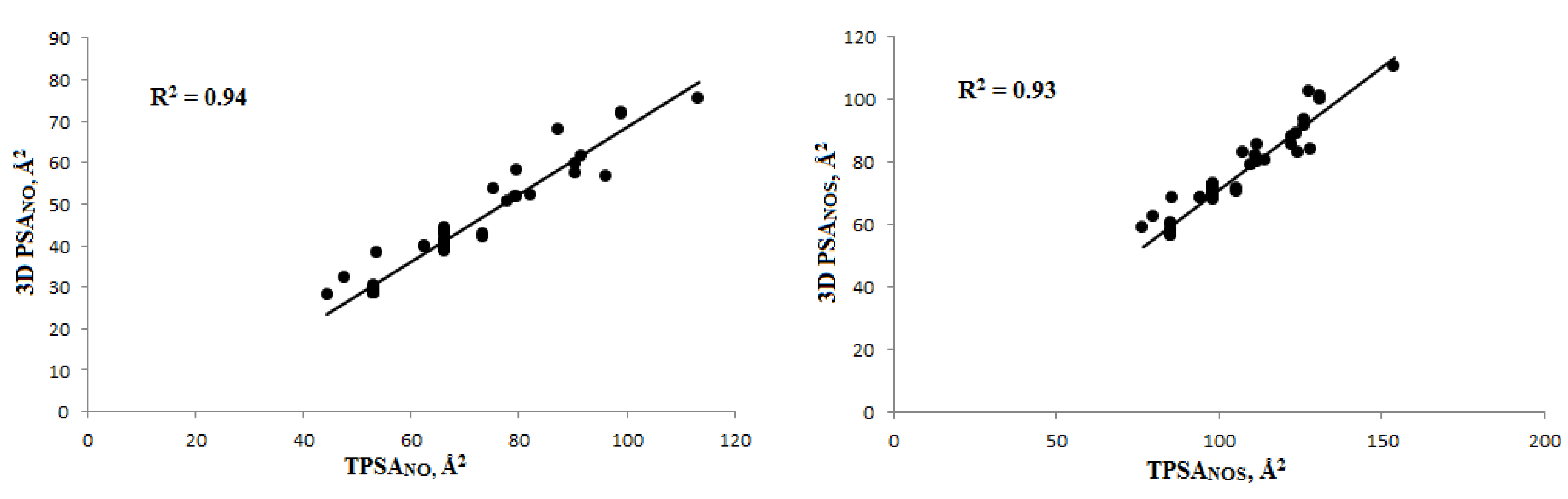
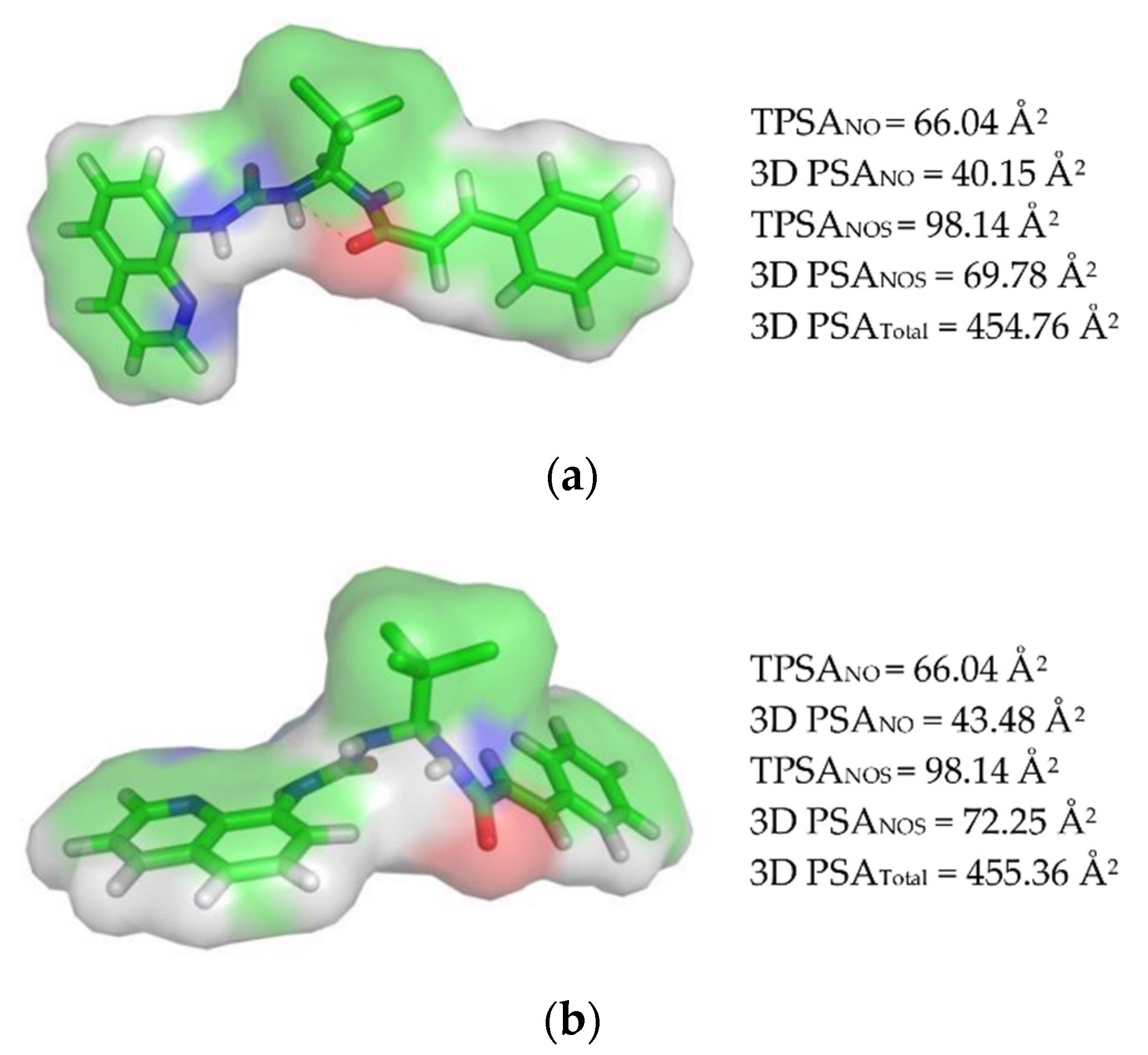
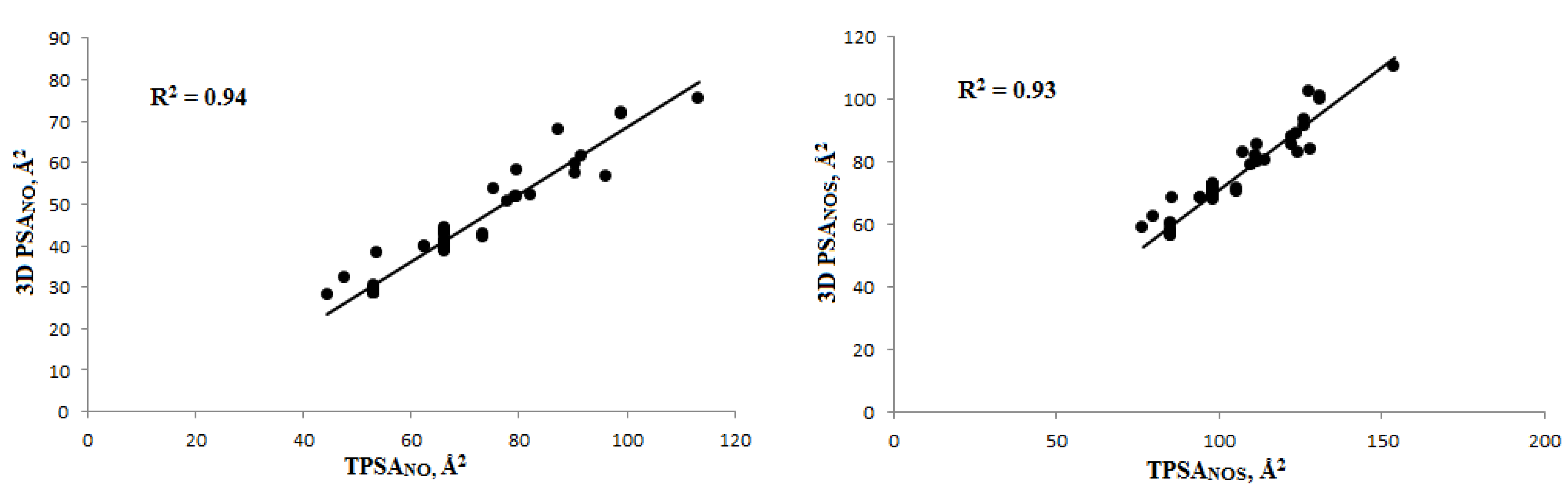
2.1.2. Calculating the Polar Surface Area (PSA)
2.1.3. Interaction with P-glycoprotein (P-gp)
2.2. Distribution
2.2.1. Volume of Distribution
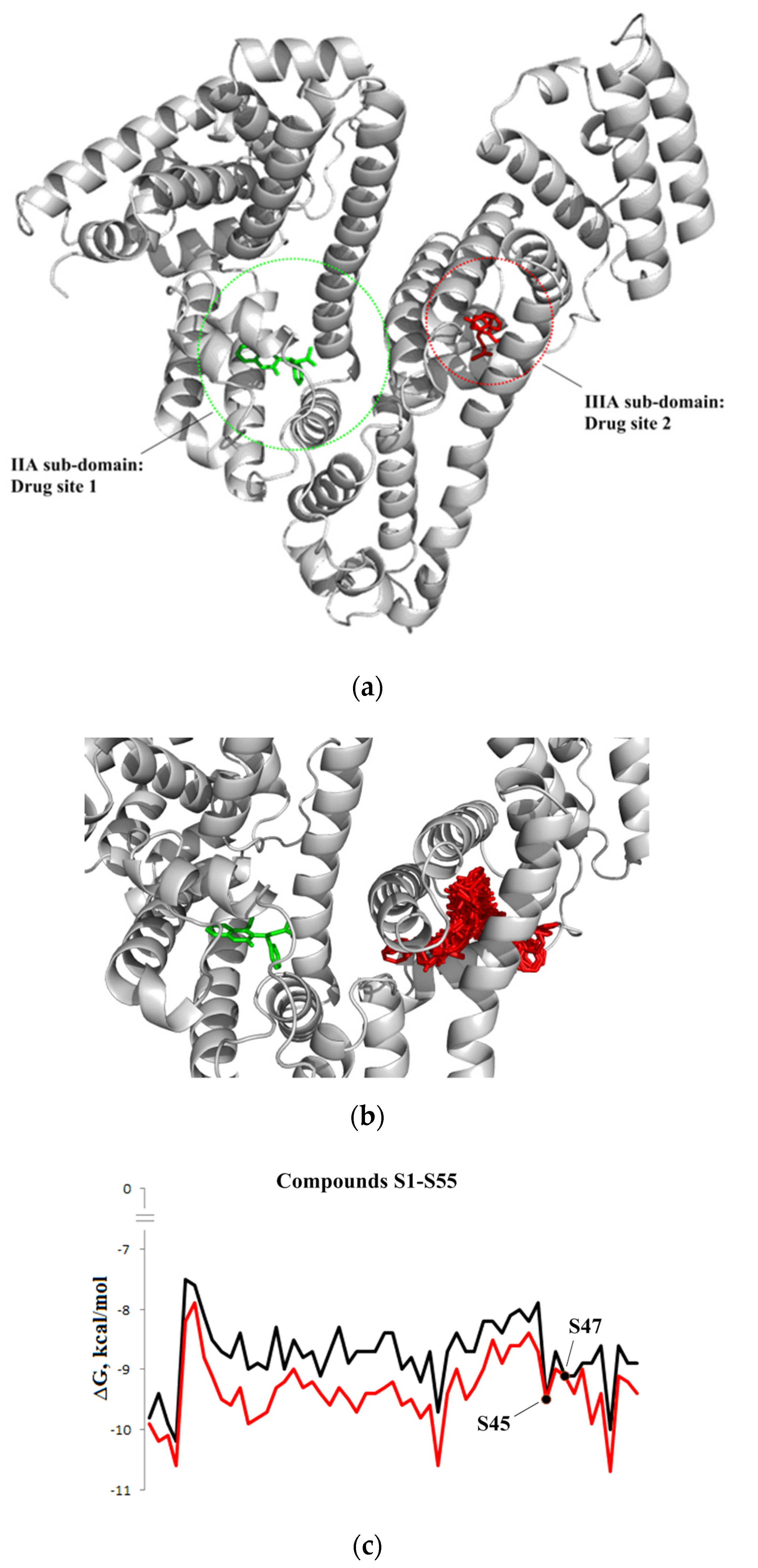
2.2.2. Blood Plasma Protein Binding
2.2.3. Overcoming the Blood–Brain Barrier
2.3. Metabolism
2.4. Elimination
3. Results and Discussion
3.1. Absorption
3.1.1. Calculation of the Lipophilicity Coefficient (LogP) and Ro 5
3.1.2. Polar Surface Area (PSA) Calculation and Veber’s Rule
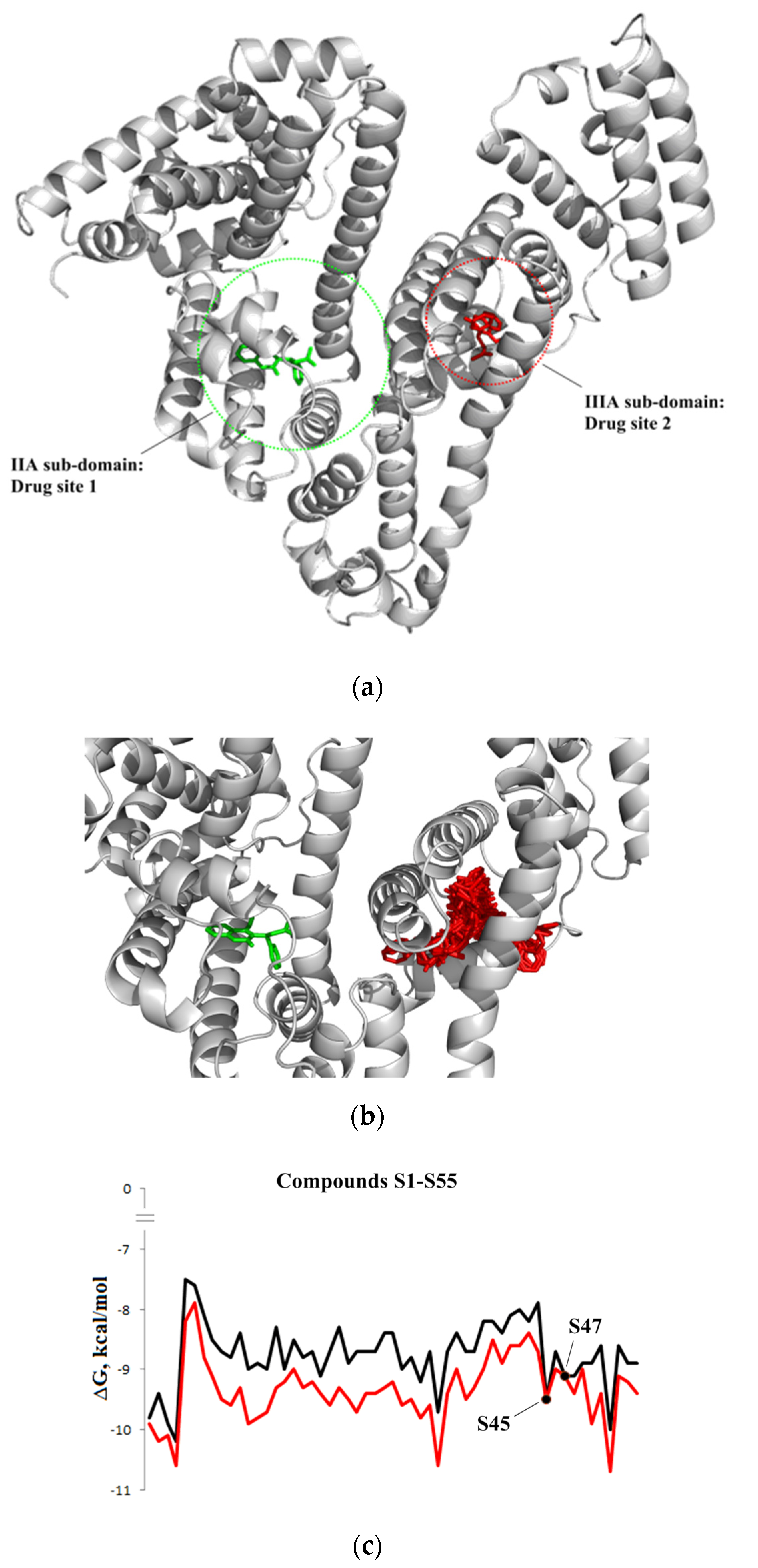
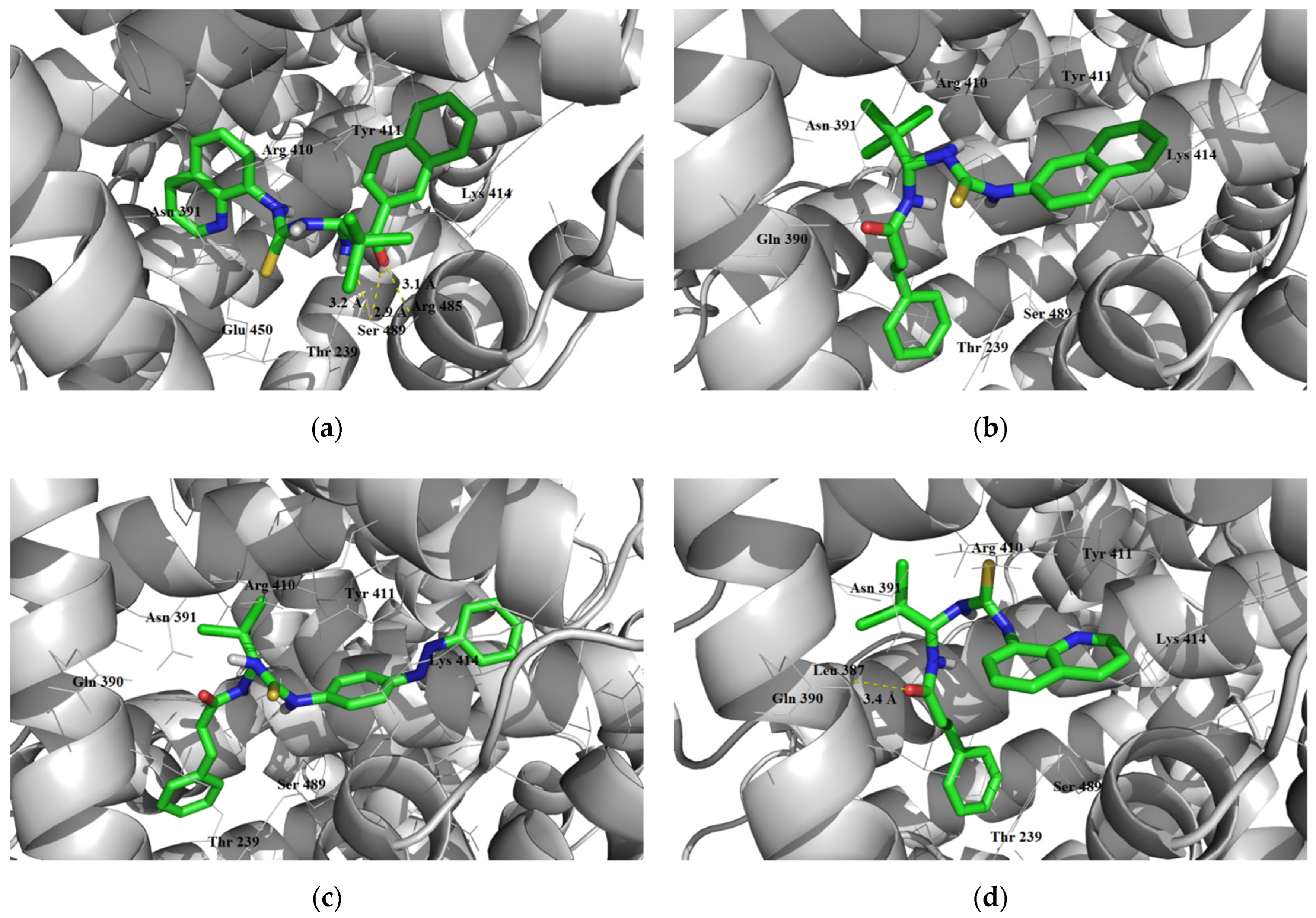
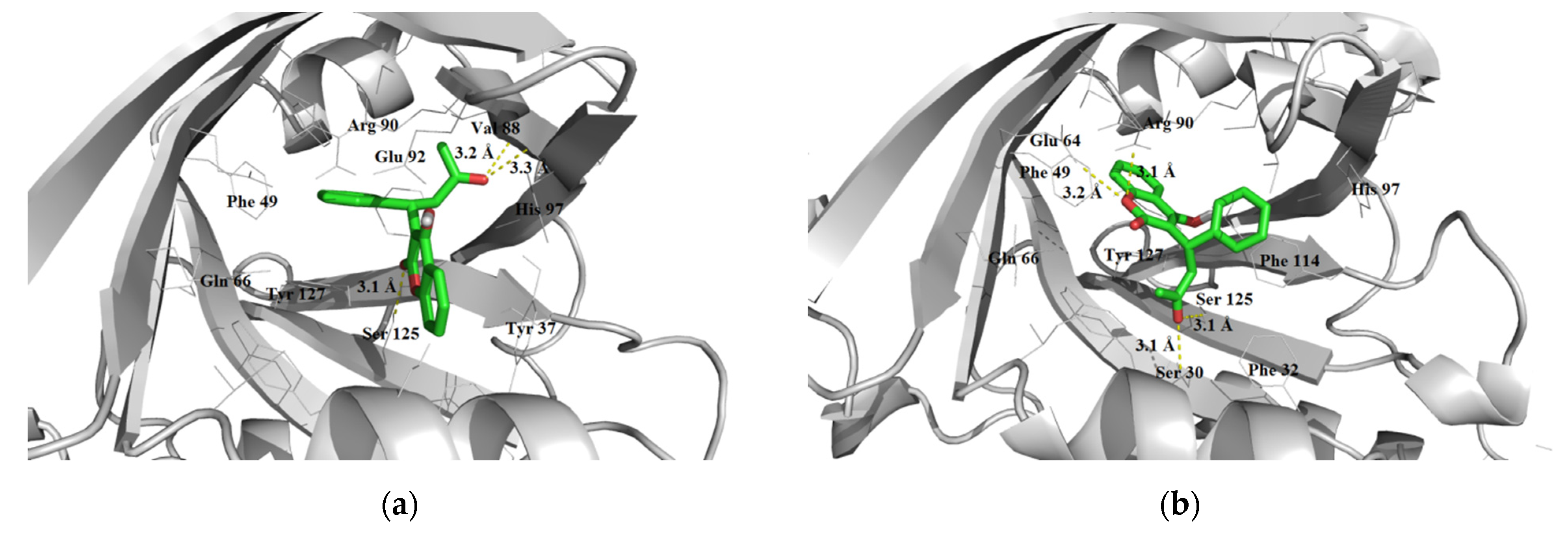
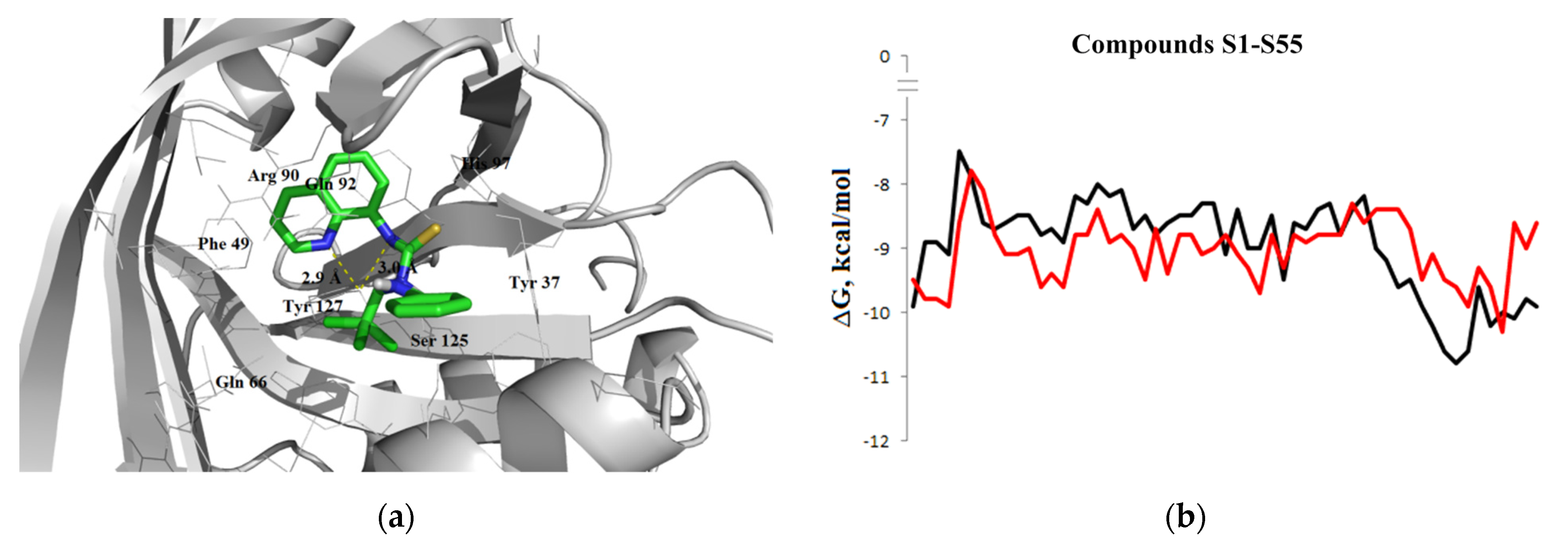
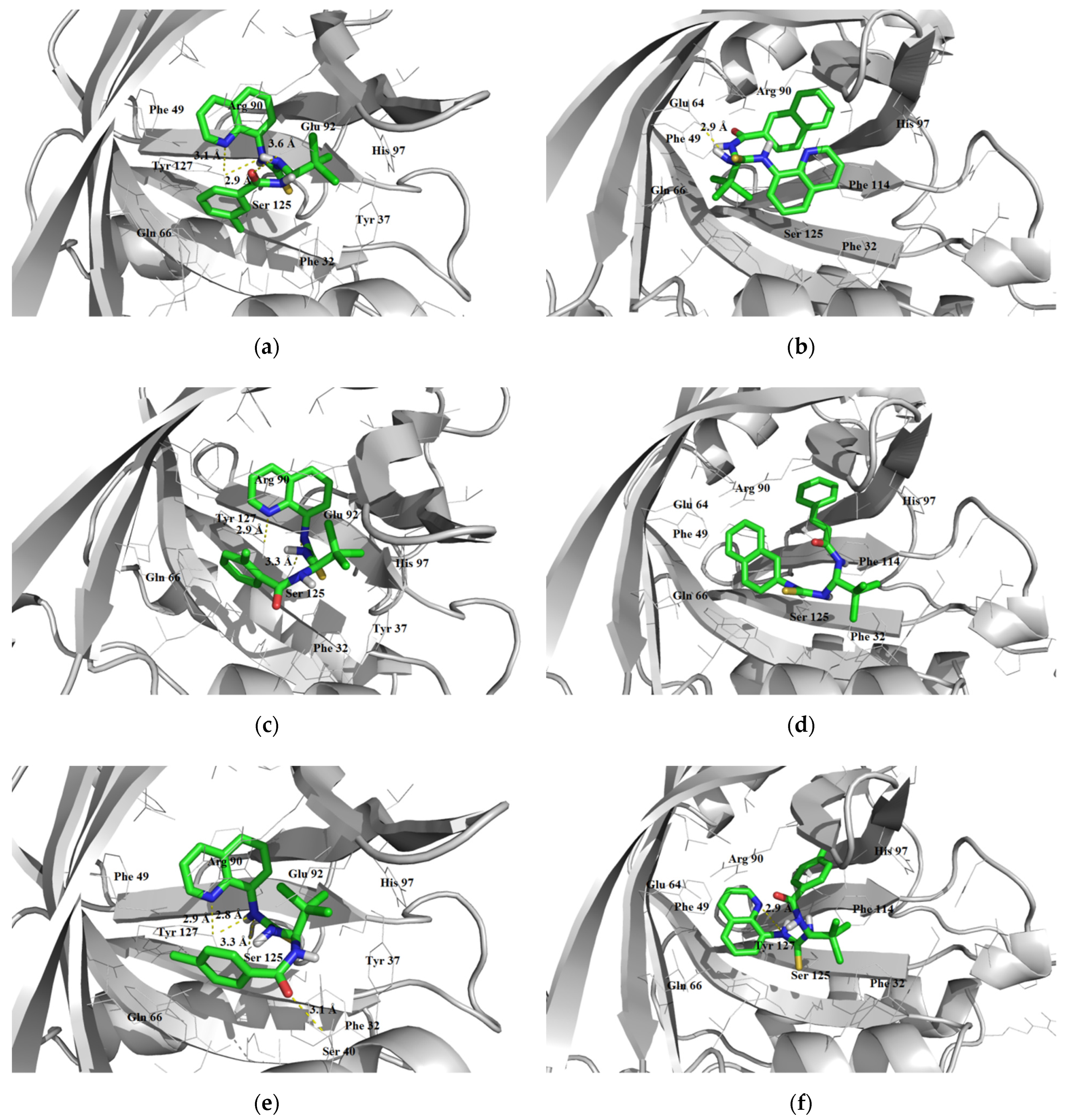
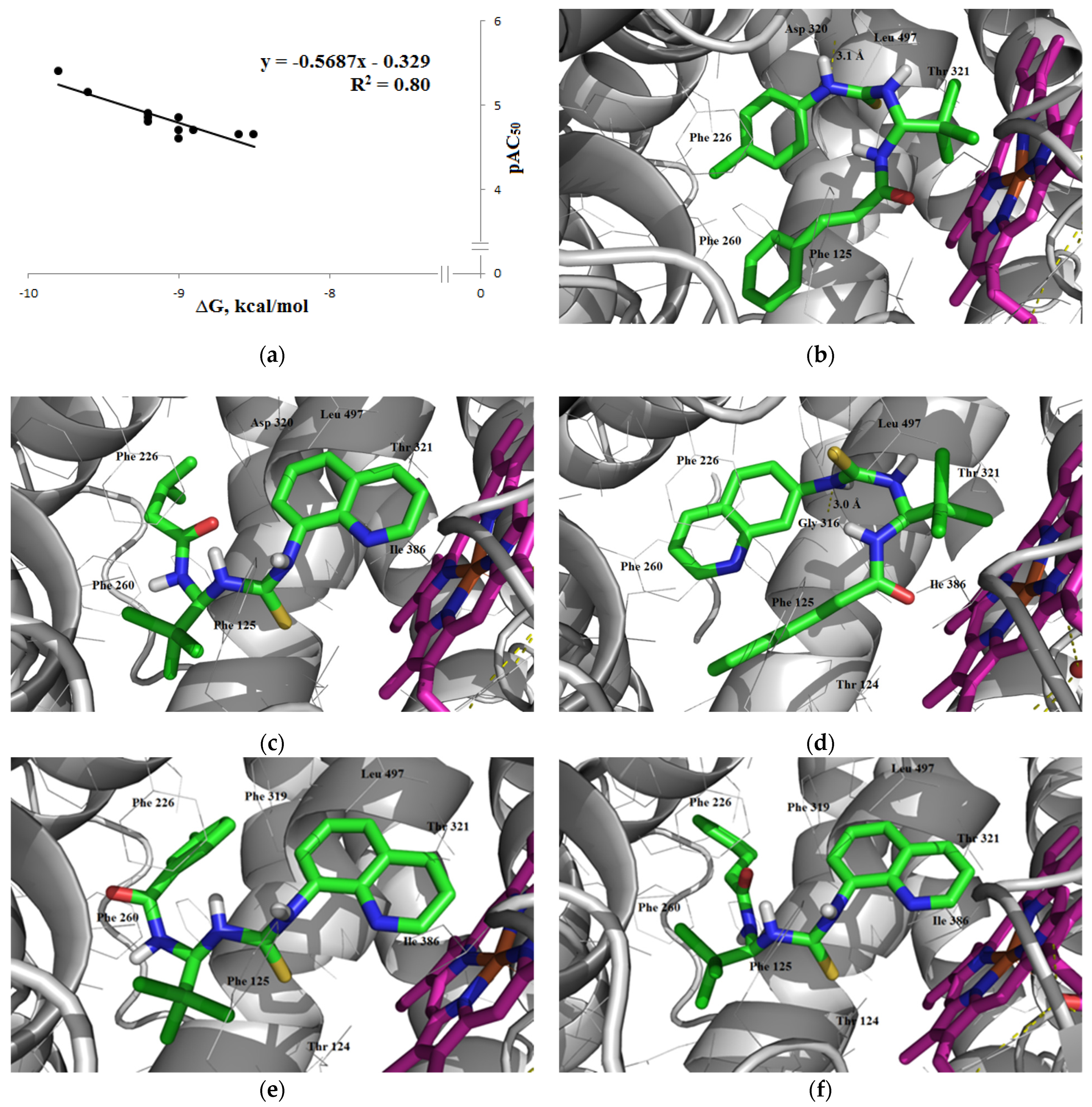
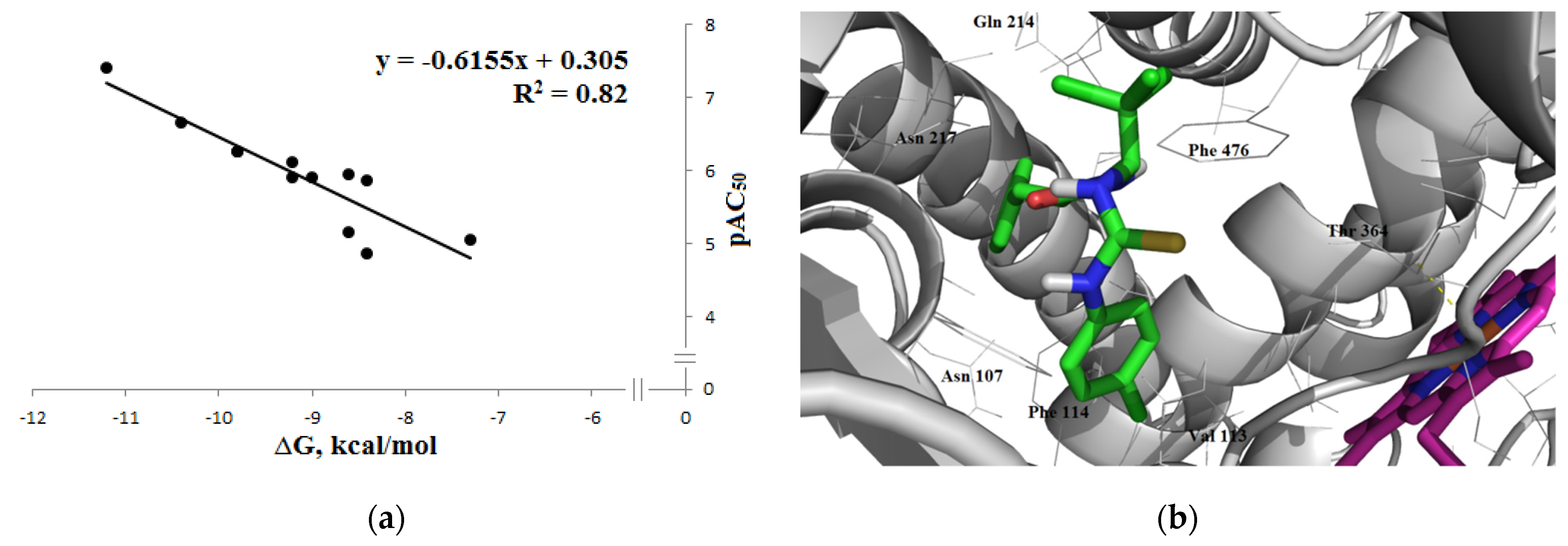
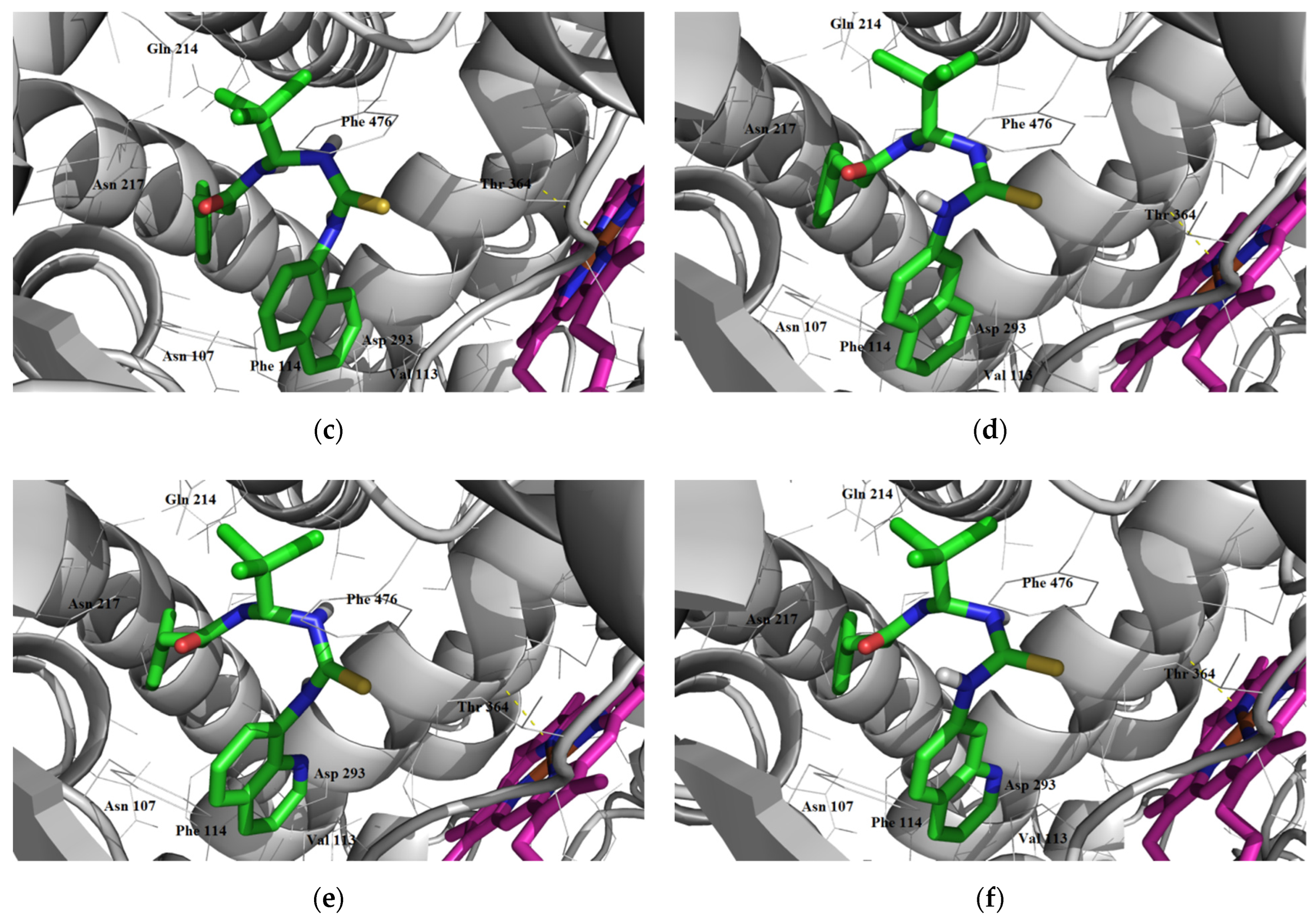
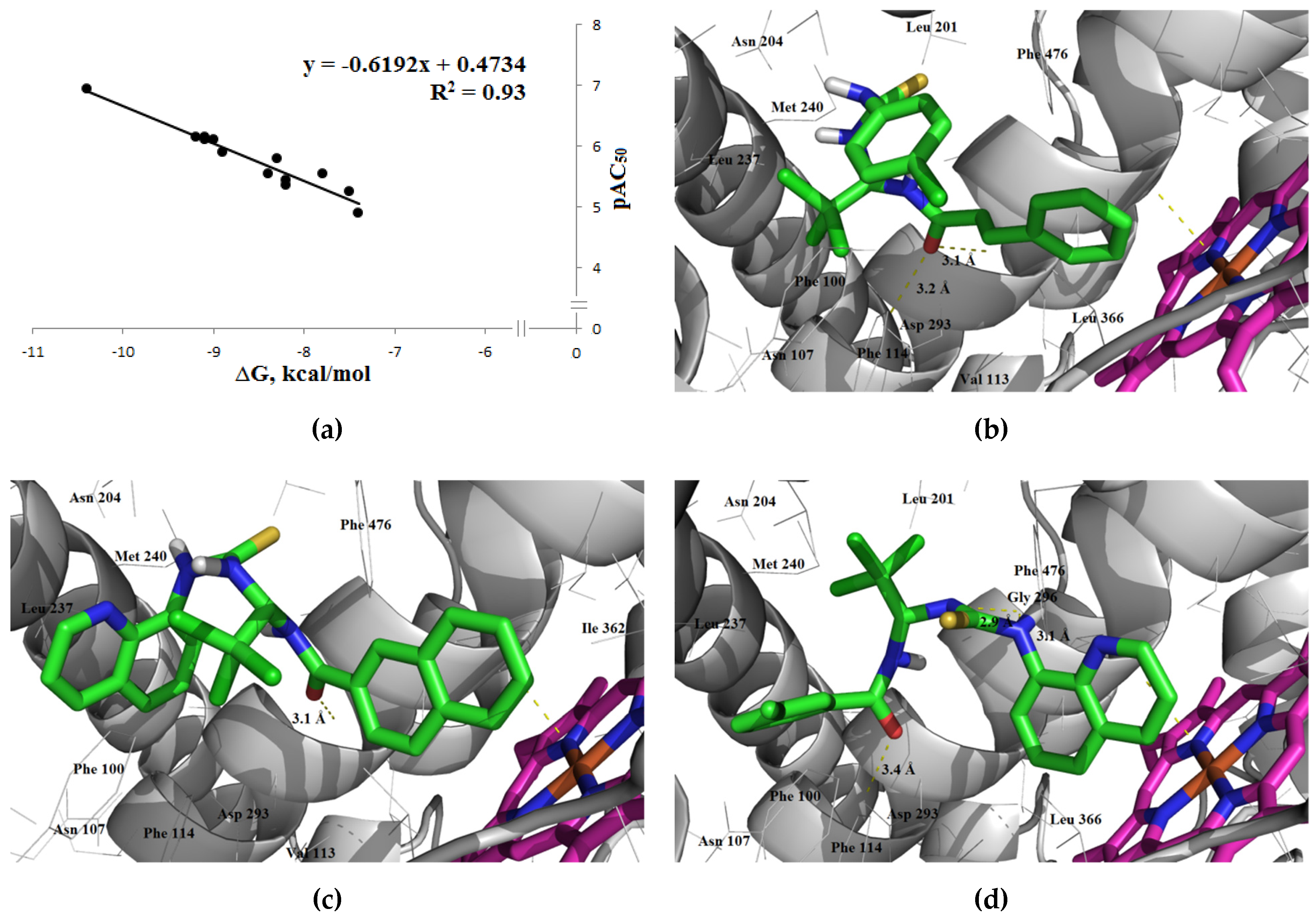
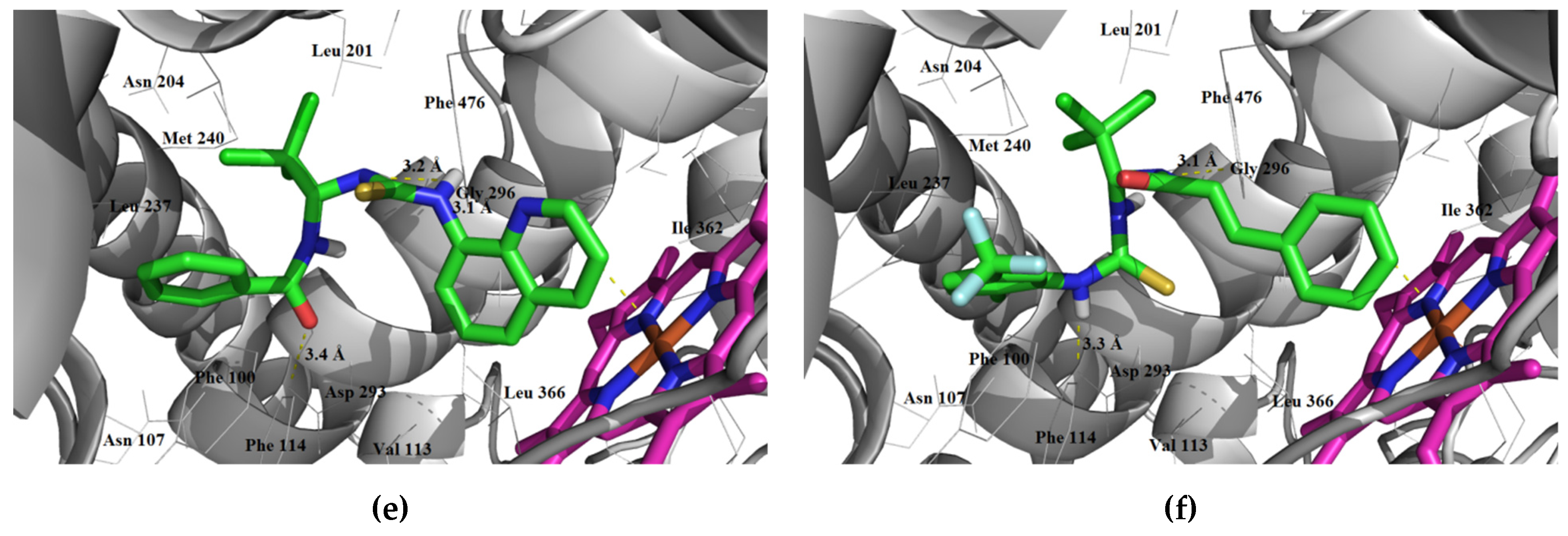
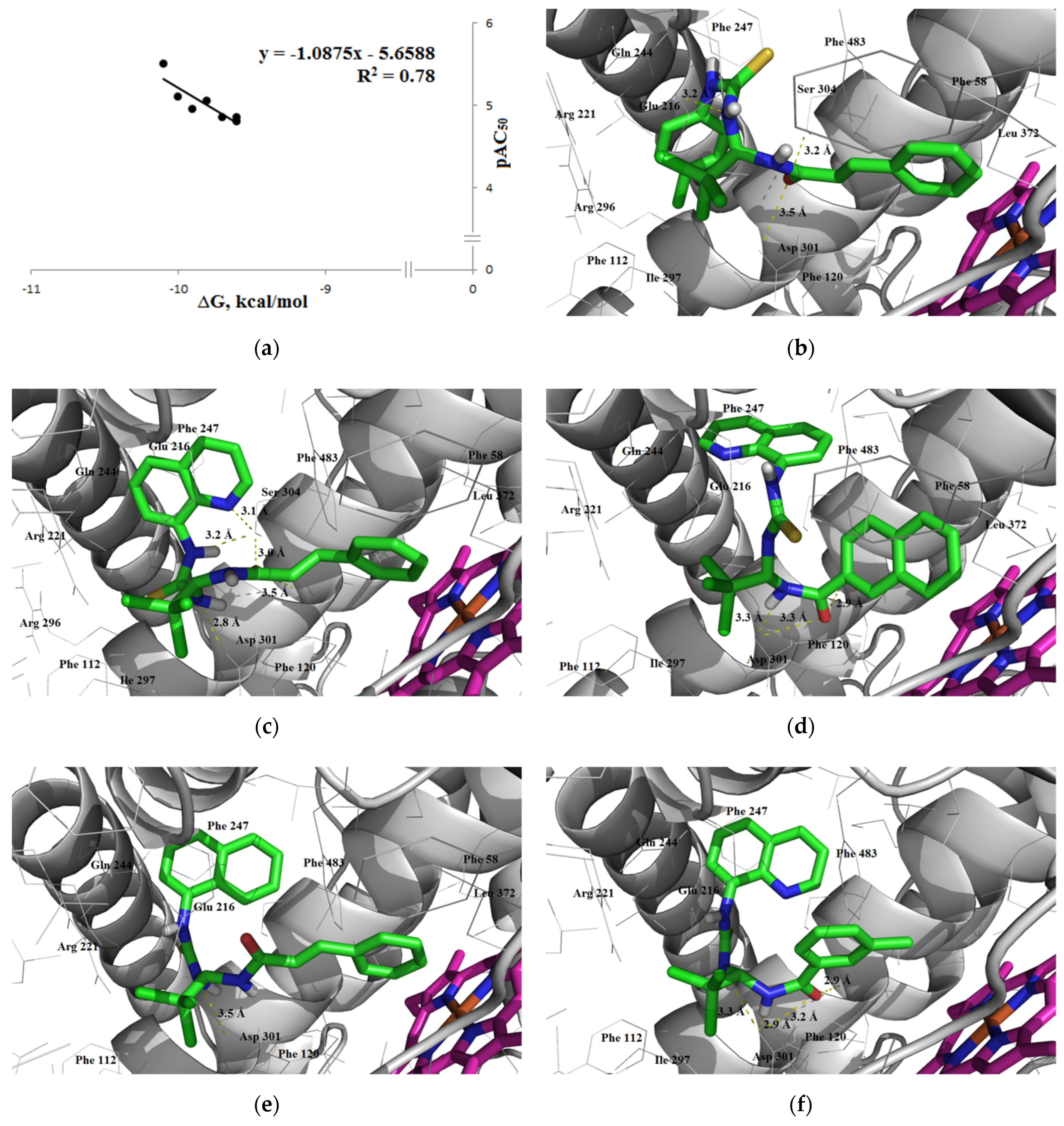
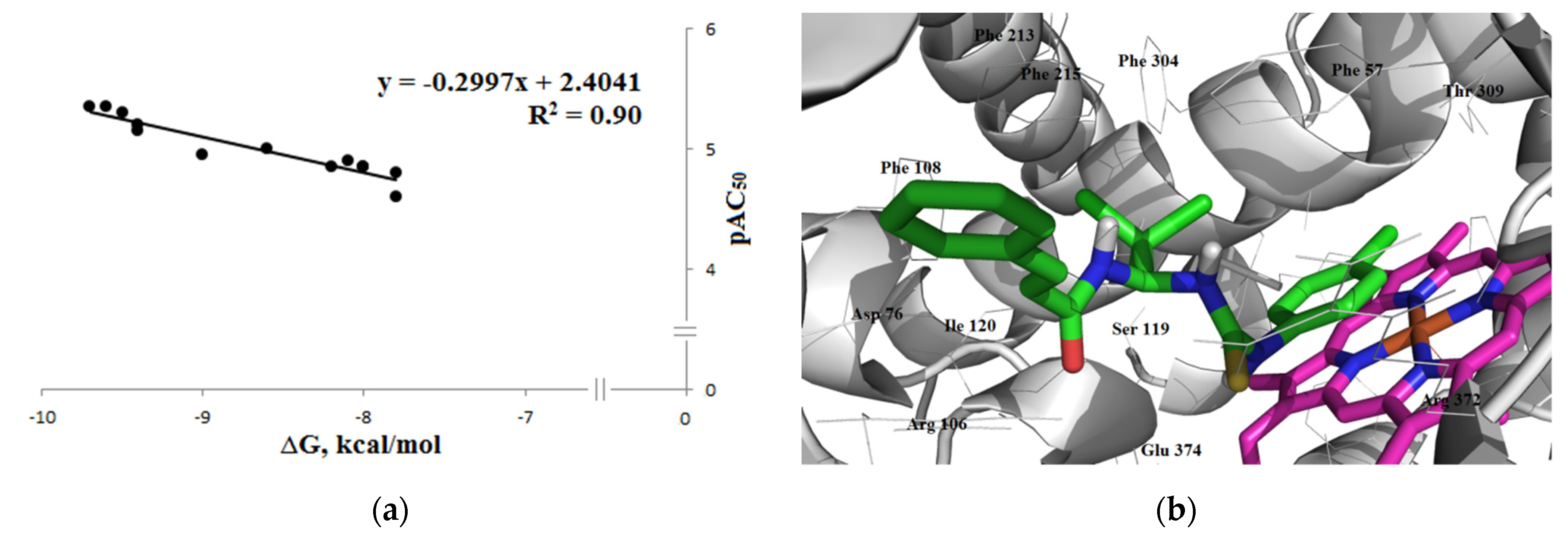
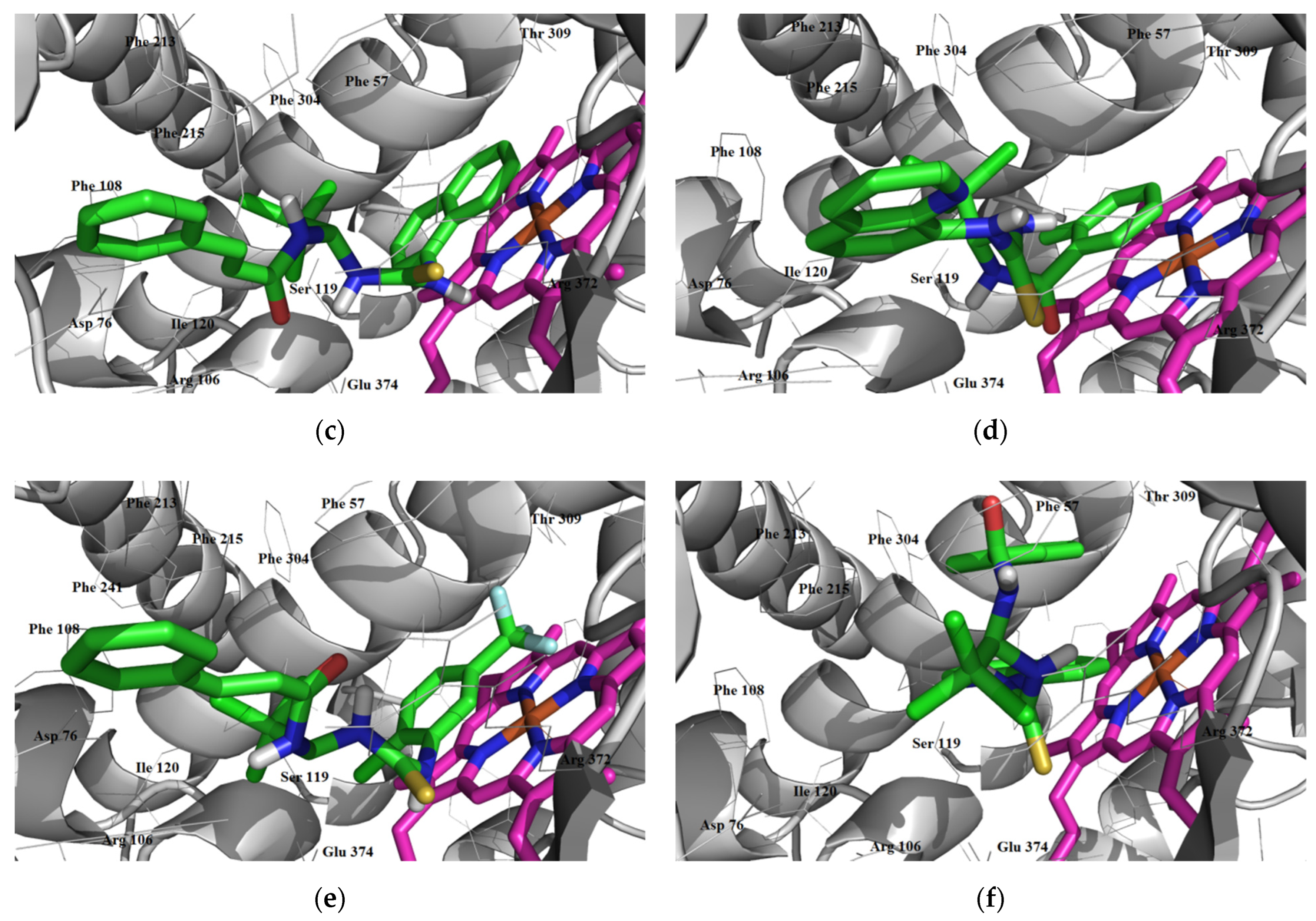
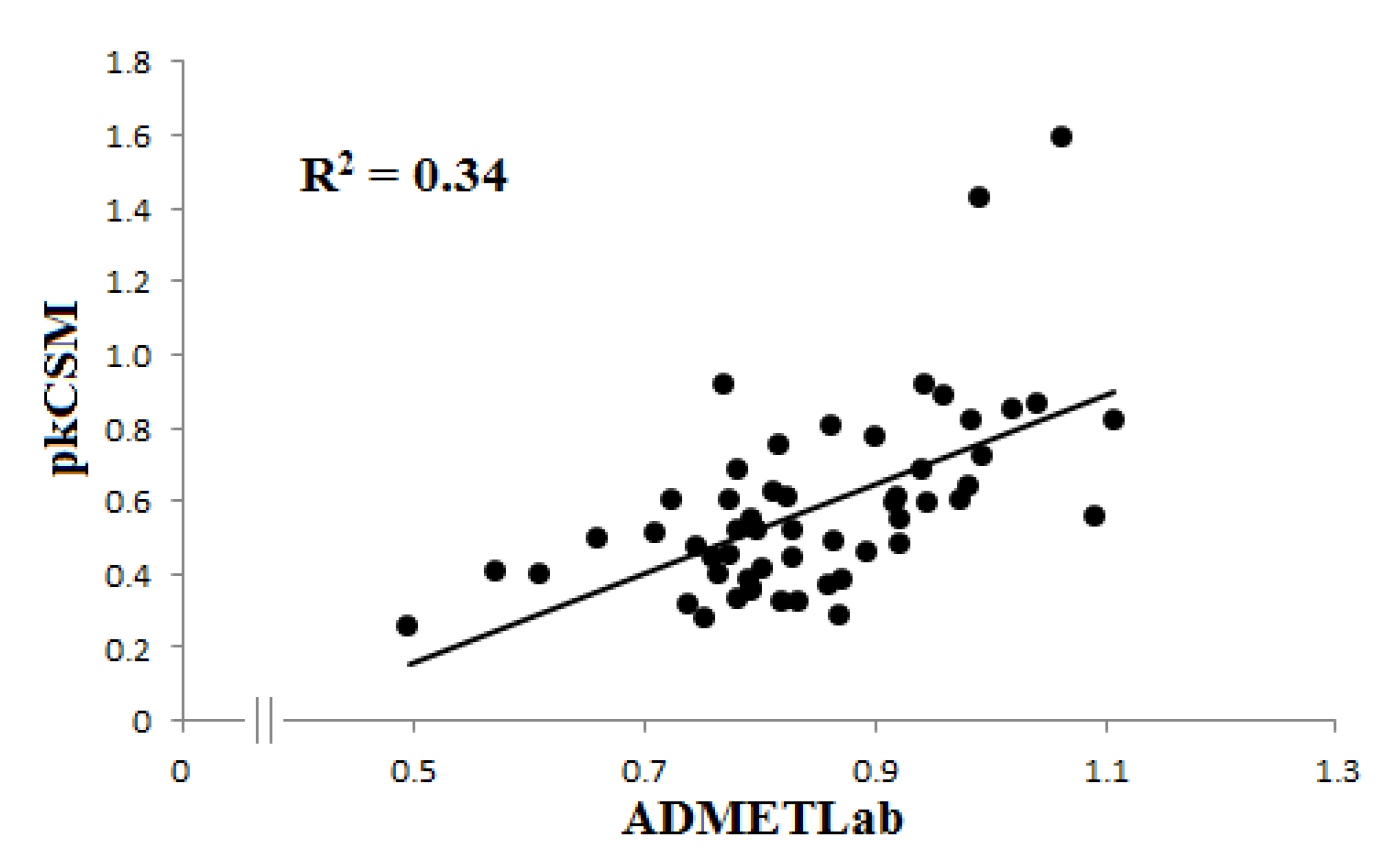
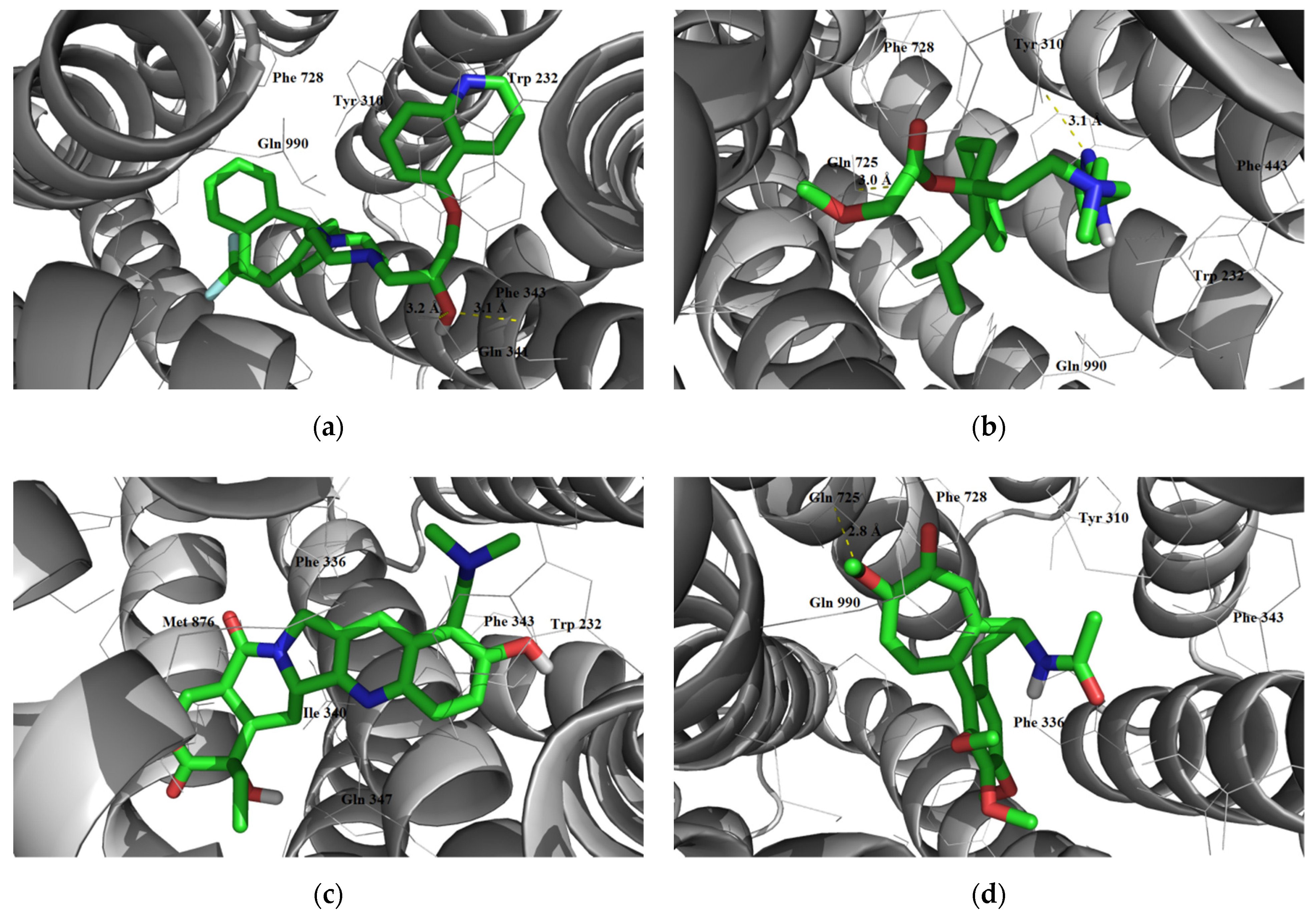
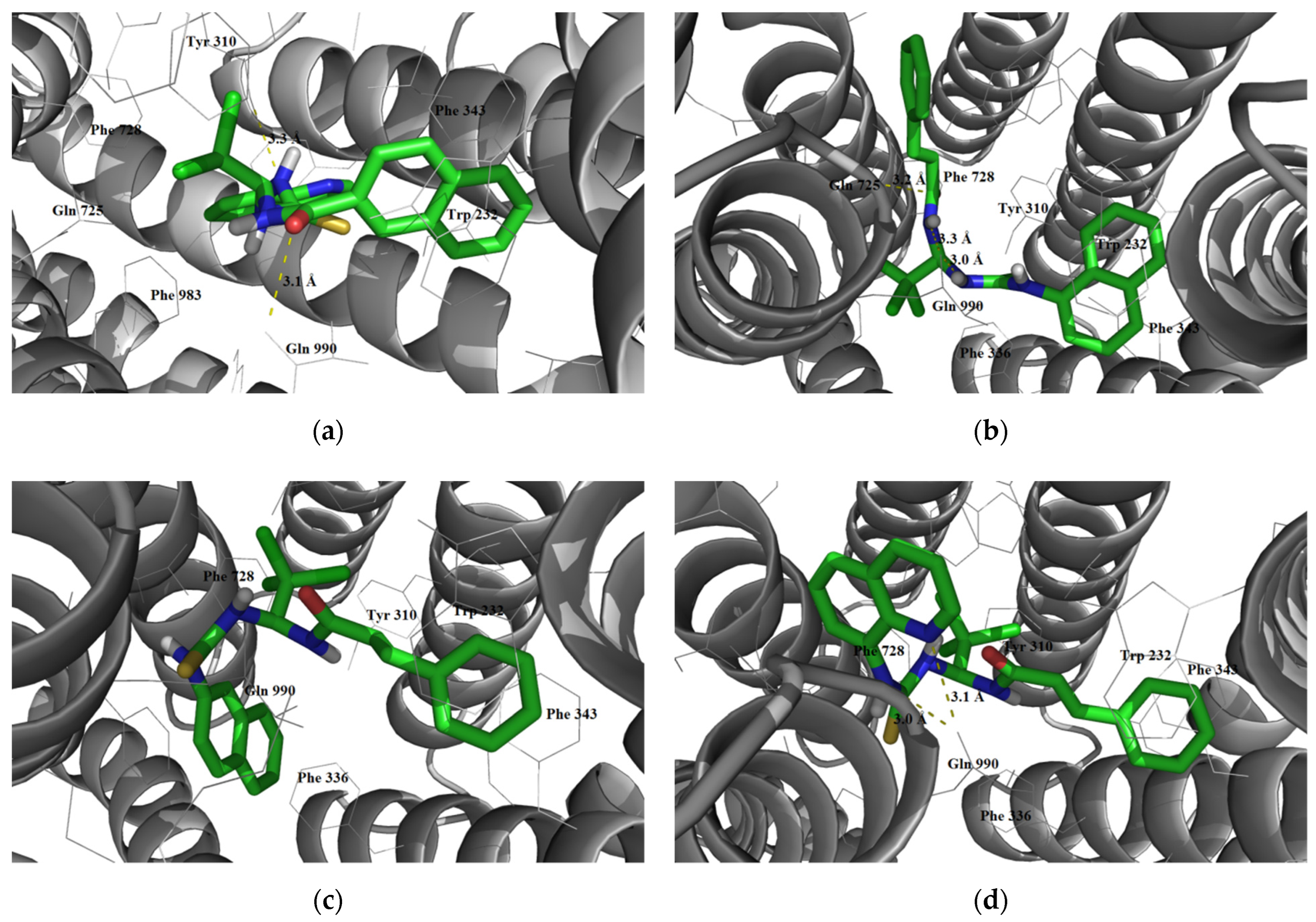
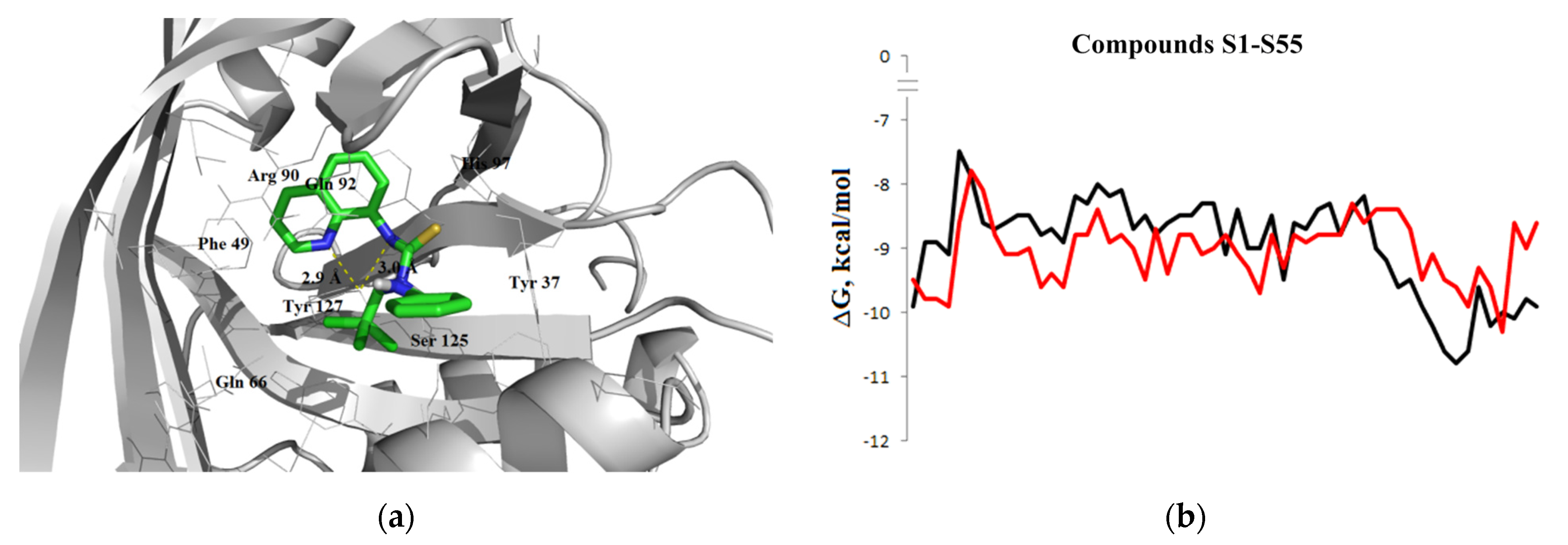
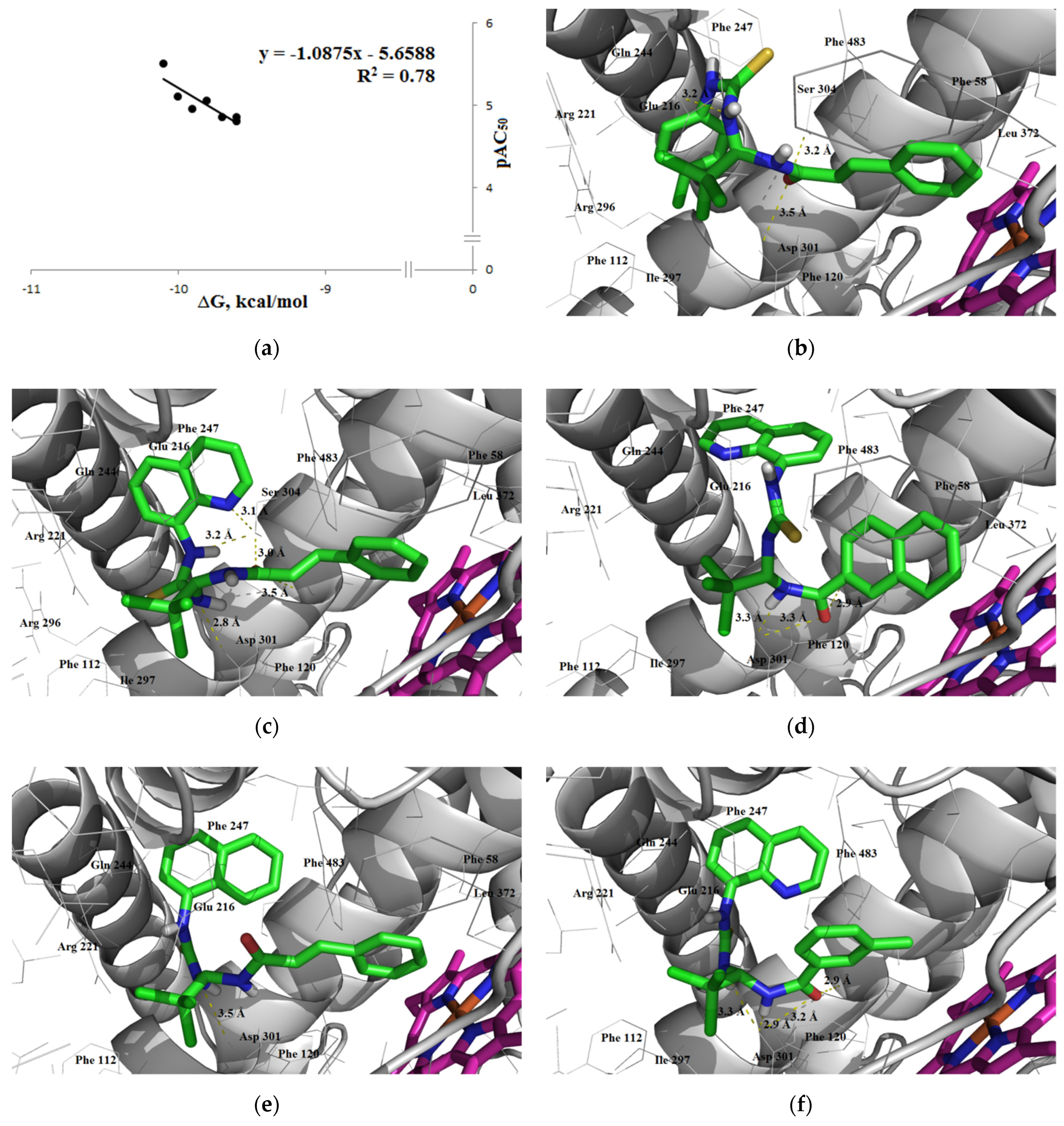
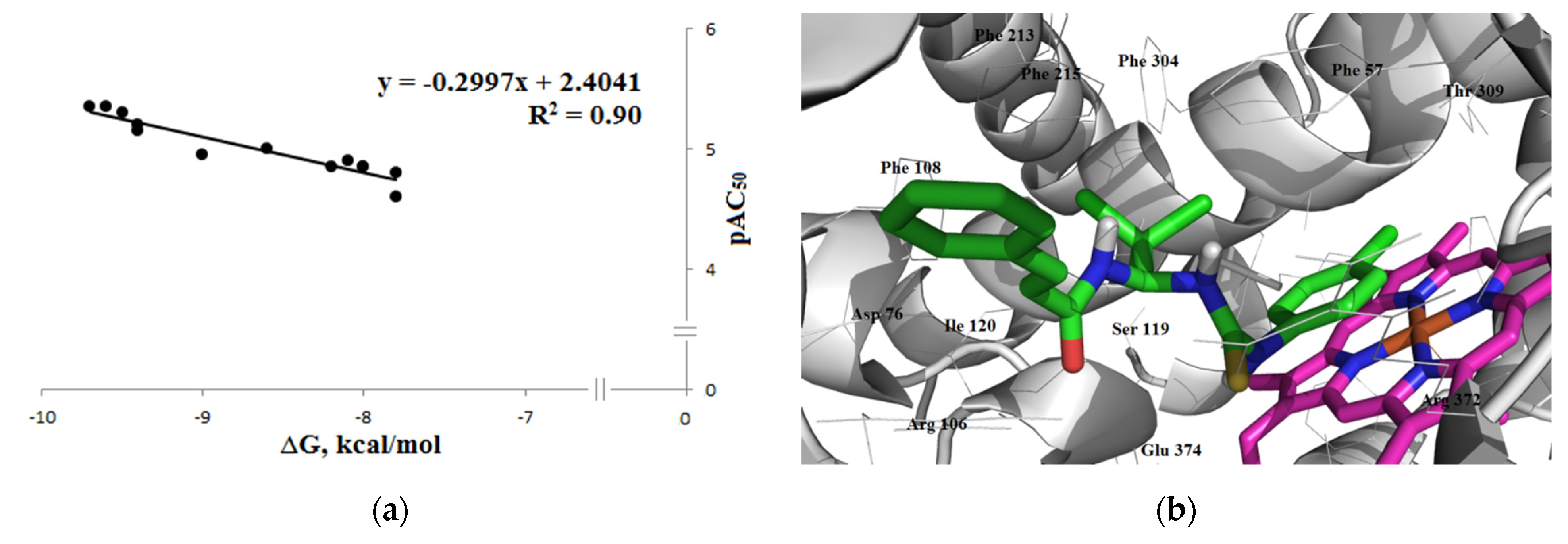
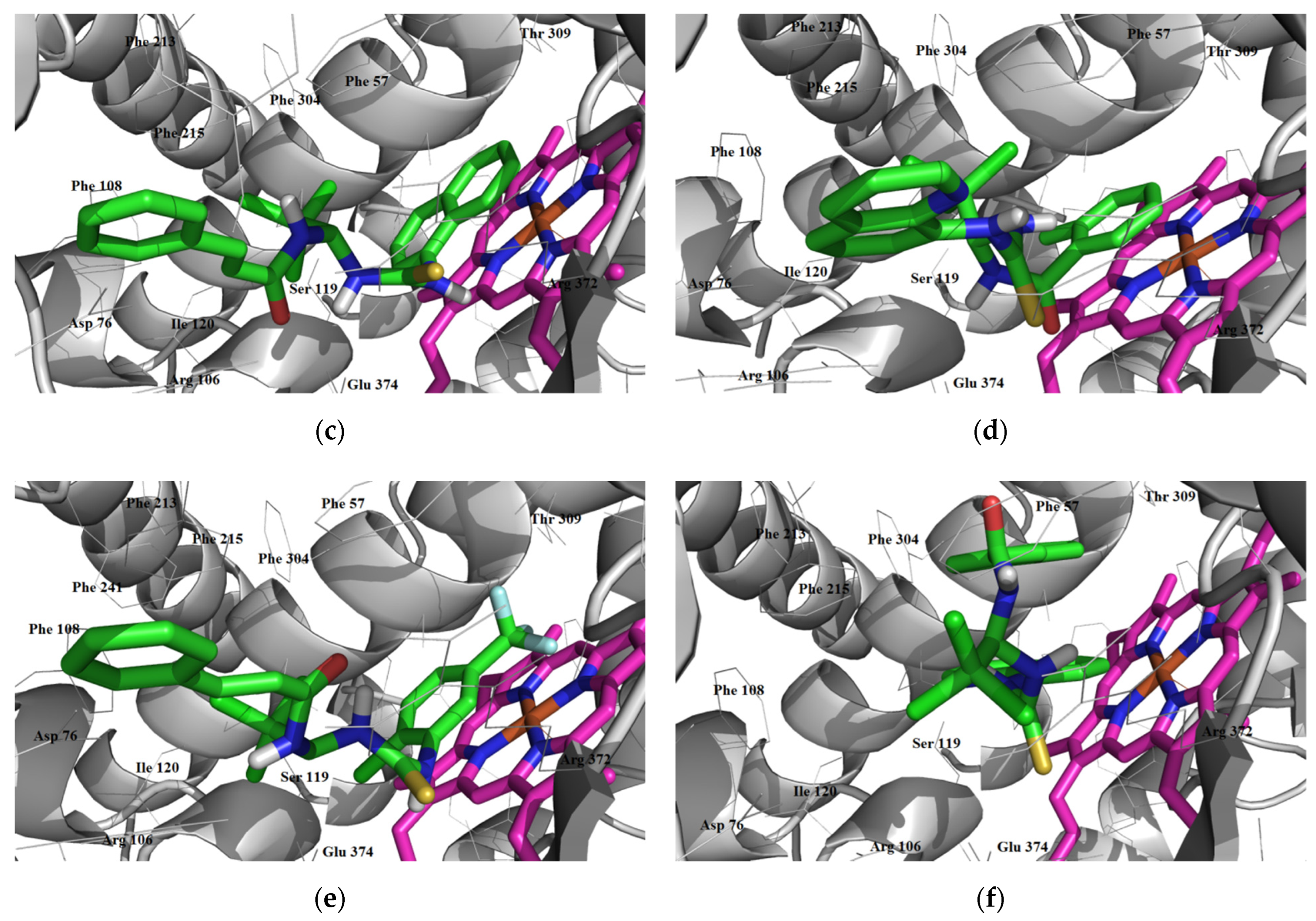
3.1.3. Interaction with P-glycoprotein (P-gp)
3.2. Distribution
3.2.1. Volume of Distribution
3.2.2. Blood Plasma Protein Binding
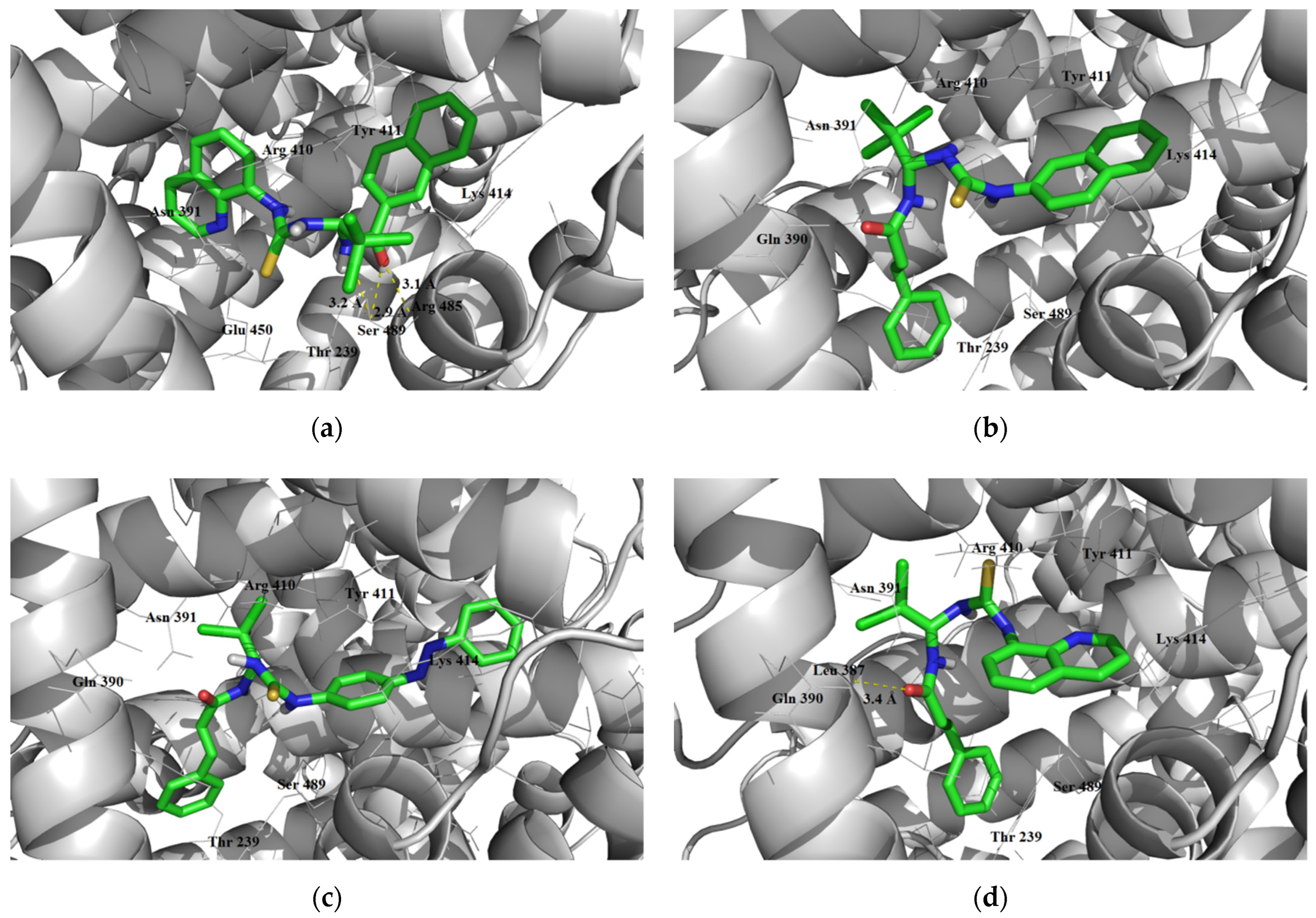
3.2.3. Overcoming the Blood–Brain Barrier
3.3. Metabolism
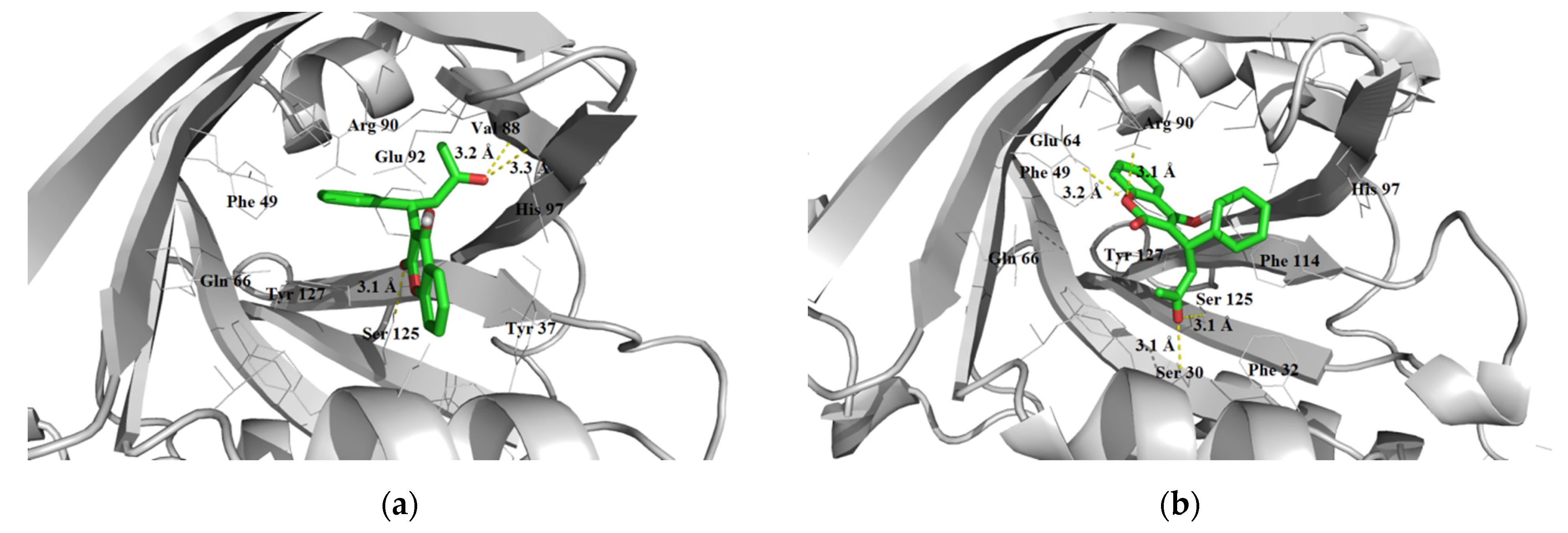
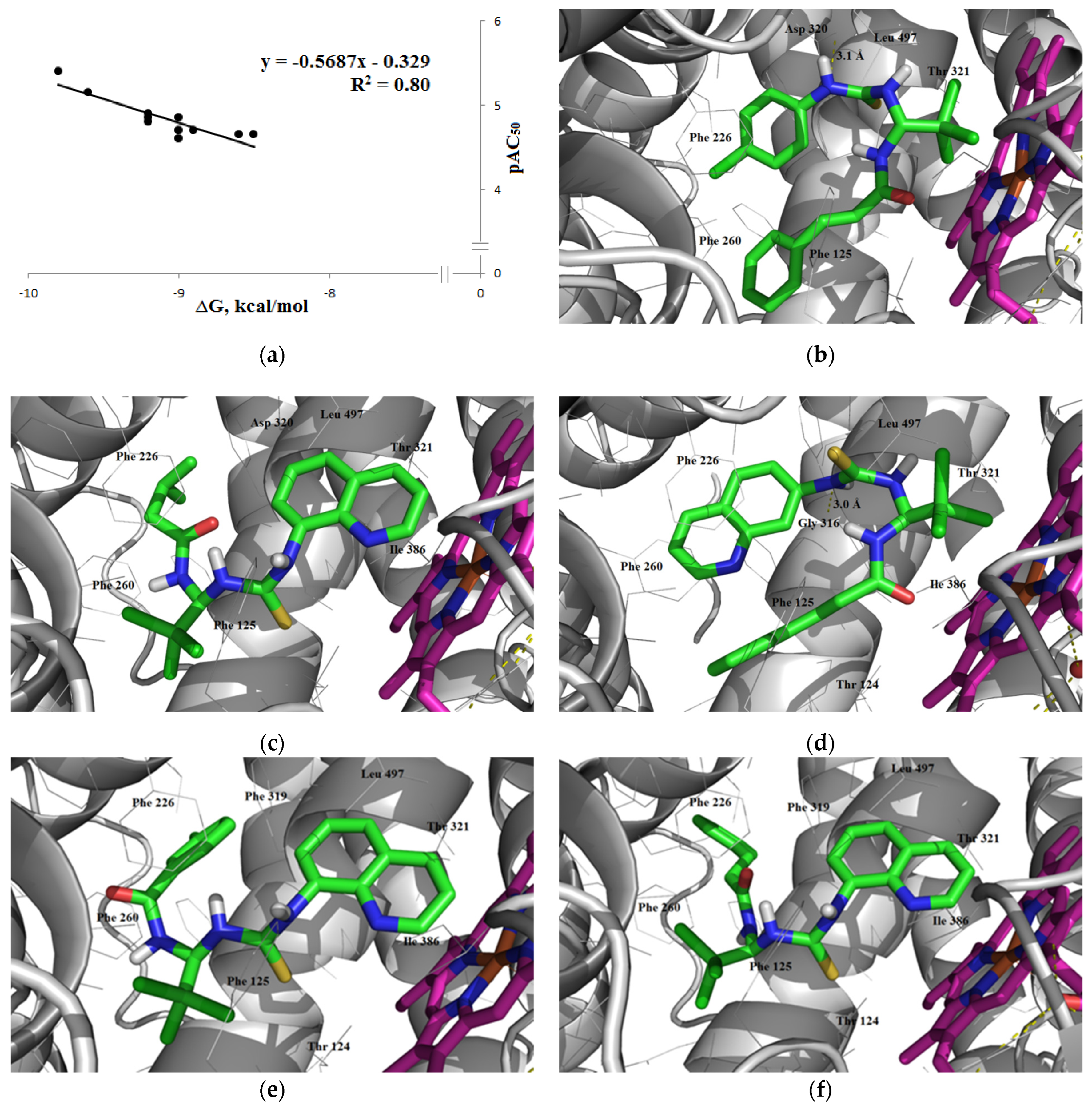
3.3.1. Inhibitory Activity
Inhibition of CYP1A2
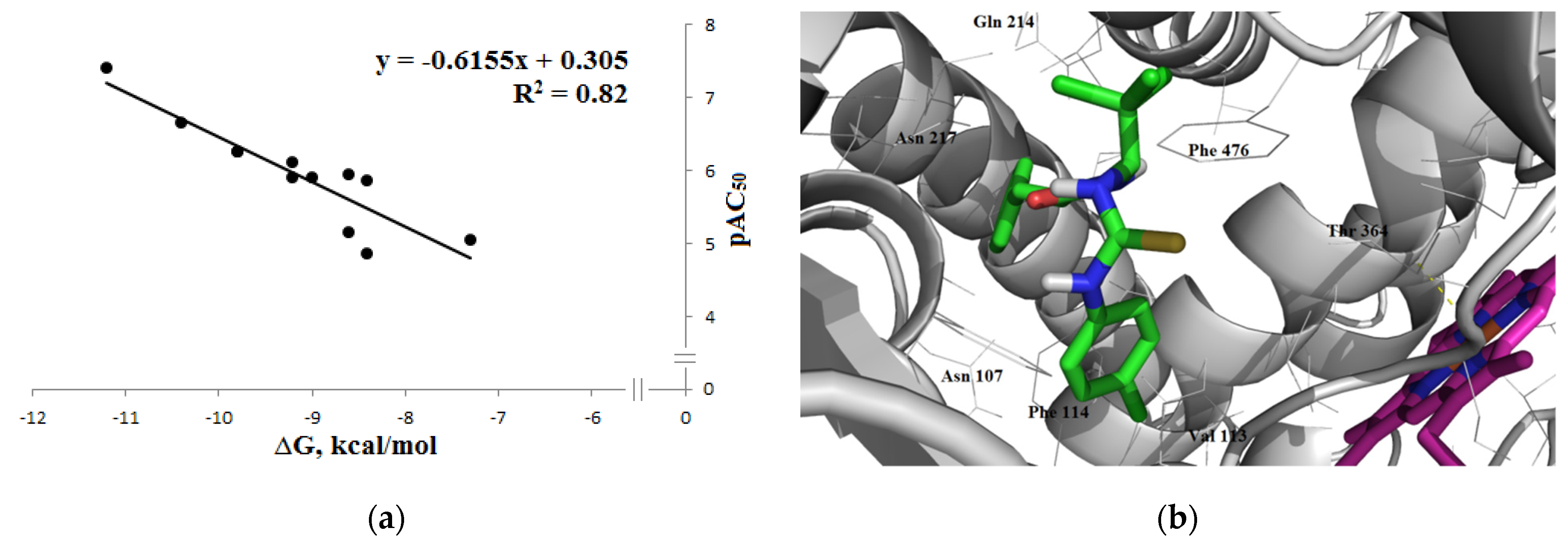
Inhibition of CYP2C9
Inhibition of CYP2C19
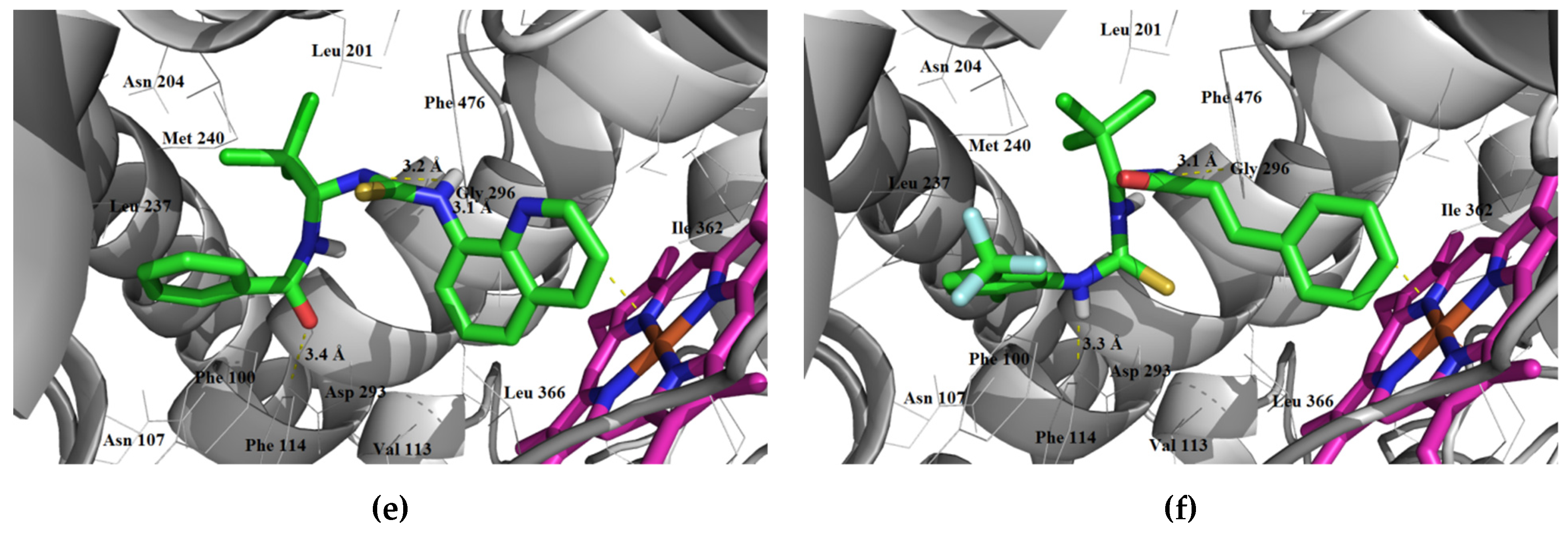
Inhibition of CYP2D6
Inhibition of CYP3A4
Substrate Activity
3.4. Elimination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette; |
| ADME/T | absorption, distribution, metabolism, excretion / toxicity; |
| AGP | alpha-1-acid glycoprotein; |
| ATF4/6 | activating transcription factor 4/6; |
| BBB | blood–brain barrier; |
| BOILED-Egg | Brain Or IntestinaL EstimateD; |
| CNS | central nervous system; |
| CYP | cytochrome P450; |
| eIF2α | eukaryotic translation initiation factor 2α; |
| ER | endoplasmic reticulum; |
| GIT | gastrointestinal tract; |
| HIA | human intestinal absorption; |
| HSA | human serum albumin; |
| MACCS | molecular ACCess system; |
| P-gp | P-glycoprotein; |
| (Q)SAR | (quantitative) structure-activity relationship; |
| RF | random forest; |
| Ro 5 | Lipinski’s rule or rule of 5; |
| SVM | support vector machine; |
| (T)PSA | (topological) polar surface area. |
References
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, V.; Mailankody, S. Research and Development Spending to Bring a Single Cancer Drug to Market and Revenues after Approval. JAMA Intern. Med. 2017, 177, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Khanna, I. Drug discovery in pharmaceutical industry: Productivity challenges and trends. Drug Discov. Today 2012, 17, 1088–10102. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Basavaraj, S.; Betageri, G.V. Can formulation and drug delivery reduce attrition during drug discovery and development—Review of feasibility, benefits and challenges. Acta Pharm. Sin. B 2014, 4, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Shou, W.Z. Current status and future directions of high-throughput ADME screening in drug discovery. J. Pharm. Anal. 2020, 10, 201–208. [Google Scholar] [CrossRef]
- Pellegatti, M. The debate on animal ADME studies in drug development: An update. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1615–1620. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef]
- Aysha, A. The Flaws and Human Harms of Animal Experimentation. Camb. Q. Healthc. Ethics. 2015, 24, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Ferdowsian, H.R.; Beck, N. Ethical and Scientific Considerations Regarding Animal Testing and Research. PLoS ONE 2011, 6, e24059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiriiri, G.K.; Njogu, P.M.; Mwangi, A.N. Exploring different approaches to improve the success of drug discovery and development projects: A review. Future J. Pharm. Sci. 2020, 6, 27. [Google Scholar] [CrossRef]
- Wang, Y.; Xing, J.; Xu, Y.; Zhou, N.; Peng, J.; Xiong, Z.; Liu, X.; Luo, X.; Luo, C.; Chen, K.; et al. In silico ADME/T modelling for rational drug design. Q. Rev. Biophys. 2015, 48, 488–515. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H. Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 573–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef]
- Kar, S.; Leszczynski, J. Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin. Drug Discov. 2020, 15, 1473–1487. [Google Scholar] [CrossRef]
- Jia, C.-Y.; Li, J.-Y.; Hao, G.-F.; Yang, G.-F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today 2020, 25, 248–258. [Google Scholar] [CrossRef]
- Madden, J.C.; Enoch, S.J.; Paini, A.; Cronin, M.T.D. A Review of In Silico Tools as Alternatives to Animal Testing: Principles, Resources and Applications. Altern. Lab. Anim. 2020, 48, 146–172. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Part 2: Potential Alternatives to the Use of Animals in Preclinical Trials. JACC Basic Transl. Sci. 2020, 5, 387–397. [Google Scholar] [CrossRef]
- Matsuoka, M.; Komoike, Y. Experimental Evidence Shows Salubrinal, an eIF2α Dephosphorylation Inhibitor, Reduces Xenotoxicant-Induced Cellular Damage. Int. J. Mol. Sci. 2015, 16, 16275–16287. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, D.C.; Valentão, P.; Andrade, P.B.; Pereira, D.M. Endoplasmic reticulum stress signaling in cancer and neurodegenerative disorders: Tools and strategies to understand its complexity. Pharmacol. Res. 2020, 155, 104702. [Google Scholar] [CrossRef]
- Fu, J.; Tao, T.; Li, Z.; Chen, Y.; Li, J.; Peng, L. The roles of ER stress in epilepsy: Molecular mechanisms and therapeutic implications. Biomed. Pharmacother. 2020, 131, 110658. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Choi, J.A.; Song, C.H. Insights into the Role of Endoplasmic Reticulum Stress in Infectious Diseases. Front. Immunol. 2020, 10, 3147. [Google Scholar] [CrossRef] [Green Version]
- Manalo, R.V.M.; Medina, P.M.B. The endoplasmic reticulum stress response in disease pathogenesis and pathophysiology. Egypt. J. Med. Hum. Genet. 2018, 19, 59–68. [Google Scholar] [CrossRef]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A Selective Inhibitor of eIF2α Dephosphorylation Protects Cells from ER Stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Role of endoplasmic reticulum stress in atherosclerosis and diabetic macrovascular complications. BioMed Res. Int. 2014, 2014, 610140. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Teuber, V.; Albert-Gasco, H.; Auyeung, V.C.; Papa, F.R.; Mallucci, G.R.; Hetz, C. Small Molecules to Improve ER Proteostasis in Disease. Trends Pharmacol. Sci. 2019, 40, 684–695. [Google Scholar] [CrossRef]
- Grandjean, J.M.D.; Wiseman, R.L. Small molecule strategies to harness the unfolded protein response: Where do we go from here? J. Biol. Chem. 2020, 295, 15692–15711. [Google Scholar] [CrossRef] [PubMed]
- Emincik, A.; Gungor, Z.B.; Guzel, E. Effects of salubrinal on ER stress in an experimental model of polycystic ovary syndrome. Ultrastruct. Pathol. 2020, 44, 422–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.F.; Gao, C.; Chen, W.; Gao, Y.; Wang, H.C.; Meng, Y.; Luo, C.-L.; Zhang, M.-Y.; Chen, G.; Chen, X.-P.; et al. Salubrinal offers neuroprotection through suppressing endoplasmic reticulum stress, autophagy and apoptosis in a mouse traumatic brain injury model. Neurobiol. Learn. Mem. 2019, 161, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.; Afjal, M.A.; Akhter, J.; Mangla, A.; Khan, J.; Parvez, S.; Raisuddin, S. Involvement of endoplasmic reticulum stress in amyloid β(1-42)-induced Alzheimer’s like neuropathological process in rat brain. Brain Res. Bull. 2020, 165, 108–117. [Google Scholar] [CrossRef]
- Borreca, A.; Valeri, F.; De Luca, M.; Ernst, L.; Russo, A.; Nobili, A.; Cordella, A.; Corsetti, V.; Amadoro, G.; Mercuri, N.B.; et al. Transient upregulation of translational efficiency in prodromal and early symptomatic Tg2576 mice contributes to Aβ pathology. Neurobiol. Dis. 2020, 139, 104787. [Google Scholar] [CrossRef]
- Anuncibay-Soto, B.; Santos-Galdiano, M.; Fernández-López, A. Neuroprotection by salubrinal treatment in global cerebral ischemia. Neural. Regen. Res. 2016, 11, 1744–1745. [Google Scholar] [CrossRef]
- Romero-Ramírez, L.; Nieto-Sampedro, M.; Barreda-Manso, M.A. Integrated Stress Response as a Therapeutic Target for CNS Injuries. BioMed Res. Int. 2017, 2017, 6953156. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, S.J.; Cao, S.L.; Guo, W.Z.; Yan, B.; Fang, H.B. Protective effects of salubrinal on liver injury in rat models of brain death. Chin. Med. J. 2015, 128, 1523–1528. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Liu, D.; Zhang, S.; Tan, N.; Yokota, H.; Zhang, P. Phosphorylation of eIF2α signaling pathway attenuates obesity-induced non-alcoholic fatty liver disease in an ER stress and autophagy-dependent manner. Cell Death Dis. 2020, 11, 1069. [Google Scholar] [CrossRef]
- Yang, B.; Lu, X.; Li, Y.; Li, Y.; Yu, D.; Zhang, W.; Duan, C.; Taguchi, K.; Yasui, T.; Kohri, K.; et al. A Proteomic Network Approach across the Kidney Stone Disease Reveals Endoplasmic Reticulum Stress and Crystal-Cell Interaction in the Kidney. Oxidative Med. Cell. Longev. 2019, 2019, 9307256. [Google Scholar] [CrossRef]
- Wu, C.T.; Sheu, M.L.; Tsai, K.S.; Chiang, C.K.; Liu, S.H. Salubrinal, an eIF2α dephosphorylation inhibitor, enhances cisplatin-induced oxidative stress and nephrotoxicity in a mouse model. Free Radic. Biol. Med. 2011, 51, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Wu, C.H.; Chiang, C.K. Therapeutic Approaches Targeting Proteostasis in Kidney Disease and Fibrosis. Int. J. Mol. Sci. 2021, 22, 8674. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Sreenivasaiah, P.K.; Cho, C.; Kim, D.H. Salubrinal Alleviates Pressure Overload-Induced Cardiac Hypertrophy by Inhibiting Endoplasmic Reticulum Stress Pathway. Mol. Cells 2017, 40, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, N.; Wu, J.H.; Liang, Z.S.; Jiang, W.H.; Wang, X.W. Role of salubrinal in protecting cardiomyocytes from doxorubicin-induced apoptosis. Genet. Mol. Res. 2015, 14, 12377–12385. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Qi, S.Y.; Ru, L.S.; Ding, C.; Wang, H.J.; Zhao, J.-S.; Li, J.-J.; Li, A.-Y.; Wang, D.-M. Reduced endoplasmic reticulum stress might alter the course of heart failure via caspase-12 and JNK pathways. Can. J. Cardiol. 2014, 30, 368–375. [Google Scholar] [CrossRef]
- Cnop, M.; Toivonen, S.; Igoillo-Esteve, M.; Salpea, P. Endoplasmic reticulum stress and eIF2α phosphorylation: The Achilles heel of pancreatic β cells. Mol. Metab. 2017, 6, 1024–1039. [Google Scholar] [CrossRef]
- Aoi, K.; Nishio, A.; Okazaki, T.; Takeo, M.; Masuda, M.; Fukui, T.; Uchida, K.; Okazaki, K. Inhibition of the dephosphorylation of eukaryotic initiation factor 2α ameliorates murine experimental pancreatitis. Pancreatology 2019, 19, 548–556. [Google Scholar] [CrossRef]
- Takigawa, S.; Frondorf, B.; Liu, S.; Liu, Y.; Li, B.; Sudo, A.; Wallace, J.M.; Yokota, H.; Hamamura, K. Salubrinal improves mechanical properties of the femur in osteogenesis imperfecta mice. J. Pharmacol. Sci. 2016, 132, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, X.; Liu, D.; Hamamura, K.; Wan, Q.; Na, S.; Yokota, H.; Zhang, P. eIF2α signaling regulates autophagy of osteoblasts and the development of osteoclasts in OVX mice. Cell Death Dis. 2019, 10, 921. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, Y.; Li, X.; Li, J.; Yang, S.; Xing, X.; Fan, G.; Yokota, H.; Zhang, P. eIF2α signaling regulates ischemic osteonecrosis through endoplasmic reticulum stress. Sci. Rep. 2017, 7, 5062. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sun, C.; Gu, H.; Wang, H.; Li, S.; Ma, Y.; Wang, J. Salubrinal protects against Clostridium difficile toxin B-induced CT26 cell death. Acta Biochim. Biophys. Sin. 2017, 49, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsterda, A.; Asha, K.; Powrozek, O.; Repak, M.; Goswami, S.; Dunn, A.M.; Memmel, H.C.; Sharma-Walia, N. Salubrinal Exposes Anticancer Properties in Inflammatory Breast Cancer Cells by Manipulating the Endoplasmic Reticulum Stress Pathway. Front. Oncol. 2021, 11, 654940. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liang, C.; Huang, X.; Deng, X.; Jiang, J.; Luo, Z. Salubrinal Regulates the Apoptosis of Adrenocortical Carcinoma Cells via the PERK/eIF2α/ATF4 Signaling Pathway. Int. J. Endocrinol. 2021, 2021, 5038130. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauléon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer, and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, C.; Zhou, H.; Xiao, B.; Cheng, Y.; Wang, J.; Yao, F.; Duan, C.; Chen, R.; Liu, Y.; et al. Synergistic antitumor activity of the combination of salubrinal and rapamycin against human cholangiocarcinoma cells. Oncotarget 2016, 7, 85492–85501. [Google Scholar] [CrossRef] [Green Version]
- Jeon, Y.-J.; Kim, J.H.; Shin, J.-I.; Jeong, M.; Cho, J.; Lee, K. Salubrinal-Mediated Upregulation of eIF2α Phosphorylation Increases Doxorubicin Sensitivity in MCF-7/ADR Cells. Mol. Cells 2016, 39, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Kardos, G.R.; Gowda, R.; Dinavahi, S.S.; Kimball, S.; Robertson, G.P. Salubrinal in Combination with 4E1RCat Synergistically Impairs Melanoma Development by Disrupting the Protein Synthetic Machinery. Front. Oncol. 2020, 10, 834. [Google Scholar] [CrossRef]
- Bastola, P.; Neums, L.; Schoenen, F.J.; Chien, J. VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase-mediated cell death, and synergistically kill ovarian cancer cells in combination with Salubrinal. Mol. Oncol. 2016, 10, 1559–1574. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; He, K.L.; Li, X.; Li, R.J.; Liu, C.L.; Zhong, W.; Li, S. SAR, Cardiac Myocytes Protection Activity and 3D-QSAR Studies of Salubrinal and its Potent Derivatives. Curr. Med. Chem. 2012, 19, 6072–6079. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Gobert, D.; Stern, E.; Gamache, K.; Colina, R.; Cuello, C.; Sossin, W.; Kaufman, R.; Pelletier, J.; Rosenblum, K.; et al. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 2007, 129, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, K.; Boyce, M.; Lin, H.; Yuan, J.; Ma, D. Structure-activity relationship studies of salubrinal lead to its active biotinylated derivative. Bioorg. Med. Chem. Lett. 2005, 15, 3849–3852. [Google Scholar] [CrossRef] [PubMed]
- Zadorozhnii, P.V.; Pokotylo, I.O.; Kiselev, V.V.; Okhtina, O.V.; Kharchenko, A.V. Molecular docking studies of salubrinal and its analogs as inhibitors of the GADD34:PP1 enzyme. ADMET DMPK 2019, 7, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Zadorozhnii, P.V.; Kiselev, V.V.; Kharchenko, A.V. In silico toxicity evaluation of Salubrinal and its analogues. Eur. J. Pharm. Sci. 2020, 155, 105538. [Google Scholar] [CrossRef]
- Barreda-Manso, M.A.; Yanguas-Casás, N.; Nieto-Sampedro, M.; Romero-Ramírez, L. Neuroprotection and Blood-Brain Barrier Restoration by Salubrinal After a Cortical Stab Injury. J. Cell. Physiol. 2017, 232, 1501–1510. [Google Scholar] [CrossRef]
- Anuncibay-Soto, B.; Pérez-Rodríguez, D.; Santos-Galdiano, M.; Font, E.; Regueiro-Purriños, M.; Fernández-López, A. Post-ischemic salubrinal treatment results in a neuroprotective role in global cerebral ischemia. J. Neurochem. 2016, 138, 295–306. [Google Scholar] [CrossRef]
- Anuncibay-Soto, B.; Pérez-Rodriguez, D.; Santos-Galdiano, M.; Font-Belmonte, E.; Ugidos, I.F.; Gonzalez-Rodriguez, P.; Regueiro-Purriños, M.; Fernández-López, A. Salubrinal and robenacoxib treatment after global cerebral ischemia. Exploring the interactions between ER stress and inflammation. Biochem. Pharmacol. 2018, 151, 26–37. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, X.; Xu, Y.; Lu, L.; Yu, S.; Cheng, Q.; Yang, B.; Tsui, C.-K.; Ye, D.; Huang, J.; et al. Salubrinal attenuated retinal neovascularization by inhibiting CHOP-HIF1α-VEGF pathways. Oncotarget 2017, 8, 77219–77232. [Google Scholar] [CrossRef] [Green Version]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Wang, N.N.; Yao, Z.J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.-P.; Cao, D.-S. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Lu, A.-P.; Cao, D.-S. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for evaluating chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics. 1986. Available online: https://www.molinspiration.com/ (accessed on 4 April 2020).
- Tetko, I.V.; Tanchuk, V.Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Banerjee, P.; Dunkel, M.; Kemmler, E.; Preissner, R. SuperCYPsPred—A web server for the prediction of cytochrome activity. Nucleic Acids Res. 2020, 48, W580–W585. [Google Scholar] [CrossRef]
- Montanari, F.; Knasmüller, B.; Kohlbacher, S.; Hillisch, C.; Baierová, C.; Grandits, M.; Ecker, G.F. Vienna LiverTox Workspace—A Set of Machine Learning Models for Prediction of Interactions Profiles of Small Molecules with Transporters Relevant for Regulatory Agencies. Front. Chem. 2020, 7, 899. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.A.; Zerner, M.C. A theoretical examination of the electronic structure and spectroscopy of the photosynthetic reaction center from Rhodopseudomonas viridis. J. Am. Chem. Soc. 1991, 113, 8210–8215. [Google Scholar] [CrossRef]
- Thompson, M.A.; Glendening, E.D.; Feller, D. The Nature of K+/Crown Ether Interactions: A Hybrid Quantum Mechanical-Molecular Mechanical Study. J. Phys. Chem. 1994, 98, 10465–10476. [Google Scholar] [CrossRef]
- Thompson, M.A.; Schenter, G.K. Excited States of the Bacteriochlorophyll b Dimer of Rhodopseudomonas viridis: A QM/MM Study of the Photosynthetic Reaction Center That Includes MM Polarization. J. Phys. Chem. 1995, 99, 6374–6386. [Google Scholar] [CrossRef]
- Thompson, M.A. QM/MMpol: A Consistent Model for Solute/Solvent Polarization. Application to the Aqueous Solvation and Spectroscopy of Formaldehyde, Acetaldehyde, and Acetone. J. Phys. Chem. 1996, 100, 14492–14507. [Google Scholar] [CrossRef]
- Thompson, M. ArgusLab 4.0.1; Planaria Software LLC: Seattle, WA, USA, 2004; Available online: http://www.arguslab.com (accessed on 24 November 2021).
- Thiel, W. Semiempirical quantum-chemical methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 145–157. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: Palo Alto, CA, USA, 2003; Available online: http://www.pymol.org (accessed on 27 August 2015).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Wang, N.-N.; Huang, C.; Dong, J.; Yao, Z.-J.; Zhu, M.-F.; Deng, Z.-K.; Lv, B.; Lu, A.-P.; Chen, A.F.; Cao, D.-S. Predicting human intestinal absorption with modified random forest approach: A comprehensive evaluation of molecular representation, unbalanced data, and applicability domain issues. RSC Adv. 2017, 7, 19007–19018. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.B.O. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of Molecular Lipophilicity: State-of-the-Art and Comparison of Log P Methods on More Than 96,000 Compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef] [PubMed]
- Zakeri-Milani, P.; Valizadeh, H. Intestinal transporters: Enhanced absorption through P-glycoprotein-related drug interactions. Expert Opin. Drug Metab. Toxicol. 2014, 10, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, A.Y.; Wang, R.; Lai, L. Computation of Octanol-Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, I.; Hirono, S.; Nakagome, I.; Hirano, H. Comparison of Reliability of log P Values for Drugs Calculated by Several Methods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple Method of Calculating Octanol/Water Partition Coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Huuskonen, J.J.; Villa, A.E.; Tetko, I.V. Prediction of partition coefficient based on atom-type electrotopological state indices. J. Pharm. Sci. 1999, 88, 229–233. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. What has polar surface area ever done for drug discovery? Future Med. Chem. 2011, 3, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J. Pharm. Sci. 1999, 88, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Rossi Sebastiano, M.; Doak, B.C.; Backlund, M.; Poongavanam, V.; Over, B.; Ermondi, G.; Caron, G.; Matsson, P.; Kihlberg, J. Impact of Dynamically Exposed Polarity on Permeability and Solubility of Chameleonic Drugs Beyond the Rule of 5. J. Med. Chem. 2018, 61, 4189–4202. [Google Scholar] [CrossRef] [PubMed]
- He, S.M.; Li, R.; Kanwar, J.R.; Zhou, S.F. Structural and functional properties of human multidrug resistance protein 1 (MRP1/ABCC1). Curr. Med. Chem. 2011, 18, 439–481. [Google Scholar] [CrossRef]
- Saaby, L.; Brodin, B. A Critical View on In Vitro Analysis of P-glycoprotein (P-gp) Transport Kinetics. J. Pharm. Sci. 2017, 106, 2257–2264. [Google Scholar] [CrossRef] [Green Version]
- Broccatelli, F.; Carosati, E.; Neri, A.; Frosini, M.; Goracci, L.; Oprea, T.I.; Cruciani, G. A novel approach for predicting P-glycoprotein (ABCB1) inhibition using molecular interaction fields. J. Med. Chem. 2011, 54, 1740–1751. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, Y.; Zhao, Q.; Peng, H.; Hou, T. ADME evaluation in drug discovery. 10. Predictions of P-glycoprotein inhibitors using recursive partitioning and naive Bayesian classification techniques. Mol. Pharm. 2011, 8, 889–900. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Sayle, R.A. Comparing structural fingerprints using a literature-based similarity benchmark. J. Cheminform. 2016, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, Y.; Liang, H.; Bender, A.; Glen, R.C.; Yan, A. P-glycoprotein substrate models using support vector machines based on a comprehensive data set. J. Chem. Inf. Model. 2011, 51, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chen, L.; Li, Y.; Tian, S.; Sun, H.; Hou, T. ADMET evaluation in drug discovery. 13. Development of in silico prediction models for P-glycoprotein substrates. Mol. Pharm. 2014, 11, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Szakács, G.; Annereau, J.P.; Lababidi, S.; Shankavaram, U.; Arciello, A.; Bussey, K.J.; Reinhold, W.; Guo, Y.; Kruh, G.D.; Reimers, M.; et al. Predicting drug sensitivity and resistance: Profiling ABC transporter genes in cancer cells. Cancer Cell 2004, 6, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Grandits, M.; Ecker, G.F. Interspecies comparison of putative ligand binding sites of human, rat and mouse P-glycoprotein. Eur. J. Pharm. Sci. 2018, 122, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, J.K.; Huang, C.H.; Benet, L.Z. Volume of Distribution is Unaffected by Metabolic Drug–Drug Interactions. Clin. Pharmacokinet. 2021, 60, 205–222. [Google Scholar] [CrossRef]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Volume of Distribution in Drug Design. J. Med. Chem. 2015, 58, 5691–5698. [Google Scholar] [CrossRef]
- Obach, R.S.; Lombardo, F.; Waters, N.J. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab. Dispos. 2008, 36, 1385–1405. [Google Scholar] [CrossRef] [Green Version]
- Lambrinidis, G.; Vallianatou, T.; Tsantili-Kakoulidou, A. In vitro, in silico and integrated strategies for the estimation of plasma protein binding. A review. Adv. Drug Deliv. Rev. 2015, 86, 27–45. [Google Scholar] [CrossRef]
- Wanat, K. Biological barriers, and the influence of protein binding on the passage of drugs across them. Mol. Biol. Rep. 2020, 47, 3221–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.W.; Sedykh, A.; Zhu, H.; Liu, S.S.; Tropsha, A. The use of pseudo-equilibrium constant affords improved QSAR models of human plasma protein binding. Pharm. Res. 2013, 30, 1790–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, L.M.; Hall, L.H.; Kier, L.B. QSAR modeling of beta-lactam binding to human serum proteins. J. Comput. Aided Mol. Des. 2003, 17, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Zhivkova, Z.; Doytchinova, I. Quantitative structure—Plasma protein binding relationships of acidic drugs. J. Pharm. Sci. 2012, 101, 4627–4641. [Google Scholar] [CrossRef]
- Ghafourian, T.; Amin, Z. QSAR Models for the Prediction of Plasma Protein Binding. Bioimpacts 2013, 3, 21–27. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Sun, L.; Yang, H.; Li, J.; Wang, T.; Li, W.; Liu, G.; Tang, Y. In Silico Prediction of Compounds Binding to Human Plasma Proteins by QSAR Models. ChemMedChem 2018, 13, 572–581. [Google Scholar] [CrossRef]
- Bteich, M. An overview of albumin and alpha-1-acid glycoprotein main characteristics: Highlighting the roles of amino acids in binding kinetics and molecular interactions. Heliyon 2019, 5, e02879. [Google Scholar] [CrossRef] [Green Version]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamasaki, K.; Seo, H.; Otagiri, M. Potential Use of Biological Proteins for Liver Failure Therapy. Pharmaceutics 2015, 7, 255–274. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.X.; Cooper, M.A.; Baker, M.A.; Azad, M.A.; Nation, R.L.; Li, J.; Velkov, T. Drug-binding energetics of human α-1-acid glycoprotein assessed by isothermal titration calorimetry and molecular docking simulations. J. Mol. Recognit. 2012, 25, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Nishi, K.; Ono, T.; Nakamura, T.; Fukunaga, N.; Izumi, M.; Watanabe, H.; Suenaga, A.; Maruyama, T.; Yamagata, Y.; Curry, S.; et al. Structural insights into differences in drug-binding selectivity between two forms of human alpha1-acid glycoprotein genetic variants, the A and F1*S forms. J. Biol. Chem. 2011, 286, 14427–14434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönfeld, D.L.; Ravelli, R.B.; Mueller, U.; Skerra, A. The 1.8-A crystal structure of alpha1-acid glycoprotein (Orosomucoid) solved by UV RIP reveals the broad drug-binding activity of this human plasma lipocalin. J. Mol. Biol. 2008, 384, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-vitro blood-brain barrier models for drug screening and permeation studies: An overview. Drug Des. Dev. Ther. 2019, 13, 3591–3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, R.; Chen, L.; Götz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. 2020, 165–166, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yap, C.W.; Ung, C.Y.; Xue, Y.; Cao, Z.W.; Chen, Y.Z. Effect of selection of molecular descriptors on the prediction of blood-brain barrier penetrating and nonpenetrating agents by statistical learning methods. J. Chem. Inf. Model. 2005, 45, 1376–1384. [Google Scholar] [CrossRef]
- Yan, A.; Liang, H.; Chong, Y.; Nie, X.; Yu, C. In-silico prediction of blood-brain barrier permeability. SAR QSAR Environ. Res. 2013, 24, 61–74. [Google Scholar] [CrossRef]
- Vilar, S.; Chakrabarti, M.; Costanzi, S. Prediction of passive blood-brain partitioning: Straightforward and effective classification models based on in silico derived physicochemical descriptors. J. Mol. Graph. Model. 2010, 28, 899–903. [Google Scholar] [CrossRef] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Beck, T.C.; Beck, K.R.; Morningstar, J.; Benjamin, M.M.; Norris, R.A. Descriptors of Cytochrome Inhibitors and Useful Machine Learning Based Methods for the Design of Safer Drugs. Pharmaceuticals 2021, 14, 472. [Google Scholar] [CrossRef]
- Rostkowski, M.; Spjuth, O.; Rydberg, P. WhichCyp: Prediction of cytochromes P450 inhibition. Bioinformatics 2013, 29, 2051–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veith, H.; Southall, N.; Huang, R.; James, T.; Fayne, D.; Artemenko, N.; Shen, M.; Inglese, J.; Austin, C.P.; Lloyd, D.G.; et al. Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat. Biotechnol. 2009, 27, 1050–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Yu, Y.; Shen, J.; Yang, L.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. Classification of cytochrome P450 inhibitors and noninhibitors using combined classifiers. J. Chem. Inf. Model. 2011, 51, 996–1011. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Da Silveira, C.H.; Pires, D.E.; Minardi, R.C.; Ribeiro, C.; Veloso, C.J.; Lopes, J.C.; Meira, W.; Neshich, G.; Ramos, C.H.I.; Habesch, R.; et al. Protein cutoff scanning: A comparative analysis of cutoff dependent and cutoff free methods for prospecting contacts in proteins. Proteins 2009, 74, 727–743. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bryant, S.H.; Cheng, T.; Wang, J.; Gindulyte, A.; Shoemaker, B.A.; Thiessen, P.A.; He, S.; Zhang, J. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2017, 45, D955–D963. [Google Scholar] [CrossRef]
- Preissner, S.; Kroll, K.; Dunkel, M.; Senger, C.; Goldsobel, G.; Kuzman, D.; Guenther, S.; Winnenburg, R.; Schroeder, M.; Preissner, R. SuperCYP: A comprehensive database on Cytochrome P450 enzymes including a tool for analysis of CYP-drug interactions. Nucleic Acids Res. 2010, 38, D237–D243. [Google Scholar] [CrossRef]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Matak Vinković, D.; Jhoti, H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, 424, 464–468. [Google Scholar] [CrossRef]
- Reynald, R.L.; Sansen, S.; Stout, C.D.; Johnson, E.F. Structural characterization of human cytochrome P450 2C19: Active site differences between P450s 2C8, 2C9, and 2C19. J. Biol. Chem. 2012, 287, 44581–44591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Savas, U.; Hsu, M.H.; Stout, C.D.; Johnson, E.F. Crystal structure of human cytochrome P450 2D6 with prinomastat bound. J. Biol. Chem. 2012, 287, 10834–10843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, P.A.; Cosme, J.; Vinkovic, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Carbon-Mangels, M.; Hutter, M.C. Selecting relevant descriptors for classification by bayesian estimates: A comparison with decision trees and support vector machines approaches for disparate data sets. Mol. Inf. 2011, 30, 885–895. [Google Scholar] [CrossRef]
- Zaretzki, J.; Matlock, M.; Swamidass, S.J. XenoSite: Accurately predicting CYP-mediated sites of metabolism with neural networks. J. Chem. Inform. Model. 2013, 53, 3373–3383. [Google Scholar] [CrossRef]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Clearance in Drug Design. J. Med. Chem. 2019, 62, 2245–2255. [Google Scholar] [CrossRef]
- Jansen, K.; Pou Casellas, C.; Groenink, L.; Wever, K.E.; Masereeuw, R. Humans are animals, but are animals human enough? A systematic review and meta-analysis on interspecies differences in renal drug clearance. Drug Discov. Today 2020, 25, 706–717. [Google Scholar] [CrossRef]
- Camenisch, G.; Riede, J.; Kunze, A.; Huwyler, J.; Poller, B.; Umehara, K. The extended clearance model and its use for the interpretation of hepatobiliary elimination data. ADMET DMPK 2015, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yap, C.W.; Li, Z.R.; Chen, Y.Z. Quantitative structure-pharmacokinetic relationships for drug clearance by using statistical learning methods. J. Mol. Graph. Model. 2006, 24, 383–395. [Google Scholar] [CrossRef]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Relevance of Half-Life in Drug Design. J. Med. Chem. 2018, 61, 4273–4282. [Google Scholar] [CrossRef]
- Hua, S. Advances in Oral Drug Delivery for Regional Targeting in the Gastrointestinal Tract—Influence of Physiological, Pathophysiological and Pharmaceutical Factors. Front. Pharmacol. 2020, 11, 524. [Google Scholar] [CrossRef]
- Masuri, S.; Cadoni, E.; Cabiddu, M.G.; Isaia, F.; Demuru, M.G.; Moráň, L.; Buček, D.; Vaňhara, P.; Havel, J.; Pivetta, T. The first copper (II) complex with 1,10-phenanthroline and salubrinal with interesting biochemical properties. Metallomics 2020, 12, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, O.; Efferth, T. A Machine Learning-Based Prediction Platform for P-Glycoprotein Modulators and Its Validation by Molecular Docking. Cells 2019, 8, 1286. [Google Scholar] [CrossRef] [Green Version]
- Poirier, A.; Cascais, A.C.; Bader, U.; Portmann, R.; Brun, M.E.; Walter, I.; Hillebrecht, A.; Ullah, M.; Funk, C. Calibration of in vitro multidrug resistance protein 1 substrate and inhibition assays as a basis to support the prediction of clinically relevant interactions in vivo. Drug Metab. Dispos. 2014, 42, 1411–1422. [Google Scholar] [CrossRef]
- Tian, Q.; Zhang, J.; Chan, S.Y.; Tan, T.M.; Duan, W.; Huang, M.; Zhu, Y.; Chan, E.; Yu, Q.; Nie, Y.; et al. Topotecan is a substrate for multidrug resistance associated protein 4. Curr. Drug Metab. 2006, 7, 105–118. [Google Scholar] [CrossRef]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; Barbosa, D.J.; Palmeira, A.; Sousa, E.; Carvalho, F.; Bastos, M.D.L.; Remião, F. Colchicine effect on P-glycoprotein expression and activity: In silico and in vitro studies. Chem. Biol. Interact. 2014, 218, 50–62. [Google Scholar] [CrossRef]
- Nanayakkara, A.K.; Vogel, P.D.; Wise, J.G. Prolonged inhibition of P-glycoprotein after exposure to chemotherapeutics increases cell mortality in multidrug resistant cultured cancer cells. PLoS ONE 2019, 14, e0217940. [Google Scholar] [CrossRef]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.A.; Waters, N.J. Pharmacokinetic and Pharmacodynamic Considerations for Drugs Binding to Alpha-1-Acid Glycoprotein. Pharm. Res. 2019, 36, 30. [Google Scholar] [CrossRef]
- Otagiri, M.; Maruyama, T.; Imai, T.; Suenaga, A.; Imamura, Y. A comparative study of the interaction of warfarin with human α1-acid glycoprotein and human albumin. J. Pharm. Pharmacol. 1987, 39, 416–420. [Google Scholar] [CrossRef]
- Petitpas, I.; Bhattacharya, A.A.; Twine, S.; East, M.; Curry, S. Crystal structure analysis of warfarin binding to human serum albumin: Anatomy of drug site I. J. Biol. Chem. 2001, 276, 22804–22809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senol, N.; Oguzoglu, A.S.; Erzurumlu, Y.; Ascı, H.; Savran, M.; Gulle, K.; Ilhan, I.; Sadef, M.; Hasseyid, N.; Goksel, H.M. Modulation of Salubrinal-mediated Endoplasmic Reticulum Stress in Experimental Subarachnoid Hemorrhage Model. World Neurosurg. 2021, 153, e488–e496. [Google Scholar] [CrossRef] [PubMed]
- Font-Belmonte, E.; Ugidos, I.F.; Santos-Galdiano, M.; González-Rodríguez, P.; Anuncibay-Soto, B.; Pérez-Rodríguez, D.; Gonzalo-Orden, J.M.; Fernández-López, A. Post-ischemic salubrinal administration reduces necroptosis in a rat model of global cerebral ischemia. J. Neurochem. 2019, 151, 777–794. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. Phospho-eIF2α level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS ONE 2010, 5, e12974. [Google Scholar] [CrossRef] [Green Version]
- Shaik, A.N. Changing trends in use of hepatocytes and microsomes for evaluating metabolism studies in drug discovery. ADMET DMPK 2016, 4, 60–61. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Fukuyoshi, S.; Kato, K.; Hiratsuka, M.; Yamaotsu, N.; Hirono, S.; Gouda, H.; Oda, A. Investigation of substrate recognition for cytochrome P450 1A2 mediated by water molecules using docking and molecular dynamics simulations. J. Mol. Graph. Model. 2017, 74, 326–336. [Google Scholar] [CrossRef]
- Dutkiewicz, Z.; Mikstacka, R. Structure-Based Drug Design for Cytochrome P450 Family 1 Inhibitors. Bioinorg. Chem. Appl. 2018, 2018, 3924608. [Google Scholar] [CrossRef]
- Wang, L.; Hai, Y.; Huang, N.; Gao, X.; Liu, W.; He, X. Human cytochrome P450 enzyme inhibition profile of three flavonoids isolated from Psoralea corylifolia: In silico predictions and experimental validation. New J. Chem. 2018, 42, 10922–10934. [Google Scholar] [CrossRef]
- Yim, S.K.; Kim, K.; Chun, S.; Oh, T.; Jung, W.; Jung, K.; Yun, C.-H. Screening of Human CYP1A2 and CYP3A4 Inhibitors from Seaweed In Silico and In Vitro. Mar. Drugs 2020, 18, 603. [Google Scholar] [CrossRef]
- Kucwaj-Brysz, K.; Latacz, G.; Podlewska, S.; Żesławska, E.; Handzlik, J.; Lubelska, A.; Satała, G.; Nitek, W.; Handzlik, J. The relationship between stereochemical and both, pharmacological and ADME-Tox, properties of the potent hydantoin 5-HT7R antagonist MF-8. Bioorg. Chem. 2021, 106, 104466. [Google Scholar] [CrossRef]
- Hung, T.C.; Kuo, C.C.; Chen, C.Y. In Silico Investigation of Cytochrome P450 2C9 in relation to Aging Using Traditional Chinese Medicine. Evid. Based. Complement. Alternat. Med. 2014, 2014, 404505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, G.S.; Srisailam, K.; Sashidhar, R.B. Metabolic inhibition of meloxicam by specific CYP2C9 inhibitors in Cunninghamella blakesleeana NCIM 687: In silico and in vitro studies. SpringerPlus 2016, 5, 166. [Google Scholar] [CrossRef] [Green Version]
- Salminen, K.A.; Rahnasto-Rilla, M.; Väänänen, R.; Imming, P.; Meyer, A.; Horling, A.; Poso, A.; Laitinen, T.; Raunio, H.; Lahtela-Kakkonen, M. Time-Dependent Inhibition of CYP2C19 by Isoquinoline Alkaloids: In Vitro and In Silico Analysis. Drug Metab. Dispos. 2015, 43, 1891–1904. [Google Scholar] [CrossRef] [Green Version]
- Špičáková, A.; Kraus, P.; Gucký, T.; Kryštof, V.; Strnad, M.; Bazgier, V.; Otyepka, M.; Kubíčková, V.; Poruba, M.; Racova, Z.; et al. In vitro and in silico studies of interaction of synthetic 2,6,9-trisubstituted purine kinase inhibitors BPA-302, BP-21 and BP-117 with liver drug-metabolizing cytochromes P450. Physiol. Res. 2020, 69, S627–S636. [Google Scholar] [CrossRef]
- Li, J.; Du, H.; Wu, Z.; Su, H.; Liu, G.; Tang, Y.; Li, W. Interactions of omeprazole-based analogues with cytochrome P450 2C19: A computational study. Mol. BioSyst. 2016, 12, 1913–1921. [Google Scholar] [CrossRef]
- Li, J.; Cai, J.; Su, H.; Du, H.; Zhang, J.; Ding, S.; Liu, G.; Tang, Y.; Li, W. Effects of protein flexibility and active site water molecules on the prediction of sites of metabolism for cytochrome P450 2C19 substrates. Mol. BioSyst. 2016, 12, 868–878. [Google Scholar] [CrossRef]
- Don, C.G.; Smieško, M. Out-compute drug side effects: Focus on cytochrome P450 2D6 modeling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1366. [Google Scholar] [CrossRef]
- Don, C.G.; Smieško, M. In Silico Pharmacogenetics CYP2D6 Study Focused on the Pharmacovigilance of Herbal Antidepressants. Front. Pharmacol. 2020, 11, 683. [Google Scholar] [CrossRef]
- Martiny, V.Y.; Carbonell, P.; Chevillard, F.; Moroy, G.; Nicot, A.B.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Integrated structure- and ligand-based in silico approach to predict inhibition of cytochrome P450 2D6. Bioinformatics 2015, 31, 3930–3937. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Wang, Y.; Or, P.M.; Wan, D.C.; Kwan, Y.W.; Yeung, J.H. Molecular docking and enzyme kinetic studies of dihydrotanshinone on metabolism of a model CYP2D6 probe substrate in human liver microsomes. Phytomedicine 2012, 19, 648–657. [Google Scholar] [CrossRef]
- Pang, X.; Zhang, B.; Mu, G.; Xia, J.; Xiang, Q.; Zhao, X.; Liu, A.; Du, G.; Cui, Y. Screening of cytochrome P450 3A4 inhibitors via in silico and in vitro approaches. RSC Adv. 2018, 8, 34783–34792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiani, Y.S.; Ranaghan, K.E.; Jabeen, I.; Mulholland, A.J. Molecular Dynamics Simulation Framework to Probe the Binding Hypothesis of CYP3A4 Inhibitors. Int. J. Mol. Sci. 2019, 20, 4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, E.C.; Chea, S.; Balupuri, A.; Kang, N.S.; Chin, Y.W.; Choi, Y.H. Enzyme Kinetics and Molecular Docking Studies on Cytochrome 2B6, 2C19, 2E1, and 3A4 Activities by Sauchinone. Molecules 2018, 23, 555. [Google Scholar] [CrossRef] [Green Version]
- Bibi, Z. Role of cytochrome P450 in drug interactions. Nutr. Metab. 2008, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisaka, A.; Ohno, Y.; Yamamoto, T.; Suzuki, H. Prediction of pharmacokinetic drug-drug interaction caused by changes in cytochrome P450 activity using in vivo information. Pharmacol. Ther. 2010, 125, 230–248. [Google Scholar] [CrossRef]
- Goud, T.; Maddi, S.; Devanna, N.; Prasad, T.R. Evaluationof pharmacokinetic and pharmacodynamic interaction between repaglinide and atazanavir in healthy, diabetic and hepatic impaired rats: Possible inhibition of CYP3A, OATP, and P-glycoprotein transporters. ADMET DMPK 2016, 4, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Maideen, N.M.P. Drug interactions of non-dihydropyridine calcium channel blockers involving CYP3A enzymes and P-gp transporter protein. Biointerface Res. Appl. Chem. 2020, 10, 6026–6032. [Google Scholar] [CrossRef]

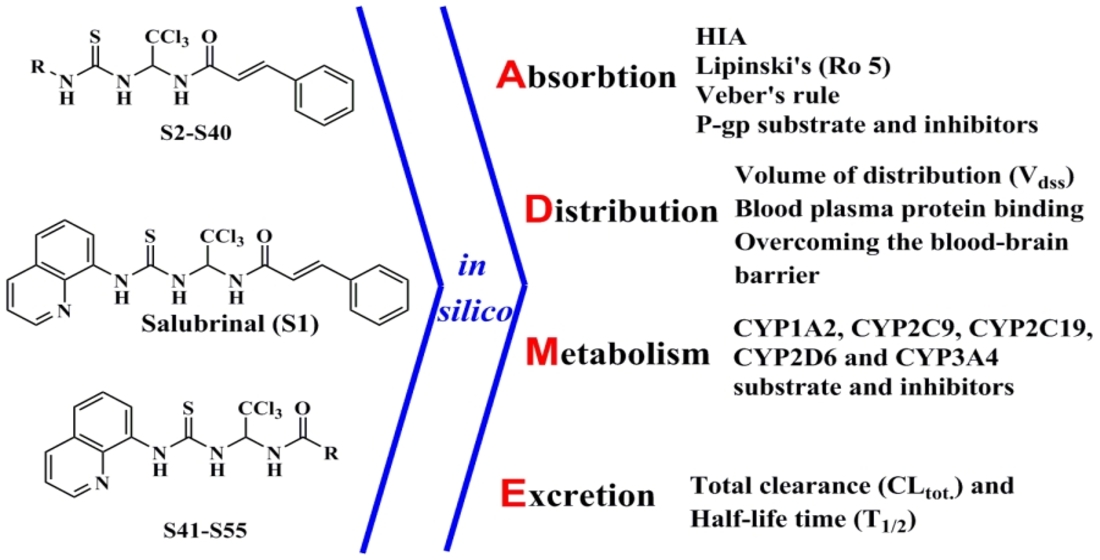{kind=link}
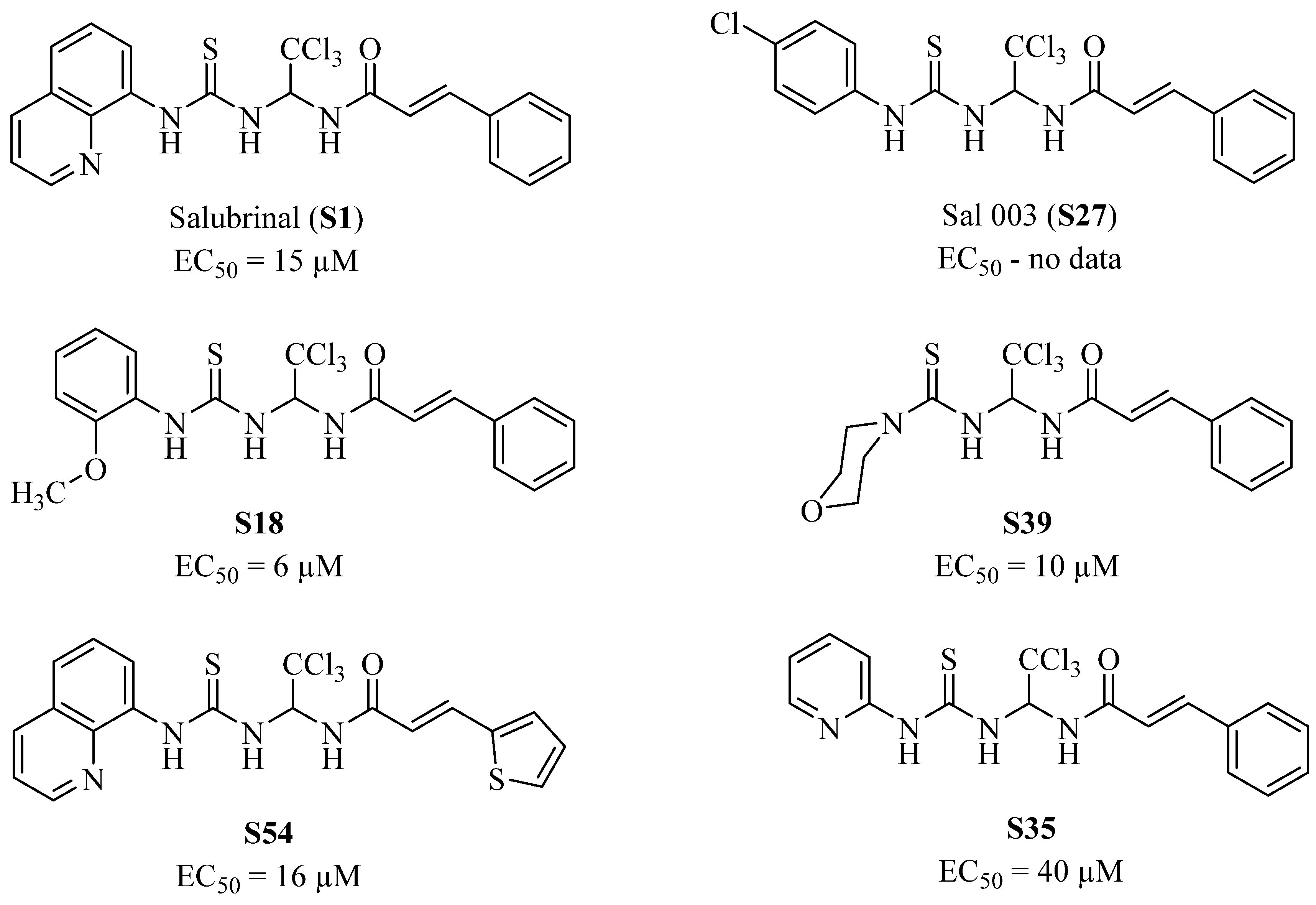{kind=link}
{kind=link}
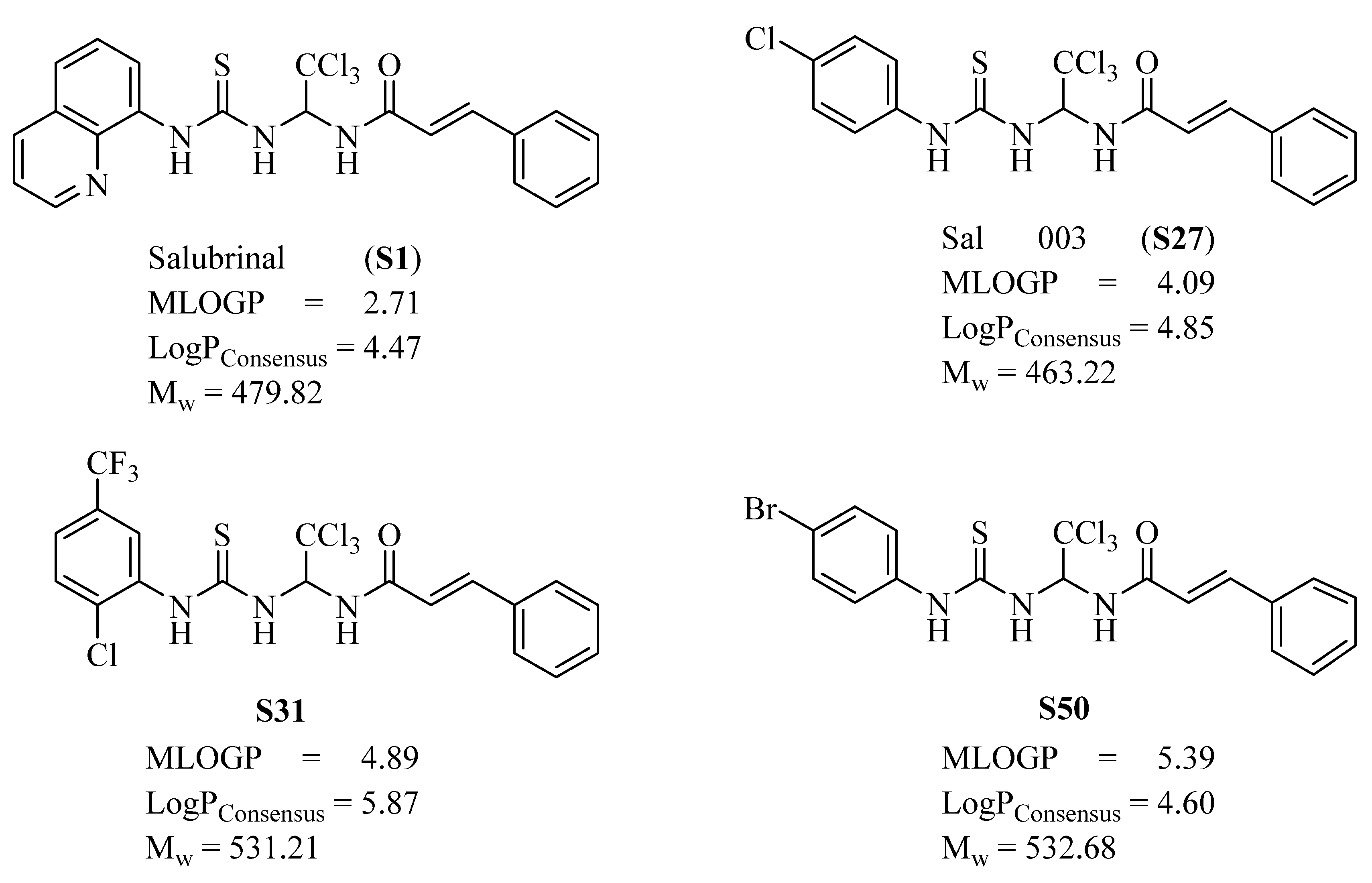{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
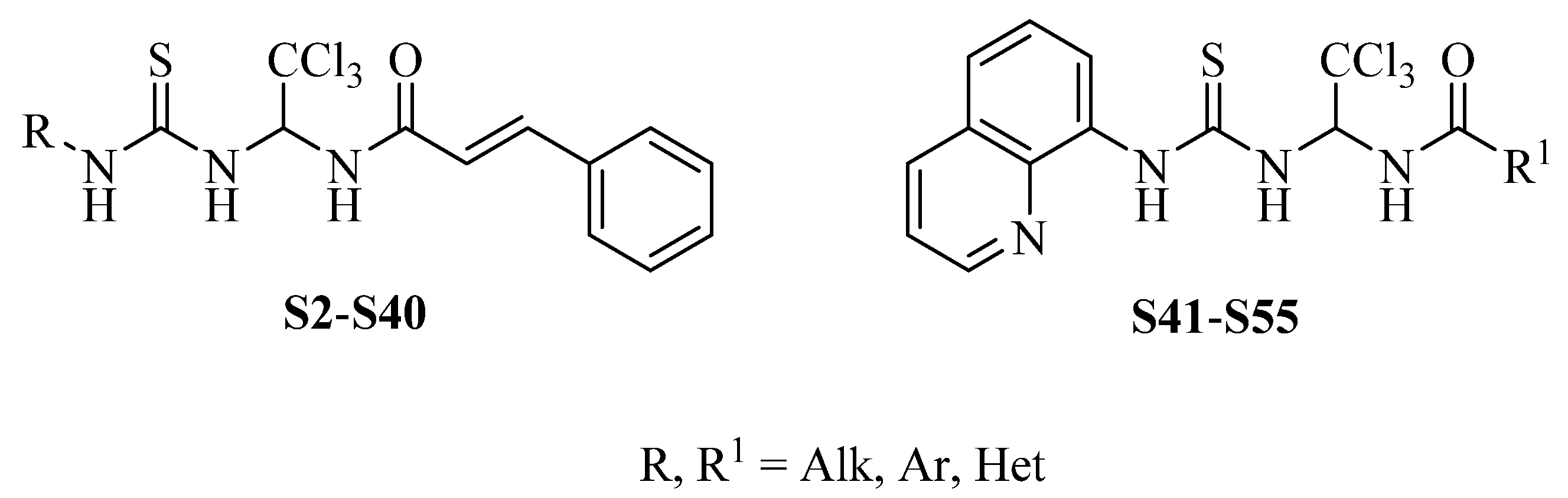





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zadorozhnii, P.V.; Kiselev, V.V.; Kharchenko, A.V. In Silico ADME Profiling of Salubrinal and Its Analogues. Future Pharmacol. 2022, 2, 160-197. https://doi.org/10.3390/futurepharmacol2020013
Zadorozhnii PV, Kiselev VV, Kharchenko AV. In Silico ADME Profiling of Salubrinal and Its Analogues. Future Pharmacology. 2022; 2(2):160-197. https://doi.org/10.3390/futurepharmacol2020013
Chicago/Turabian StyleZadorozhnii, Pavlo V., Vadym V. Kiselev, and Aleksandr V. Kharchenko. 2022. "In Silico ADME Profiling of Salubrinal and Its Analogues" Future Pharmacology 2, no. 2: 160-197. https://doi.org/10.3390/futurepharmacol2020013
APA StyleZadorozhnii, P. V., Kiselev, V. V., & Kharchenko, A. V. (2022). In Silico ADME Profiling of Salubrinal and Its Analogues. Future Pharmacology, 2(2), 160-197. https://doi.org/10.3390/futurepharmacol2020013