
Virtual Screening of Adenylate Kinase 3 Inhibitors Employing Pharmacophoric Model, Molecular Docking, and Molecular Dynamics Simulations as Potential Therapeutic Target in Chronic Lymphocytic Leukemia

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of the Structure-Based Pharmacophore Model
2.2. Pharmacophore-Based Virtual Screening of Compound Libraries
2.3. Molecular Docking Simulations
2.4. In Silico ADMET Prediction
2.5. Molecular Dynamics Simulations
2.6. Free-Energy Calculations
2.7. Free-Energy Decomposition
3. Results and Discussion
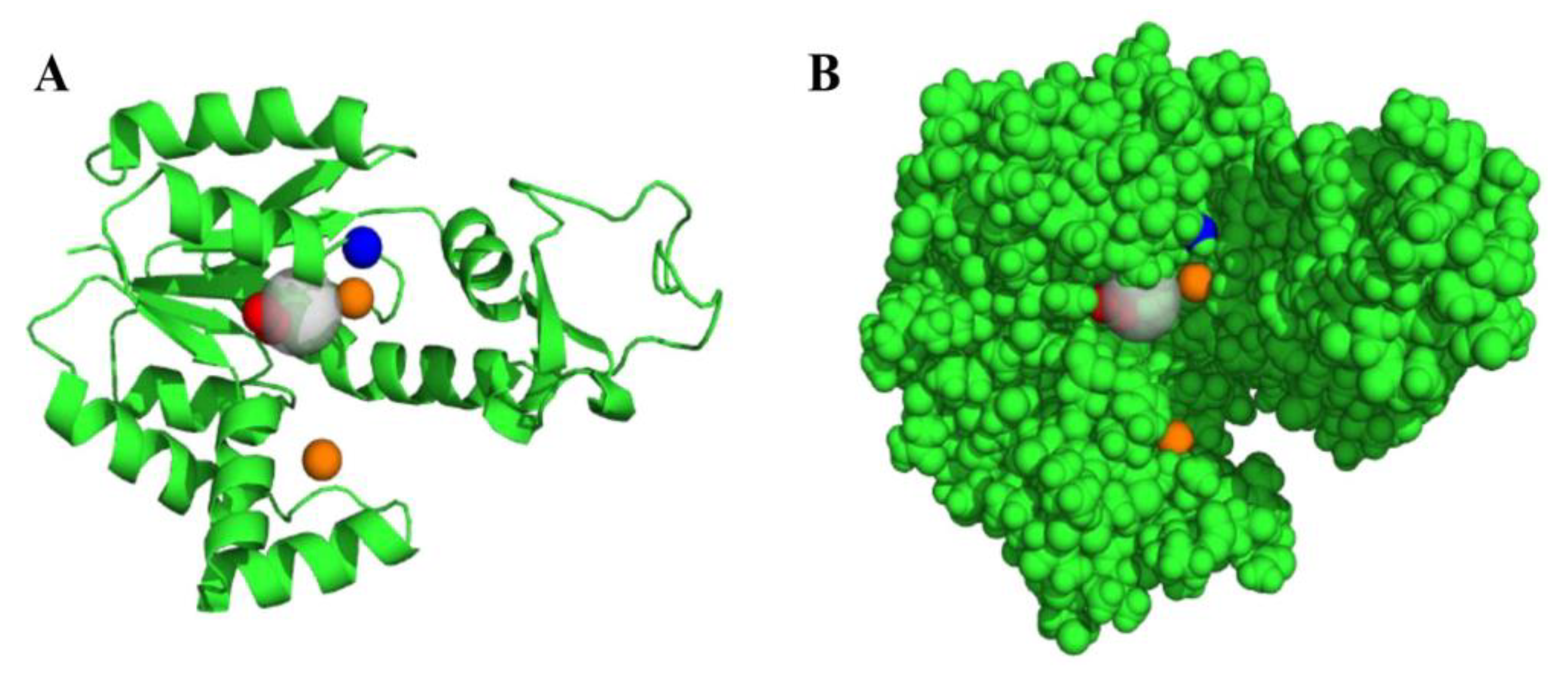
3.1. Mapping of AK3 Binding Sites and the Generation of a Pharmacophoric Model
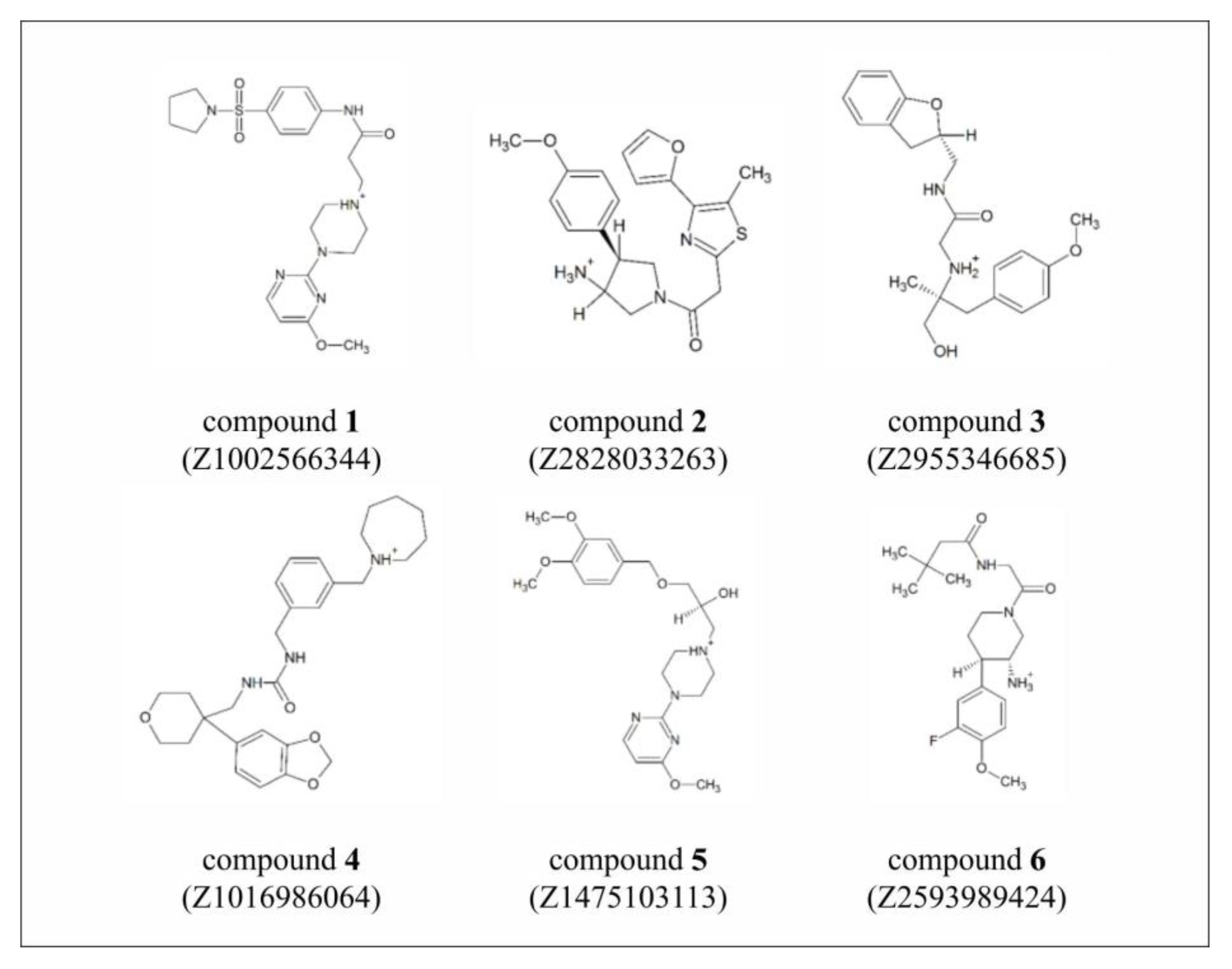
3.2. Virtual Screening
3.3. Pharmacokinetic and Toxicity Predictions
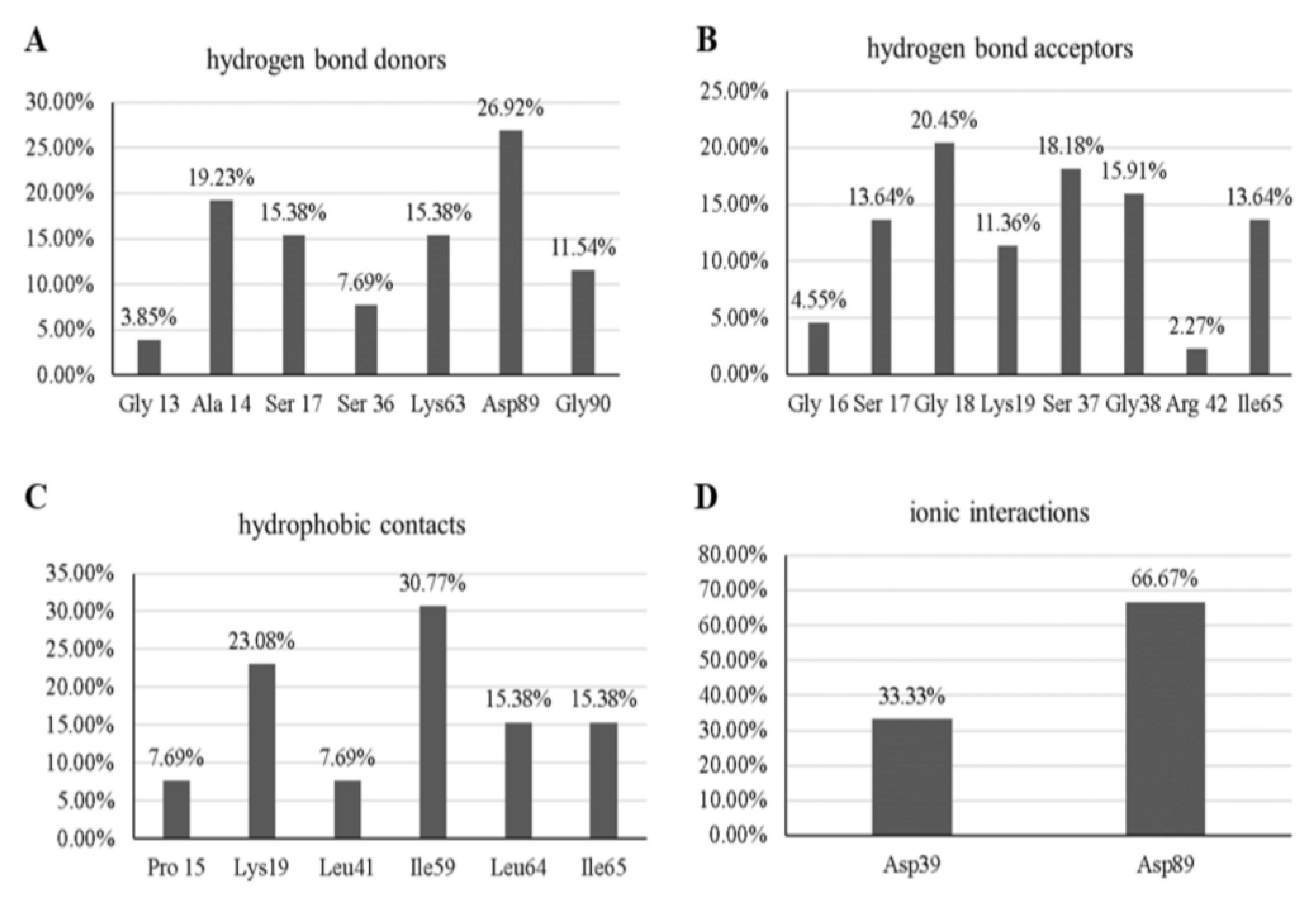
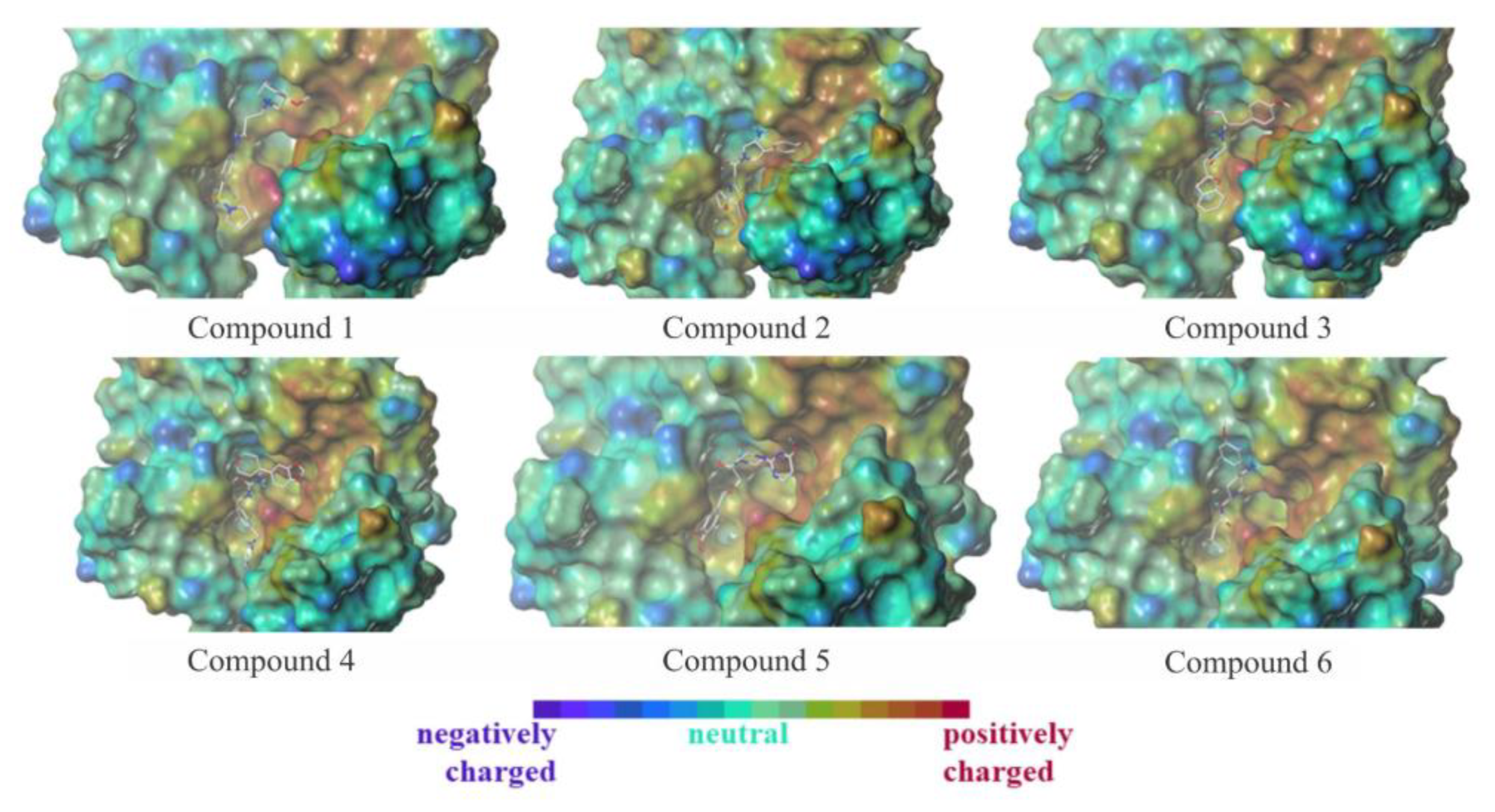
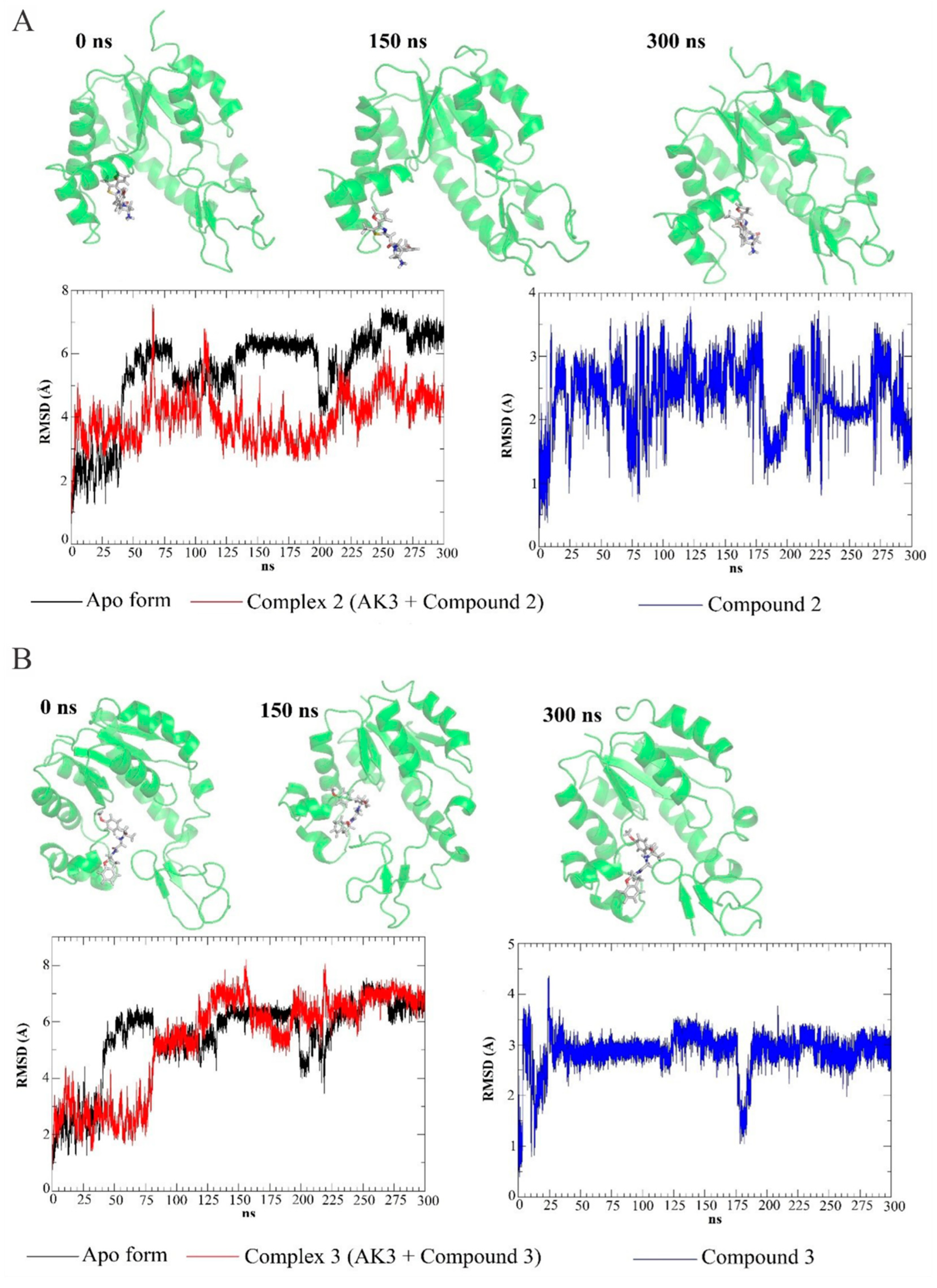
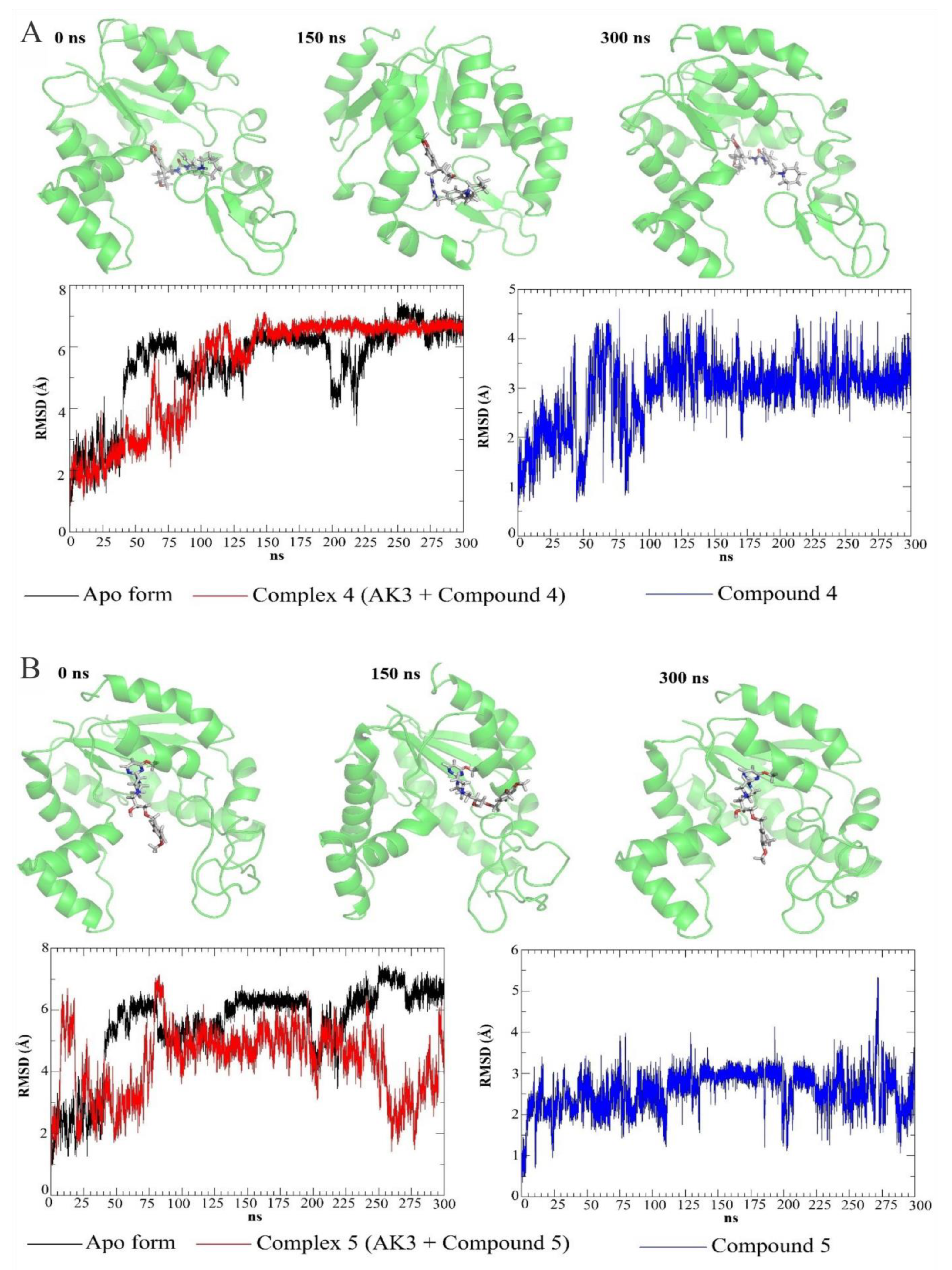
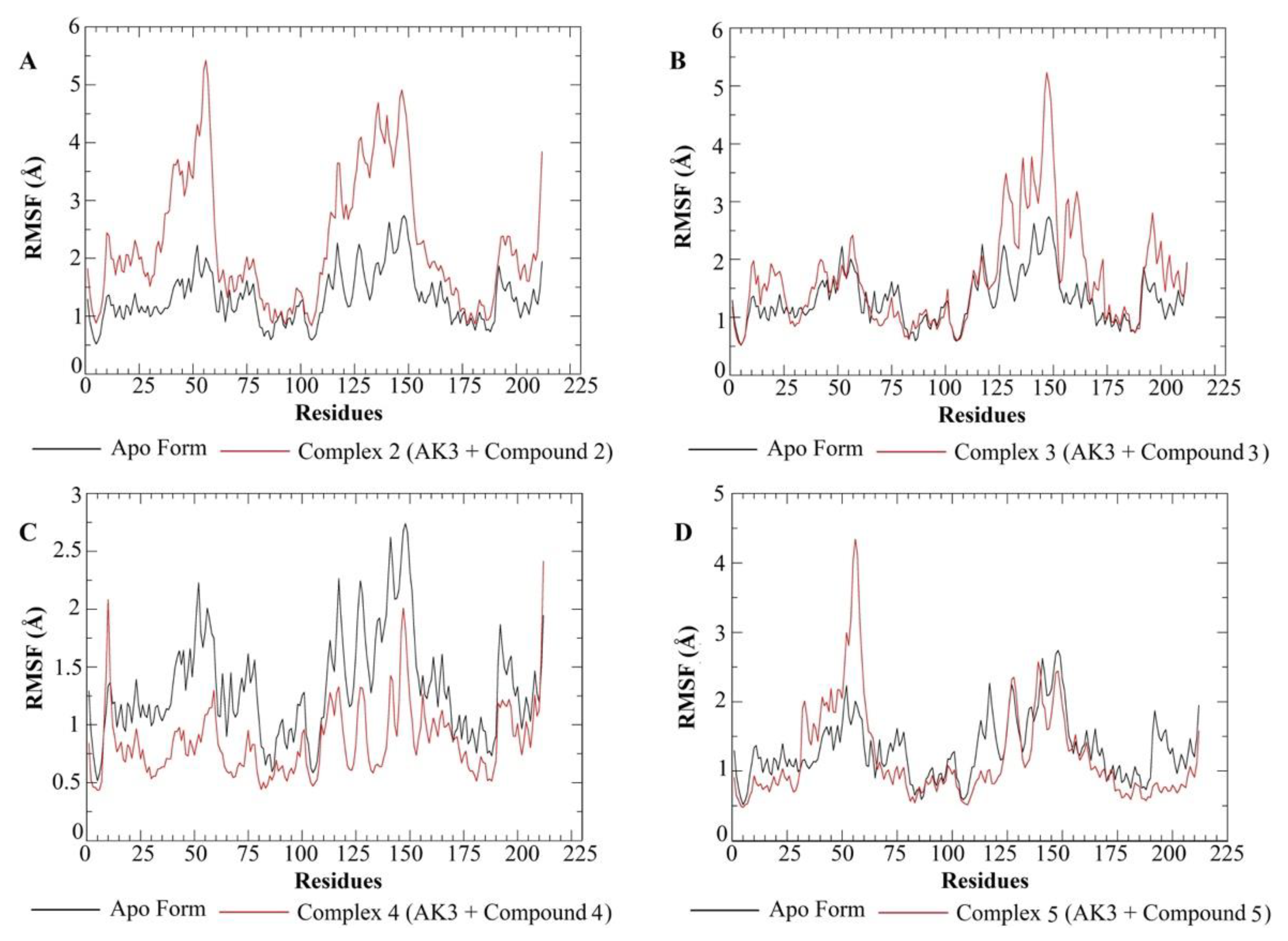
3.4. Molecular Dynamics Simulations
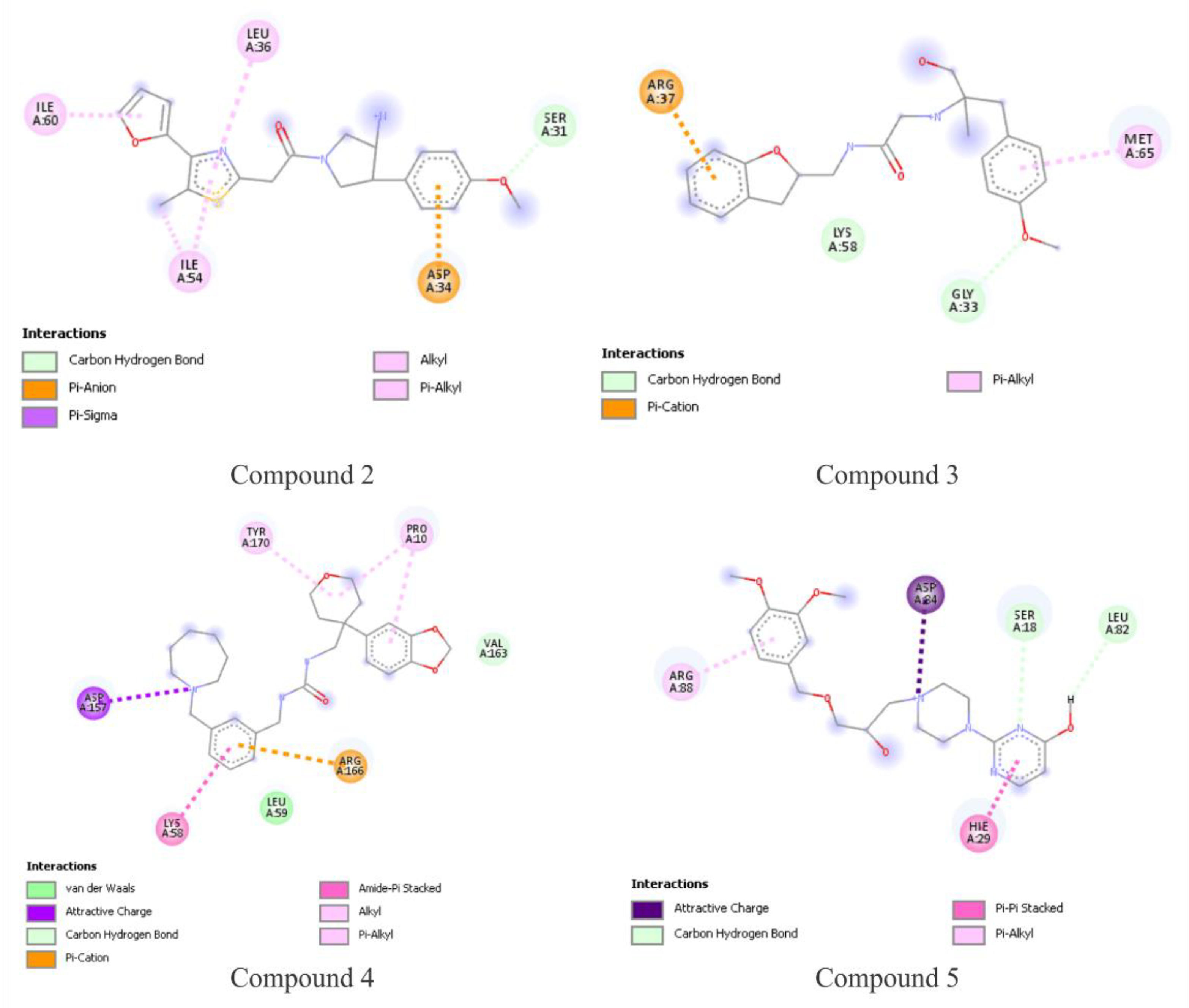
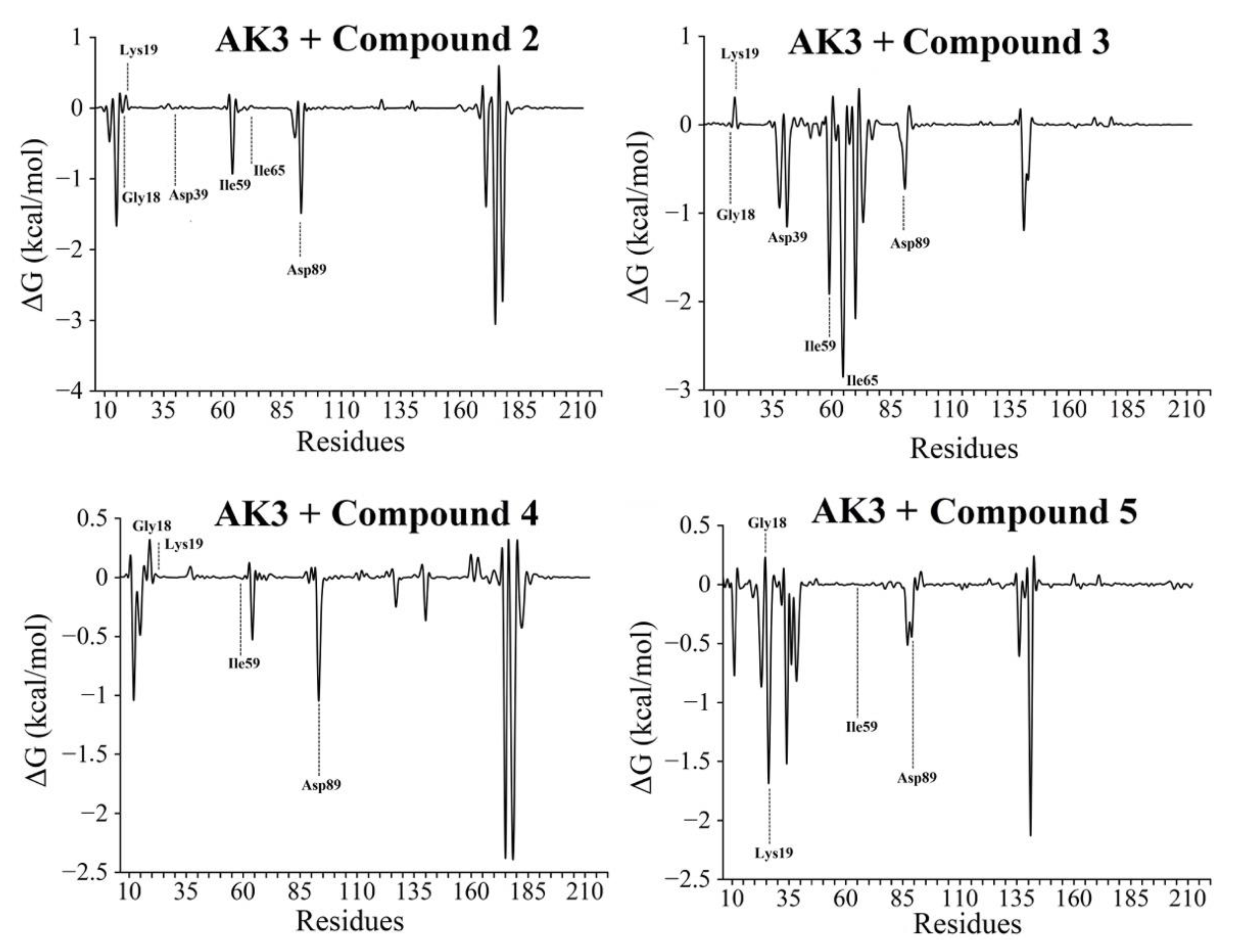
3.5. Free-Energy Calculations
4. Limitations Section
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Noma, T. Dynamics of nucleotide metabolism as a supporter of life phenomena. J. Med. Investig. 2005, 52, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Panayiotou, C.; Solaroli, N.; Karlsson, A. The many isoforms of human adenylate kinases. Int. J. Biochem. Cell Biol. 2014, 49, 75–83. [Google Scholar] [CrossRef]
- Noma, T.; Fujisawa, K.; Yamashiro, Y.; Shinohara, M.; Nakazawa, A.; Gondo, T.; Ishihara, T.; Yoshinobu, K. Structure and expression of human mitochondrial adenylate kinase targeted to the mitochondrial matrix. Biochem. J. 2001, 358, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Admoni-Elisha, L.; Nakdimon, I.; Shteinfer, A.; Prezma, T.; Arif, T.; Arbel, N.; Melkov, A.; Zelichov, O.; Levi, I.; Shoshan-Barmatz, V. Novel Biomarker Proteins in Chronic Lymphocytic Leukemia: Impact on Diagnosis, Prognosis and Treatment. PLoS ONE 2016, 11, e0148500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heraly, B.; Morrison, V.A. How I treat chronic lymphocytic leukemia in older patients. J. Geriatr. Oncol. 2015, 6, 333–340. [Google Scholar] [CrossRef]
- Sharma, S.; Rai, K.R. Chronic lymphocytic leukemia (CLL) treatment: So many choices, such great options. Cancer 2019, 125, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M.; Fischer, K.; Fingerle-Rowson, G.; Fink, A.; Busch, R.; Mayer, J.; Hensel, M.; Hopfinger, G.; Hess, G.; von Grünhagen, U.; et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: A randomised, open-label, phase 3 trial. Lancet 2010, 376, 1164–1174. [Google Scholar] [CrossRef]
- Tam, C.S.; O’Brien, S.; Wierda, W.; Kantarjian, H.; Wen, S.; Do, K.-A.; Thomas, D.A.; Cortes, J.; Lerner, S.; Keating, M.J. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008, 112, 975–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, C.; Assouline, S.; Kuruvilla, J.; Uchida, C.; Bellingham, C.; Sehn, L. Novel Therapies for Chronic Lymphocytic Leukemia: A Canadian Perspective. Clin. Lymphoma Myeloma Leuk. 2015, 15, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Arockianathan, P.M.; Rajalakshmi, K.; Nagappan, P. Proximate composition, phytochemicals, minerals and antioxidant activities of Vigna mungo L. seed coat. Bioinformation 2019, 15, 579–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prada-Gracia, D.; Huerta-Yépez, S.; Moreno-Vargas, L.M. Application of computational methods for anticancer drug discovery, design, and optimization. Boletín Médico Del Hosp. Infant. De México (Engl. Ed.) 2016, 73, 411–423. [Google Scholar] [CrossRef]
- Xu, G.; Chen, Y.; Shen, K.; Wang, X.; Li, F.; He, Y. The Discovery of Potentially Selective Human Neuronal Nitric Oxide Synthase (nNOS) Inhibitors: A Combination of Pharmacophore Modelling, CoMFA, Virtual Screening and Molecular Docking Studies. Int. J. Mol. Sci. 2014, 15, 8553–8569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peach, M.L.; Nicklaus, M.C. Combining docking with pharmacophore filtering for improved virtual screening. J. Chemin 2009, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Sun, H.; Li, Y.; Pan, P.; Li, D.; Hou, T. Development and Evaluation of an Integrated Virtual Screening Strategy by Combining Molecular Docking and Pharmacophore Searching Based on Multiple Protein Structures. J. Chem. Inf. Model. 2013, 53, 2743–2756. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- RCSB PDB—1ZD8, Structure of Human Adenylate Kinase 3 Like 1. Available online: https://www.rcsb.org/structure/1ZD8 (accessed on 27 June 2021).
- Ngan, C.-H.; Hall, D.R.; Zerbe, B.S.; Grove, L.E.; Kozakov, D.; Vajda, S. FTSite: High accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics 2011, 28, 286–287. [Google Scholar] [CrossRef]
- Ngan, C.H.; Bohnuud, T.; Mottarella, S.E.; Beglov, D.; Villar, E.A.; Hall, D.R.; Kozakov, D.; Vajda, S. FTMAP: Extended protein mapping with user-selected probe molecules. Nucleic Acids Res. 2012, 40, W271–W275. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes Biovia. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- Pantaleão, S.Q.; Philot, E.A.; De Resende-Lara, P.T.; Lima, A.N.; Perahia, D.; Miteva, M.A.; Scott, A.L.; Honorio, K.M. Structural Dynamics of DPP-4 and Its Influence on the Projection of Bioactive Ligands. Molecules 2018, 23, 490. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, D.; De Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Kronenberger, T.; Asse, L.R.J.; Wrenger, C.; Trossini, G.H.G.; Honorio, K.M.; Maltarollo, V.G. Studies of Staphylococcus aureus FabI inhibitors: Fragment-Based approach based on holographic structure–activity relationship analyses. Future Med. Chem. 2017, 9, 135–151. [Google Scholar] [CrossRef]
- Maltarollo, V.G.; Araujo, S.C.; Trossini, G.H.G.; Honorio, K.M. Understanding PPAR-δ affinity and selectivity using hologram quantitative structure–activity modeling, molecular docking and GRID calculations. Future Med. Chem. 2016, 8, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Sybyl 2.1; Tripos: St Louis, MO, USA, 2008.
- Halgren, T.A. Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Veríssimo, G.C.; dos Santos Junior, V.S.; de Almeida, I.A.D.R.; Ruas, M.S.A.M.; Coutinho, L.G.; de Oliveira, R.B.; Maltarollo, V. The Brazilian Compound Library (BraCoLi) database, a Brazilian repository of chemical and biological information for drug design. Biol. Med. Chem. 2021, 1, 1–17. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Pilon, A.; Valli, M.; Dametto, A.C.; Pinto, M.E.F.; Freire, R.T.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. NuBBEDB: An updated database to uncover chemical and biological information from Brazilian biodiversity. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Quacpac. Software Openeye Scientific Inc., Santa Fe, NM, USA. Available online: http://www.eyesopen.com/news/quacpac-v150-released (accessed on 27 November 2020).
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. OMEGA 2.5.1.4, OpenEye Scientific Software, Santa Fe, NM, USA. Available online: http://www.eyesopen.com (accessed on 15 February 2018).
- Spitzer, R.; Jain, A.N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput. Aided Mol. Des. 2012, 26, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2018, 35, 1067–1069. [Google Scholar] [CrossRef]
- Banerjee, P.; O Eckert, A.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echenique, P.; Alonso, J.L.; Echenique-Robba, P. A mathematical and computational review of Hartree–Fock SCF methods in quantum chemistry. Mol. Phys. 2007, 105, 3057–3098. [Google Scholar] [CrossRef] [Green Version]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision a. 1; Gaussian. Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Case IYB-S, D.A.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D., III; Darden, T.A.; Duke, R.E.; Gilson, M.K.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated Interaction Energy (SIE) for Scoring Protein−Ligand Binding Affinities. 1. Exploring the Parameter Space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef]
- Lill, M.A.; Thompson, J.J. Solvent Interaction Energy Calculations on Molecular Dynamics Trajectories: Increasing the Efficiency Using Systematic Frame Selection. J. Chem. Inf. Model. 2011, 51, 2680–2689. [Google Scholar] [CrossRef] [Green Version]
- Zoete, V.; Irving, M.B.; Michielin, O. MM-GBSA binding free energy decomposition and T cell receptor engineering. J. Mol. Recognit. 2010, 23, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Mark, A.; Van Gunsteren, W.F. Decomposition of the Free Energy of a System in Terms of Specific Interactions: Implications for Theoretical and Experimental Studies. J. Mol. Biol. 1994, 240, 167–176. [Google Scholar] [CrossRef]
- Silva, J.R.A.; Bishai, W.R.; Govender, T.; Lamichhane, G.; Maguire, G.E.; Kruger, H.G.; Lameira, J.; Alves, C.N. Targeting the cell wall of Mycobacterium tuberculosis: A molecular modelling investigation of the interaction of imipenem and meropenem with L, D -transpeptidase 2. J. Biomol. Struct. Dyn. 2015, 34, 1–34. [Google Scholar] [CrossRef]
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Krudy, G.; Hou, T.; Zhang, W.; Holland, G.; Xu, X. Development of Reliable Aqueous Solubility Models and Their Application in Druglike Analysis. J. Chem. Inf. Model. 2007, 47, 1395–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrio, T.; Böhme, M.; Hack, U.; Miljanic, T.; Weidner, U. Ambrosia—Eine Ursache für die Zunahme von Allergien? Allergologie 2009, 32, 453–461. [Google Scholar] [CrossRef]
- Pham-The, H.; González-Álvarez, I.; Bermejo, M.; Sanjuan, V.M.; Centelles, I.; Garrigues, T.M.; Cabrera-Pérez, M. Ángel In Silico Prediction of Caco-2 Cell Permeability by a Classification QSAR Approach. Mol. Inform. 2011, 30, 376–385. [Google Scholar] [CrossRef]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME Properties with Substructure Pattern Recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 2–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Protti, Í.F.; Rodrigues, D.R.; Fonseca, S.K.; Alves, R.J.; de Oliveira, R.B.; Maltarollo, V.G. Do Drug-likeness Rules Apply to Oral Prodrugs? ChemMedChem 2021, 16, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Corrie, P.G. Cytotoxic chemotherapy: Clinical aspects. Medicine 2011, 39, 717–722. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, Z.; Zhao, L.; Huang, L.; Yang, X.-L.; Tang, J.; Feng, S.-S. Pharmaceutical nanotechnology for oral delivery of anticancer drugs. Adv. Drug Deliv. Rev. 2013, 65, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef] [Green Version]
- Robinson, R.L.M.; Glen, R.C.; Mitchell, J.B. Winnow based identification of potent hERG inhibitors in silico: Comparative assessment on different datasets. J. Chemin. 2012, 4, O6. [Google Scholar] [CrossRef]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Du, Z.; Wang, J.; Wu, Z.; Li, W.; Liu, G.; Shen, X.; Tang, Y. In Silico Estimation of Chemical Carcinogenicity with Binary and Ternary Classification Methods. Mol. Inform. 2015, 34, 228–235. [Google Scholar] [CrossRef]
- Lamothe, S.; Guo, J.; Li, W.; Yang, T.; Zhang, S. The Human Ether-a-go-go-related Gene (hERG) Potassium Channel Represents an Unusual Target for Protease-mediated Damage. J. Biol. Chem. 2016, 291, 20387–20401. [Google Scholar] [CrossRef] [Green Version]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Teague, S.J. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discov. 2003, 2, 527–541. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 1 | Compound 2 | Compound 3 | Compound 4 | Compound 5 | Compound 6 | |
|---|---|---|---|---|---|---|
| Z1002566344 | Z2828033263 | Z2955346685 | Z1016986064 | Z1475103113 | Z2593989424 | |
| pharmacokinetics predictions | ||||||
| LogS | −3.7449 | −2.4312 | −3.2553 | −3.4296 | −2.9741 | −3.6493 |
| HIA (P) a | +(0.9828) | +(0.8411) | +(0.7078) | +(0.7093) | −(0.6149) | +(0.8960) |
| Caco-2 permeability (P) b | −0.6692) | +(0.5649) | −(0.6844) | −(0.6379) | +(0.5409) | −(0.5473) |
| Cellular distribution | mitochondria | mitochondria | mitochondria | mitochondria | mitochondria | mitochondria |
| toxicity predictions | ||||||
| hERG-1 inhibition (P) c | WI (0.7073) | WI (0.9693) | WI (0.9899) | WI (0.9047) | WI (0.6851) | WI (0.9120) |
| hERG-2 inhibition (P) d | I (0.7353) | I (0.5564) | NI (0.6431) | I (0.7064) | I (0.8003) | I (0.8882) |
| AMES (P) e | NT (0.6134) | NT (0.6926) | NT (0.6807) | NT (0.6069) | NT (0.6774) | NT (0.7279) |
| Carcinogenicity (P) f | NC (0.7627) | NC (0.8482) | NC (0.9053) | NC (0.9079) | NC (0.8816) | NC (0.7699) |
| LD50 | 729 mg/Kg | 3000 mg/Kg | 840 mg/Kg | 980 mg/Kg | 800 mg/Kg | 1000 mg/Kg |
| Toxicity class g | 4 | 5 | 4 | 4 | 4 | 4 |
| Compound | SIE |
|---|---|
| 2 | −6.53 |
| 3 | −7.55 |
| 4 | −6.95 |
| 5 | −6.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa, B.L.F.; Freitas, T.R.; Almeida, M.d.O.; Araújo, S.S.d.S.; Andrade, A.C.; Dornelas, G.G.; Fiorotto, J.G.; Maltarollo, V.G.; Sabino, A.d.P. Virtual Screening of Adenylate Kinase 3 Inhibitors Employing Pharmacophoric Model, Molecular Docking, and Molecular Dynamics Simulations as Potential Therapeutic Target in Chronic Lymphocytic Leukemia. Future Pharmacol. 2021, 1, 60-79. https://doi.org/10.3390/futurepharmacol1010006
Barbosa BLF, Freitas TR, Almeida MdO, Araújo SSdS, Andrade AC, Dornelas GG, Fiorotto JG, Maltarollo VG, Sabino AdP. Virtual Screening of Adenylate Kinase 3 Inhibitors Employing Pharmacophoric Model, Molecular Docking, and Molecular Dynamics Simulations as Potential Therapeutic Target in Chronic Lymphocytic Leukemia. Future Pharmacology. 2021; 1(1):60-79. https://doi.org/10.3390/futurepharmacol1010006
Chicago/Turabian StyleBarbosa, Bárbara Lima Fonseca, Tulio Resende Freitas, Michell de Oliveira Almeida, Sérgio Schusterschitz da Silva Araújo, Ana Clara Andrade, Geovana Gomes Dornelas, Julyana Gayva Fiorotto, Vinicius Gonçalves Maltarollo, and Adriano de Paula Sabino. 2021. "Virtual Screening of Adenylate Kinase 3 Inhibitors Employing Pharmacophoric Model, Molecular Docking, and Molecular Dynamics Simulations as Potential Therapeutic Target in Chronic Lymphocytic Leukemia" Future Pharmacology 1, no. 1: 60-79. https://doi.org/10.3390/futurepharmacol1010006
APA StyleBarbosa, B. L. F., Freitas, T. R., Almeida, M. d. O., Araújo, S. S. d. S., Andrade, A. C., Dornelas, G. G., Fiorotto, J. G., Maltarollo, V. G., & Sabino, A. d. P. (2021). Virtual Screening of Adenylate Kinase 3 Inhibitors Employing Pharmacophoric Model, Molecular Docking, and Molecular Dynamics Simulations as Potential Therapeutic Target in Chronic Lymphocytic Leukemia. Future Pharmacology, 1(1), 60-79. https://doi.org/10.3390/futurepharmacol1010006