
Screening the Oxygen Reduction Reaction Performance of Carbon-Supported Pt-M (M = Ni, Cu, Co) Binary Electrocatalysts via Tuning Metal–Support Interaction


Abstract
1. Introduction
2. Experimental Section
2.1. Preparation of Pt-M/C Binary Electrocatalysts
2.2. Structural Characterizations of Binary Electrocatalyst
2.3. Electrochemical Analysis
3. Results and Discussion
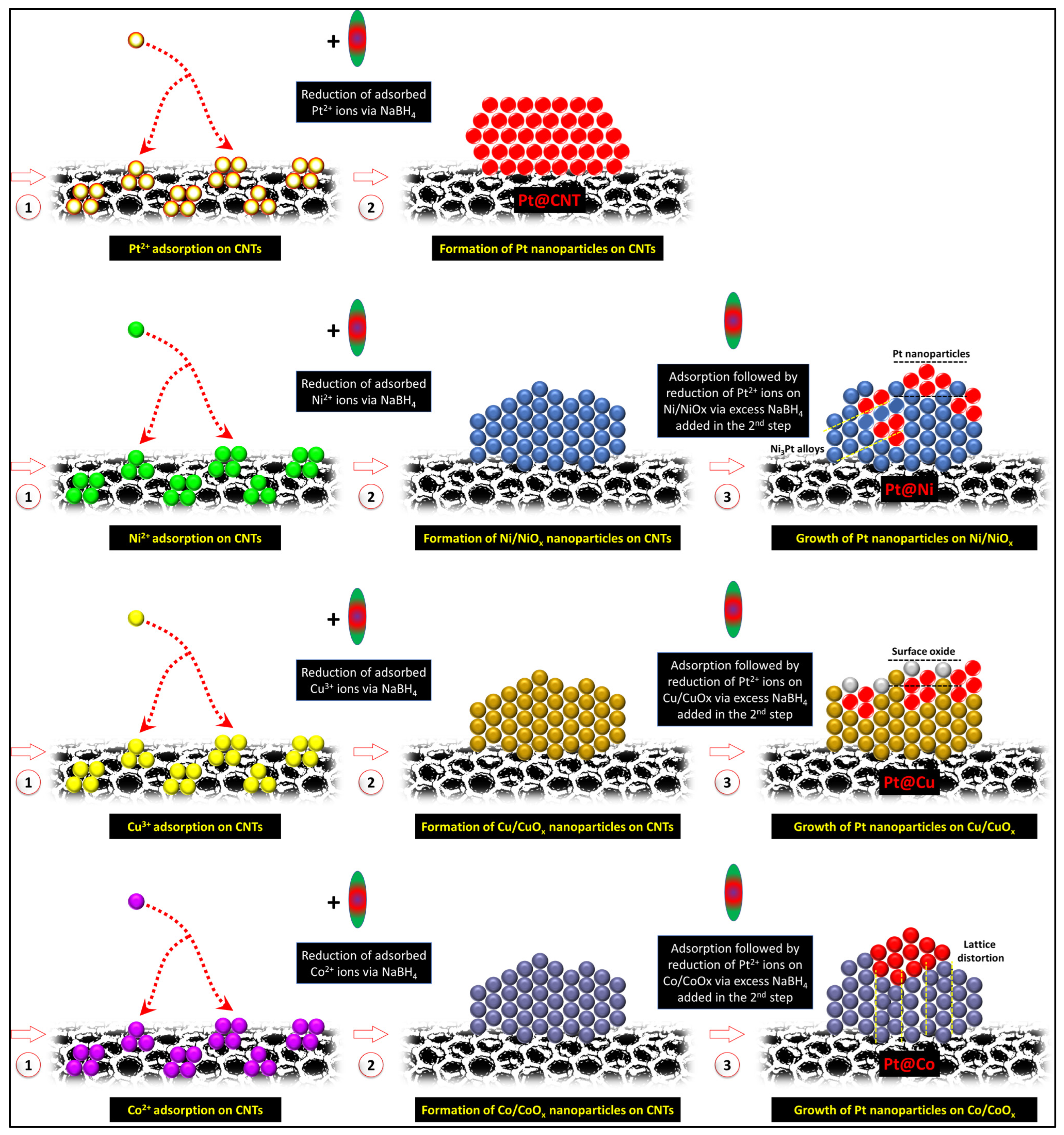
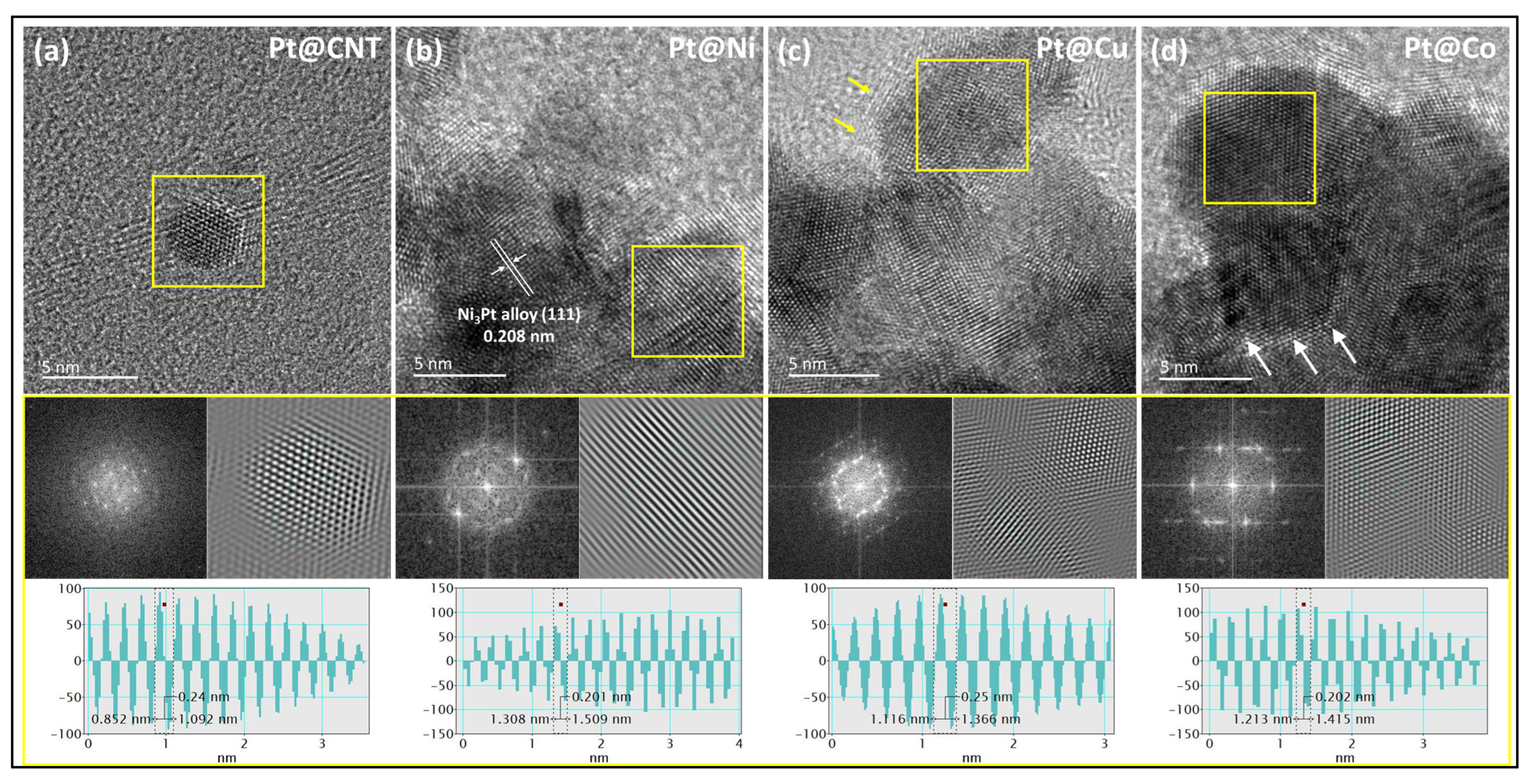
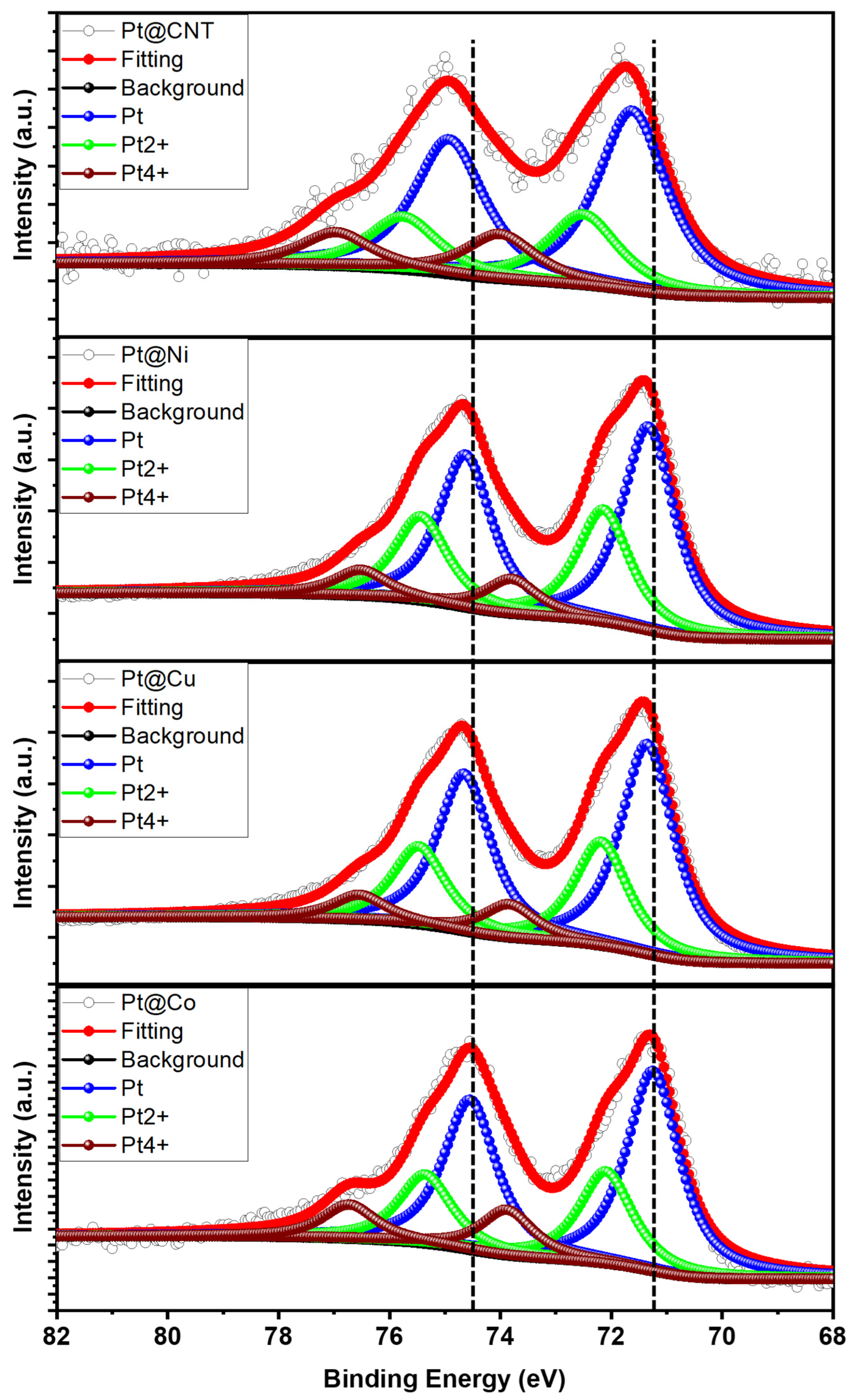
3.1. Morphological and Structural Properties
3.2. Electrochemical Performance in ORR
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qasem, N.A.A.; Abdulrahman, G.A.Q. A Recent Comprehensive Review of Fuel Cells: History, Types, and Applications. Int. J. Energy Res. 2024, 2024, 7271748. [Google Scholar] [CrossRef]
- Zaman, S.; Huang, L.; Douka, A.I.; Yang, H.; You, B.; Xia, B.Y. Oxygen Reduction Electrocatalysts toward Practical Fuel Cells: Progress and Perspectives. Angew. Chem. Int. Ed. 2021, 60, 17832–17852. [Google Scholar] [CrossRef] [PubMed]
- Alfaifi, S.M.; Balu, R.; Chiang, K.; Choudhury, N.R.; Dutta, N.K. Electrocatalysts for the Oxygen Reduction Reaction in Proton Exchange Membrane Fuel Cells: Significant Advances, Major Challenges, and Future Directions. ACS Catal. 2025, 15, 9301–9345. [Google Scholar] [CrossRef]
- Huang, L.; Zaman, S.; Tian, X.; Wang, Z.; Fang, W.; Xia, B.Y. Advanced Platinum-Based Oxygen Reduction Electrocatalysts for Fuel Cells. Acc. Chem. Res. 2021, 54, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sasaki, K. Advanced Pt-Based Core–Shell Electrocatalysts for Fuel Cell Cathodes. Acc. Chem. Res. 2022, 55, 1226–1236. [Google Scholar] [CrossRef]
- Bhalothia, D.; Fan, Y.-J.; Lai, Y.-C.; Yang, Y.-T.; Yang, Y.-W.; Lee, C.-H.; Chen, T.-Y. Conformational Effects of Pt-Shells on Nanostructures and Corresponding Oxygen Reduction Reaction Activity of Au-Cluster-Decorated NiOx@Pt Nanocatalysts. Nanomaterials 2019, 9, 1003. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.; Zhang, Z.; Hou, D.; Bai, F.; Han, Y.; Zhang, C.; Zhang, Y.; Hu, J. Metal–support interactions for heterogeneous catalysis: Mechanisms, characterization techniques and applications. J. Mater. Chem. A 2023, 11, 8540–8572. [Google Scholar] [CrossRef]
- Mu, Y.; Wang, T.; Zhang, J.; Meng, C.; Zhang, Y.; Kou, Z. Single-Atom Catalysts: Advances and Challenges in Metal-Support Interactions for Enhanced Electrocatalysis. Electrochem. Energy Rev. 2022, 5, 145–186. [Google Scholar] [CrossRef]
- Luo, Y.; Alonso-Vante, N. The Effect of Support on Advanced Pt-based Cathodes towards the Oxygen Reduction Reaction. State Art. Electrochim. Acta 2015, 179, 108–118. [Google Scholar] [CrossRef]
- Samad, S.; Loh, K.S.; Wong, W.Y.; Lee, T.K.; Sunarso, J.; Chong, S.T.; Wan Daud, W.R. Carbon and non-carbon support materials for platinum-based catalysts in fuel cells. Int. J. Hydrogen Energy 2018, 43, 7823–7854. [Google Scholar] [CrossRef]
- Cong, Y.; Wang, H.; Meng, F.; Dou, D.; Meng, X.; Zhao, Q.; Cao, D.; Wang, Y. One-pot synthesis of NiPt core–shell nanoparticles toward efficient oxygen reduction reaction. J. Solid State Electrochem. 2022, 26, 1381–1388. [Google Scholar] [CrossRef]
- Anwar, R.; Iqbal, N.; Hanif, S.; Noor, T.; Shi, X.; Zaman, N.; Haider, D.; Rizvi, S.A.M.; Kannan, A.M. MOF-Derived CuPt/NC Electrocatalyst for Oxygen Reduction Reaction. Catalysts 2020, 10, 799. [Google Scholar] [CrossRef]
- Bhalothia, D.; Fan, Y.-J.; Huang, T.-H.; Lin, Z.-J.; Yang, Y.-T.; Wang, K.-W.; Chen, T.-Y. Local Structural Disorder Enhances the Oxygen Reduction Reaction Activity of Carbon-Supported Low Pt Loading CoPt Nanocatalysts. J. Phys. Chem. C 2019, 123, 19013–19021. [Google Scholar] [CrossRef]
- Liu, X.; Liang, J.; Li, Q. Design principle and synthetic approach of intermetallic Pt-M alloy oxygen reduction catalysts for fuel cells. Chin. J. Catal. 2023, 45, 17–26. [Google Scholar] [CrossRef]
- Wang, S.; Wang, M.; Zhang, Y.; Wang, H.; Fei, H.; Liu, R.; Kong, H.; Gao, R.; Zhao, S.; Liu, T.; et al. Metal Oxide-Supported Metal Catalysts for Electrocatalytic Oxygen Reduction Reaction: Characterization Methods, Modulation Strategies, and Recent Progress. Small Methods 2023, 7, 2201714. [Google Scholar] [CrossRef]
- Xiong, L.; Manthiram, A. Effect of Atomic Ordering on the Catalytic Activity of Carbon Supported PtM (M = Fe, Co, Ni, and Cu) Alloys for Oxygen Reduction in PEMFCs. J. Electrochem. Soc. 2005, 152, A697. [Google Scholar] [CrossRef]
- Zhang, C.; Hwang, S.Y.; Peng, Z. Size-dependent oxygen reduction property of octahedral Pt-Ni nanoparticle electrocatalysts. J. Mater. Chem. A 2014, 2, 19778–19787. [Google Scholar] [CrossRef]
- Li, X.; He, Y.; Cheng, S.; Li, B.; Zeng, Y.; Xie, Z.; Meng, Q.; Ma, L.; Kisslinger, K.; Tong, X.; et al. Atomic Structure Evolution of Pt-Co Binary Catalysts: Single Metal Sites versus Intermetallic Nanocrystals. Adv. Mater. 2021, 33, 2106371. [Google Scholar] [CrossRef]
- Jia, Q.; Caldwell, K.; Ziegelbauer, J.M.; Kongkanand, A.; Wagner, F.T.; Mukerjee, S.; Ramaker, D.E. The Role of OOH Binding Site and Pt Surface Structure on ORR Activities. J. Electrochem. Soc. 2014, 161, F1323. [Google Scholar] [CrossRef]
- Yan, Q.-Q.; Wu, D.-X.; Chu, S.-Q.; Chen, Z.-Q.; Lin, Y.; Chen, M.-X.; Zhang, J.; Wu, X.-J.; Liang, H.-W. Reversing the charge transfer between platinum and sulfur-doped carbon support for electrocatalytic hydrogen evolution. Nat. Commun. 2019, 10, 4977. [Google Scholar] [CrossRef]
- Bhalothia, D.; Yan, C.; Hiraoka, N.; Ishii, H.; Liao, Y.-F.; Chen, P.-C.; Wang, K.-W.; Chou, J.-P.; Dai, S.; Chen, T.-Y. Pt-Mediated Interface Engineering Boosts the Oxygen Reduction Reaction Performance of Ni Hydroxide-Supported Pd Nanoparticles. ACS Appl. Mater. Interfaces 2023, 15, 16177–16188. [Google Scholar] [CrossRef] [PubMed]
- Moreira Da Silva, C.; Girard, A.; Dufond, M.; Fossard, F.; Andrieux-Ledier, A.; Huc, V.; Loiseau, A. Nickel platinum (NixPt1−x) nanoalloy monodisperse particles without the core–shell structure by colloidal synthesis. Nanoscale Adv. 2020, 2, 3882–3889. [Google Scholar] [CrossRef]
- Bhalothia, D.; Chen, P.-C.; Yan, C.; Yeh, W.; Tsai, D.-L.; Chan, T.-S.; Wang, K.-W.; Chen, T.-Y. Heterogeneous assembly of Pt-clusters on hierarchically structured CoOx@SnPd2@SnO2 quaternary nanocatalysts manifesting oxygen reduction reaction performance. New J. Chem. 2020, 44, 9712–9724. [Google Scholar] [CrossRef]
- Bhalothia, D.; Chen, P.-C.; Yan, C.; Wang, K.-W.; Chen, T.-Y. Heterogeneous NiO2-to-Pd Epitaxial Structure Performs Outstanding Oxygen Reduction Reaction Activity. J. Phys. Chem. C 2020, 124, 2295–2306. [Google Scholar] [CrossRef]
- Luo, Y.; Lou, W.; Feng, H.; Liu, Z.; Chen, Q.; Liao, G.; Huang, X.; Tsiakaras, P.; Shen, P. Ultra-Small Nanoparticles of Pd-Pt-Ni Alloy Octahedra with High Lattice Strain for Efficient Oxygen Reduction Reaction. Catalysts 2023, 13, 97. [Google Scholar] [CrossRef]
- Bhalothia, D.; Beniwal, A.; Yan, C.; Chen, T.-Y. Collective efforts of oxygen vacancies, Ni, Pd and Pt ensemble sites promote the oxygen reduction reaction performance of a hierarchical structured quaternary electrocatalyst. Mater. Today Energy 2025, 48, 101776. [Google Scholar] [CrossRef]
- Bhalothia, D.; Beniwal, A.; Yan, C.; Wang, K.-C.; Wang, C.-H.; Chen, T.-Y. Potential synergy between Pt2Ni4 Atomic-Clusters, oxygen vacancies and adjacent Pd nanoparticles outperforms commercial Pt nanocatalyst in alkaline fuel cells. Chem. Eng. J. 2024, 483, 149421. [Google Scholar] [CrossRef]
- Bhalothia, D.; Lin, C.-Y.; Yan, C.; Yang, Y.-T.; Chen, T.-Y. Effects of Pt metal loading on the atomic restructure and oxygen reduction reaction performance of Pt-cluster decorated Cu@Pd electrocatalysts. Sustain. Energy Fuels 2019, 3, 1668–1681. [Google Scholar] [CrossRef]
- Bhalothia, D.; Yan, C.; Hiraoka, N.; Ishii, H.; Liao, Y.F.; Dai, S.; Chen, P.-C.; Chen, T.-Y. Iridium Single Atoms to Nanoparticles: Nurturing the Local Synergy with Cobalt-Oxide Supported Palladium Nanoparticles for Oxygen Reduction Reaction. Adv. Sci. 2024, 11, 2404076. [Google Scholar] [CrossRef]
- Rahul, R.; Singh, R.K.; Neergat, M. Effect of oxidative heat-treatment on electrochemical properties and oxygen reduction reaction (ORR) activity of Pd–Co alloy catalysts. J. Electroanal. Chem. 2014, 712, 223–229. [Google Scholar] [CrossRef]
- Chang, H.-W.; Yang, T.; Yan, C.; Chiu, P.-H.; Wu, C.-Y.; Yen, H.-W.; Bhalothia, D.; Wang, K.-W.; Chen, P.-C.; Chen, T.-Y. Oxidized Ti Single Atoms and Co3O4 with Abundant Oxygen Vacancies Collaborating with Adjacent Pd Sites for an Efficient and Stable Oxygen Reduction Reaction. Adv. Sci. 2025, 12, 2417789. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Pt L3-Edge | ||
|---|---|---|---|
| Bond Pair | CN | R | |
| Pt@CNT | Pt-Pt | 6.95 | 2.783 |
| Pt-O | 1.46 | 2.781 | |
| Pt@Ni | Pt-Pt | 1.75 | 2.712 |
| Pt-O | 2.46 | 2.765 | |
| Pt-Ni | 2.35 | 2.523 | |
| Pt@Cu | Pt-Pt | 3.76 | 2.734 |
| Pt-O | 2.79 | 2.762 | |
| Pt-Cu | 1.87 | 2.536 | |
| Pt@Co | Pt-Pt | 4.02 | 2.722 |
| Pt-O | 2.44 | 2.768 | |
| Pt-Co | 1.12 | 2.522 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beniwal, A.; Gurjar, H.; Shekhawat, K.; Bagaria, A.; Bhalothia, D. Screening the Oxygen Reduction Reaction Performance of Carbon-Supported Pt-M (M = Ni, Cu, Co) Binary Electrocatalysts via Tuning Metal–Support Interaction. Oxygen 2025, 5, 10. https://doi.org/10.3390/oxygen5030010
Beniwal A, Gurjar H, Shekhawat K, Bagaria A, Bhalothia D. Screening the Oxygen Reduction Reaction Performance of Carbon-Supported Pt-M (M = Ni, Cu, Co) Binary Electrocatalysts via Tuning Metal–Support Interaction. Oxygen. 2025; 5(3):10. https://doi.org/10.3390/oxygen5030010
Chicago/Turabian StyleBeniwal, Amisha, Hariom Gurjar, Khushabu Shekhawat, Ashima Bagaria, and Dinesh Bhalothia. 2025. "Screening the Oxygen Reduction Reaction Performance of Carbon-Supported Pt-M (M = Ni, Cu, Co) Binary Electrocatalysts via Tuning Metal–Support Interaction" Oxygen 5, no. 3: 10. https://doi.org/10.3390/oxygen5030010
APA StyleBeniwal, A., Gurjar, H., Shekhawat, K., Bagaria, A., & Bhalothia, D. (2025). Screening the Oxygen Reduction Reaction Performance of Carbon-Supported Pt-M (M = Ni, Cu, Co) Binary Electrocatalysts via Tuning Metal–Support Interaction. Oxygen, 5(3), 10. https://doi.org/10.3390/oxygen5030010