

RONS and Oxidative Stress: An Overview of Basic Concepts

 ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS)
2. Signaling and Physiological Functions of RONS
3. Oxidative Stress
3.1. Sources of Oxidative Stress
3.1.1. Endogenous Sources
Mitochondria
NADPH Oxidases (NOXs)
Peroxisomes
Endoplasmic Reticulum (ER)
Xanthine Oxidases (XOs)
Myeloperoxidase (MPO)
Endothelial Nitric Oxide Synthase (eNOS)
3.1.2. Exogenous and Environmental Sources
UV Light
Ionizing Radiation (IR)
Contaminants
Foods
Alcohol and Smoking
Medicines
4. Oxidative Stress-Induced Oxidative Damage
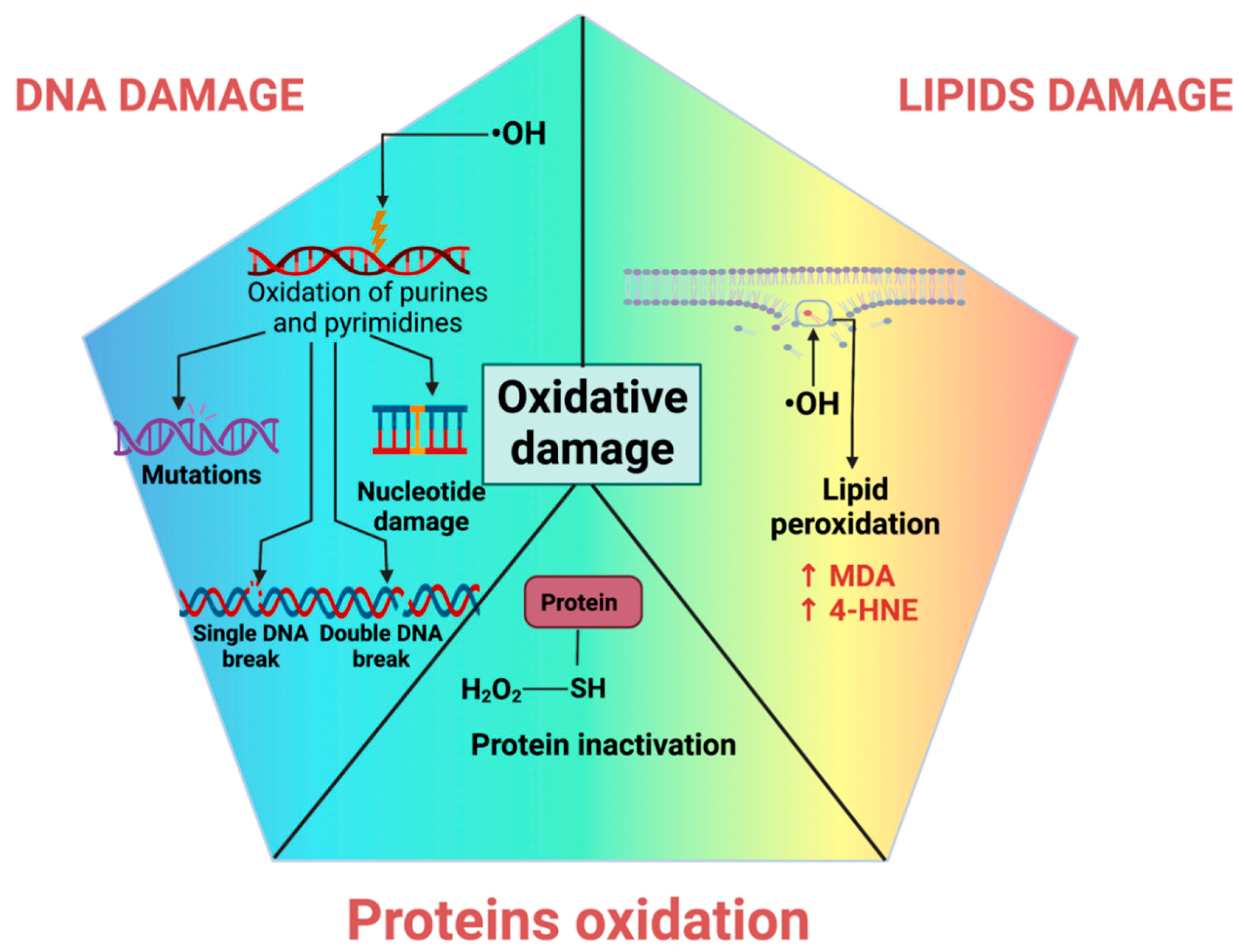
4.1. DNA Oxidative Damage
4.2. Lipid Peroxidation
4.3. Protein Oxidation
5. Antioxidants
5.1. Endogenous Antioxidants
5.1.1. Enzymatic Antioxidants
Superoxide Dismutase (SOD)
Catalase (CAT)
Peroxidase (Prx)
Glutathione Peroxidase (GPx)
Heme Oxygenase-1 (HO-1)
Glutathione Reductase (GR)
Glutathione S-Transferase (GST)
Thioredoxin (Trx)
Thioredoxin Reductase (TrxR)
5.1.2. Non-Enzymatic Antioxidants
Glutathione (GSH)
Albumin (ALB)
Bilirubin (BR)
Uric Acid
Melatonin (MEL)
Coenzyme Q10 (CoQ10)
5.2. Exogen or Dietary Antioxidants
5.2.1. Ascorbic Acid
5.2.2. Polyphenols
5.2.3. Flavonoids
5.2.4. Resveratrol
6. OS and Diseases
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, Y.R.; Trush, M. Defining ROS in Biology and Medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Neish, A.S. Nox Enzymes and New Thinking on Reactive Oxygen: A Double-Edged Sword Revisited. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Girotti, A.W. Mechanisms of lipid peroxidation. J. Free Radic. Biol. Med. 1985, 1, 87–95. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Cheema, A.K.; Zhang, L.; Suzuki, Y.J. Protein Carbonylation as a Novel Mechanism in Redox Signaling. Circ. Res. 2008, 102, 310–318. [Google Scholar] [CrossRef]
- Anand, P.; Stamler, J.S. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J. Mol. Med. 2012, 90, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Buetler, T.M.; Krauskopf, A.; Ruegg, U.T. Role of Superoxide as a Signaling Molecule. Physiology 2004, 19, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, e4350965. [Google Scholar] [CrossRef]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Carroll, K.S. Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta BBA-Gen. Subj. 2014, 1840, 847–875. [Google Scholar] [CrossRef] [PubMed]
- Madesh, M.; Hajnóczky, G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 2001, 155, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H Oxidase: Role in Cardiovascular Biology and Disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Chio, C.-C.; Chi, K.-H.; Wu, H.-M.; Lin, W.-W. Superoxide Anion-Dependent Raf/MEK/ERK Activation by Peroxisome Proliferator Activated Receptor γ Agonists 15-Deoxy-Δ12,14-prostaglandin J2, Ciglitazone, and GW1929. Exp. Cell Res. 2002, 277, 192–200. [Google Scholar] [CrossRef]
- Yang, J.-Q.; Buettner, G.R.; Domann, F.E.; Li, Q.; Engelhardt, J.F.; Weydert, C.D.; Oberley, L.W. v-Ha-ras mitogenic signaling through superoxide and derived reactive oxygen species. Mol. Carcinog. 2002, 33, 206–218. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Ann. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Pedraza-Chaverri, J.; Scholze, A. Nrf2 Activation in Chronic Kidney Disease: Promises and Pitfalls. Antioxidants 2022, 11, 1112. [Google Scholar] [CrossRef]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1–Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 576. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Borniquel, S.; García-Quintáns, N.; Valle, I.; Olmos, Y.; Wild, B.; Martínez-Granero, F.; Soria, E.; Lamas, S.; Monsalve, M. Inactivation of Foxo3a and Subsequent Downregulation of PGC-1α Mediate Nitric Oxide-Induced Endothelial Cell Migration. Mol. Cell. Biol. 2010, 30, 4035–4044. [Google Scholar] [CrossRef]
- Wu, M.; Bian, Q.; Liu, Y.; Fernandes, A.; Taylor, A.; Pereira, P.; Shang, F. Sustained oxidative stress inhibits NF-κB activation partially via inactivating the proteasome. Free Radic. Biol. Med. 2009, 46, 62–69. [Google Scholar] [CrossRef]
- Nishi, T.; Shimizu, N.; Hiramoto, M.; Sato, I.; Yamaguchi, Y.; Hasegawa, M.; Aizawa, S.; Tanaka, H.; Kataoka, K.; Watanabe, H.; et al. Spatial Redox Regulation of a Critical Cysteine Residue of NF-κB In Vivo. J. Biol. Chem. 2002, 277, 44548–44556. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Go, Y.-M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. α-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2011, 45, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Jang, H.-S.; Noh, M.R.; Kim, J.; Kong, M.J.; Kim, J.I.; Park, J.-W.; Park, K.M. Mitochondrial NADP +-Dependent Isocitrate Dehydrogenase Deficiency Exacerbates Mitochondrial and Cell Damage after Kidney Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2017, 28, 1200–1215. [Google Scholar] [CrossRef]
- Hurd, T.R.; Collins, Y.; Abakumova, I.; Chouchani, E.T.; Baranowski, B.; Fearnley, I.M.; Prime, T.A.; Murphy, M.P.; James, A.M. Inactivation of Pyruvate Dehydrogenase Kinase 2 by Mitochondrial Reactive Oxygen Species. J. Biol. Chem. 2012, 287, 35153–35160. [Google Scholar] [CrossRef] [PubMed]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef]
- Hill, B.G.; Higdon, A.N.; Dranka, B.P.; Darley-Usmar, V.M. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim. Biophys. Acta BBA-Bioenerg. 2010, 1797, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Kurahashi, T.; Saito, Y.; Otsuki, N.; Kwon, M.; Ohtake, H.; Yamakawa, M.; Yamada, K.-I.; Miyata, S.; Tomita, Y.; et al. Kidney fibrosis is independent of the amount of ascorbic acid in mice with unilateral ureteral obstruction. Free Radic. Res. 2014, 48, 1115–1124. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Ortega-Lozano, A.J.; Pedraza-Chaverri, J. Redox signaling pathways in unilateral ureteral obstruction (UUO)-induced renal fibrosis. Free Radic. Biol. Med. 2021, 172, 65–81. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef]
- Zhao, R.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Dröse, S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.J.; Huang, J.; Oselkin, M.; Tarsitano, M.J.; Wang, G.; Iadecola, C.; Pickel, V.M. Subcellular localization of nicotinamide adenine dinucleotide phosphate oxidase subunits in neurons and astroglia of the rat medial nucleus tractus solitarius: Relationship with tyrosine hydroxylase immunoreactive neurons. Neuroscience 2006, 143, 547–564. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Mitochondrial bioenergetics, redox state, dynamics and turnover alterations in renal mass reduction models of chronic kidney diseases and their possible implications in the progression of this illness. Pharmacol. Res. 2018, 135, 1–11. [Google Scholar] [CrossRef]
- Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. Int. J. Mol. Sci. 2019, 20, 3673. [CrossRef]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Di Nardo, A.; Han, J.M.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. ER Stress and Its Functional Link to Mitochondria: Role in Cell Survival and Death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Hof, P.; Duarte, R.O.; Moura, J.J.; Moura, I.; Liu, M.Y.; LeGall, J.; Hille, R.; Archer, M.; Romão, M.J. A structure-based catalytic mechanism for the xanthine oxidase family of molybdenum enzymes. Proc. Natl. Acad. Sci. USA 1996, 93, 8846–8851. [Google Scholar] [CrossRef] [PubMed]
- Ives, A.; Nomura, J.; Martinon, F.; Roger, T.; LeRoy, D.; Miner, J.N.; Simon, G.; Busso, N.; So, A. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat. Commun. 2015, 6, 6555. [Google Scholar] [CrossRef]
- Bredemeier, M.; Lopes, L.M.; Eisenreich, M.A.; Hickmann, S.; Bongiorno, G.K.; d’Avila, R.; Morsch, A.L.B.; da Silva Stein, F.; Campos, G.G.D. Xanthine oxidase inhibitors for prevention of cardiovascular events: A systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2018, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Osarogiagbon, U.R.; Choong, S.; Belcher, J.D.; Vercellotti, G.M.; Paller, M.S.; Hebbel, R.P. Reperfusion injury pathophysiology in sickle transgenic mice. Blood 2000, 96, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Ndrepepa, G. Myeloperoxidase—A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin. Chim. Acta 2019, 493, 36–51. [Google Scholar] [CrossRef]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef]
- Anatoliotakis, N.; Deftereos, S.; Bouras, G.; Giannopoulos, G.; Tsounis, D.; Angelidis, C.; Kaoukis, A.; Stefanadis, C. Myeloperoxidase: Expressing Inflammation and Oxidative Stress in Cardiovascular Disease. Curr. Top. Med. Chem. 2013, 13, 115–138. [Google Scholar] [CrossRef]
- Lehners, A.; Lange, S.; Niemann, G.; Rosendahl, A.; Meyer-Schwesinger, C.; Oh, J.; Stahl, R.; Ehmke, H.; Benndorf, R.; Klinke, A.; et al. Myeloperoxidase deficiency ameliorates progression of chronic kidney disease in mice. Am. J. Physiol.-Ren. Physiol. 2014, 307, F407–F417. [Google Scholar] [CrossRef]
- Haegens, A.; van der Vliet, A.; Butnor, K.J.; Heintz, N.; Taatjes, D.; Hemenway, D.; Vacek, P.; Freeman, B.A.; Hazen, S.L.; Brennan, M.L.; et al. Asbestos-Induced Lung Inflammation and Epithelial Cell Proliferation Are Altered in Myeloperoxidase-Null Mice. Cancer Res. 2005, 65, 9670–9677. [Google Scholar] [CrossRef] [PubMed]
- Lowry, J.L.; Brovkovych, V.; Zhang, Y.; Skidgel, R.A. Endothelial Nitric-oxide Synthase Activation Generates an Inducible Nitric-oxide Synthase-like Output of Nitric Oxide in Inflamed Endothelium. J. Biol. Chem. 2013, 288, 4174–4193. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Münzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease: From Marvel to Menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- de Jager, T.L.; Cockrell, A.E.; Du Plessis, S.S. Ultraviolet Light Induced Generation of Reactive Oxygen Species. In Ultraviolet Light in Human Health, Diseases and Environment; Ahmad, S.I., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Germany, 2017; Volume 996, pp. 15–23. ISBN 978-3-319-56016-8. [Google Scholar]
- Holman, D.M.; Ding, H.; Guy, G.P.; Watson, M.; Hartman, A.M.; Perna, F.M. Prevalence of Sun Protection Use and Sunburn and Association of Demographic and Behaviorial Characteristics With Sunburn Among US Adults. JAMA Dermatol. 2018, 154, 561. [Google Scholar] [CrossRef]
- Young, A.R.; Narbutt, J.; Harrison, G.I.; Lawrence, K.P.; Bell, M.; O’Connor, C.; Olsen, P.; Grys, K.; Baczynska, K.A.; Rogowski-Tylman, M.; et al. Optimal sunscreen use, during a sun holiday with a very high ultraviolet index, allows vitamin D synthesis without sunburn. Br. J. Dermatol. 2019, 181, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.; Elhassaneen, Y. Natural dye from red onion skins and applied in dyeing cotton fabrics for the production of women’s headwear resistance to ultraviolet radiation (UVR). J. Am. Sci. 2014, 10, 129–139. [Google Scholar]
- Bergman, R.S. Germicidal UV Sources and Systems. Photochem. Photobiol. 2021, 97, 466–470. [Google Scholar] [CrossRef]
- Kimeswenger, S.; Schwarz, A.; Födinger, D.; Müller, S.; Pehamberger, H.; Schwarz, T.; Jantschitsch, C. Infrared A radiation promotes survival of human melanocytes carrying ultraviolet radiation-induced DNA damage. Exp. Dermatol. 2016, 25, 447–452. [Google Scholar] [CrossRef]
- Heck, D.E.; Vetrano, A.M.; Mariano, T.M.; Laskin, J.D. UVB Light Stimulates Production of Reactive Oxygen Species. J. Biol. Chem. 2003, 278, 22432–22436. [Google Scholar] [CrossRef]
- Deliconstantinos, G.; Villiotou, V.; Stavrides, J.C. Alterations of nitric oxide synthase and xanthine oxidase activities of human keratinocytes by ultraviolet B radiation. Biochem. Pharmacol. 1996, 51, 1727–1738. [Google Scholar] [CrossRef]
- Bossi, O.; Gartsbein, M.; Leitges, M.; Kuroki, T.; Grossman, S.; Tennenbaum, T. UV irradiation increases ROS production via PKCδ signaling in primary murine fibroblasts. J. Cell. Biochem. 2008, 105, 194–207. [Google Scholar] [CrossRef]
- Wei, H.; Cai, Q.; Rahn, R.; Zhang, X. Singlet Oxygen Involvement in Ultraviolet (254 nm) Radiation-Induced Formation of 8-Hydroxy-Deoxyguanosine in DNA. Free Radic. Biol. Med. 1997, 23, 148–154. [Google Scholar] [CrossRef]
- Ray, A.J.; Turner, R.; Nikaido, O.; Rees, J.L.; Birch-Machin, M.A. The Spectrum of Mitochondrial DNA Deletions is a Ubiquitous Marker of Ultraviolet Radiation Exposure in Human Skin. J. Invest. Dermatol. 2000, 115, 674–679. [Google Scholar] [CrossRef]
- Bernard, J.J.; Gallo, R.L.; Krutmann, J. Photoimmunology: How ultraviolet radiation affects the immune system. Nat. Rev. Immunol. 2019, 19, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Bouillaguet, S.; Owen, B.; Wataha, J.C.; Campo, M.A.; Lange, N.; Schrenzel, J. Intracellular reactive oxygen species in monocytes generated by photosensitive chromophores activated with blue light. Dent. Mater. 2008, 24, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A. Ultraweak photon emission induced by visible light and ultraviolet A radiation via photoactivated skin chromophores: In vivo charge coupled device imaging. J. Biomed. Opt. 2012, 17, 085004. [Google Scholar] [CrossRef] [PubMed]
- Boo, Y.C. Emerging Strategies to Protect the Skin from Ultraviolet Rays Using Plant-Derived Materials. Antioxidants 2020, 9, 637. [Google Scholar] [CrossRef]
- Bickers, D.R.; Athar, M. Oxidative Stress in the Pathogenesis of Skin Disease. J. Invest. Dermatol. 2006, 126, 2565–2575. [Google Scholar] [CrossRef]
- Reelfs, O.; Tyrrell, R.M.; Pourzand, C. Ultraviolet A Radiation-Induced Immediate Iron Release Is a Key Modulator of the Activation of NF-κB in Human Skin Fibroblasts. J. Invest. Dermatol. 2004, 122, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Bassez, M.-P. Water, Air, Earth and Cosmic Radiation. Orig. Life Evol. Biosph. 2015, 45, 5–13. [Google Scholar] [CrossRef]
- Indriolo, N.; Neufeld, D.A.; Gerin, M.; Schilke, P.; Benz, A.O.; Winkel, B.; Menten, K.M.; Chambers, E.T.; Black, J.H.; Bruderer, S.; et al. Herschel Survey of Galactic OH+, H2O+, and H3O+: Probing the Molecular Hydrogen Fraction and Cosmic-Ray Ionization Rate. Astrophys. J. 2015, 800, 40. [Google Scholar] [CrossRef]
- Zdrojewicz, Z.; Szlagor, A.; Wielogórska, M.; Nowakowska, D.; Nowakowski, J. Influence of ionizing radiation on human body. Fam. Med. Prim. Care Rev. 2016, 2, 174–179. [Google Scholar] [CrossRef]
- Alizadeh, E.; Orlando, T.M.; Sanche, L. Biomolecular Damage Induced by Ionizing Radiation: The Direct and Indirect Effects of Low-Energy Electrons on DNA. Annu. Rev. Phys. Chem. 2015, 66, 379–398. [Google Scholar] [CrossRef] [PubMed]
- ICNIRP. Principles for Non-Ionizing Radiation Protection. Health Phys. 2020, 118, 477–482. [Google Scholar] [CrossRef]
- Kirsch, D.G.; Diehn, M.; Kesarwala, A.H.; Maity, A.; Morgan, M.A.; Schwarz, J.K.; Bristow, R.; Demaria, S.; Eke, I.; Griffin, R.J.; et al. The Future of Radiobiology. JNCI J. Natl. Cancer Inst. 2018, 110, 329–340. [Google Scholar] [CrossRef]
- Cui, F.; Ma, N.; Han, X.; Chen, N.; Xi, Y.; Yuan, W.; Xu, Y.; Han, J.; Xu, X.; Tu, Y. Effects of 60Co γ Irradiation on the Reproductive Function of Caenorhabditis elegans. Dose-Response 2019, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Santacruz-Gomez, K.; Sarabia-Sainz, A.; Acosta-Elias, M.; Sarabia-Sainz, M.; Janetanakit, W.; Khosla, N.; Melendrez, R.; Montero, M.P.; Lal, R. Antioxidant activity of hydrated carboxylated nanodiamonds and its influence on water γ -radiolysis. Nanotechnology 2018, 29, 125707. [Google Scholar] [CrossRef] [PubMed]
- Rai, Y.; Anita; Kumari, N.; Singh, S.; Kalra, N.; Soni, R.; Bhatt, A.N. Mild mitochondrial uncoupling protects from ionizing radiation induced cell death by attenuating oxidative stress and mitochondrial damage. Biochim. Biophys. Acta BBA-Bioenerg. 2021, 1862, 148325. [Google Scholar] [CrossRef]
- Einor, D.; Bonisoli-Alquati, A.; Costantini, D.; Mousseau, T.A.; Møller, A.P. Ionizing radiation, antioxidant response and oxidative damage: A meta-analysis. Sci. Total Environ. 2016, 548–549, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.A.; Yoon, S.H.; Rho, J.K.; Jang, B.; Choi, D.S.; Lee, D.-E.; Byun, E.-B.; Jeon, J.; Park, S.H. Radioprotective effect of hesperetin against γ-irradiation-induced DNA damage and immune dysfunction in murine splenocytes. Food Sci. Biotechnol. 2016, 25, 163–168. [Google Scholar] [CrossRef]
- Rezaeyan, A.; Haddadi, G.; Hosseinzadeh, M.; Moradi, M.; Najafi, M. Radioprotective effects of hesperidin on oxidative damages and histopathological changes induced by X-irradiation in rats heart tissue. J. Med. Phys. 2016, 41, 182. [Google Scholar] [CrossRef]
- Shaban, N.Z.; Ahmed Zahran, A.M.; El-Rashidy, F.H.; Abdo Kodous, A.S. Protective role of hesperidin against γ-radiation-induced oxidative stress and apoptosis in rat testis. J. Biol. Res.-Thessalon. 2017, 24, 5. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, C.B.; Abe, J.; Patel, Z.S.; Grande-Allen, K.J. Radiation-Induced Cardiovascular Disease: Mechanisms and Importance of Linear Energy Transfer. Front. Cardiovasc. Med. 2018, 5, 5. [Google Scholar] [CrossRef]
- Helm, J.S.; Rudel, R.A. Adverse outcome pathways for ionizing radiation and breast cancer involve direct and indirect DNA damage, oxidative stress, inflammation, genomic instability, and interaction with hormonal regulation of the breast. Arch. Toxicol. 2020, 94, 1511–1549. [Google Scholar] [CrossRef] [PubMed]
- Doukali, H.; Ben Salah, G.; Hamdaoui, L.; Hajjaji, M.; Tabebi, M.; Ammar-Keskes, L.; Masmoudi, M.-E.; Kamoun, H. Oxidative stress and glutathione S-transferase genetic polymorphisms in medical staff professionally exposed to ionizing radiation. Int. J. Radiat. Biol. 2017, 93, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, S.J.; Bhatti, P.; Brown, E.E.; Linet, M.S.; Simon, S.L.; Weinstock, R.M.; Hutchinson, A.A.; Stovall, M.; Preston, D.L.; Alexander, B.H.; et al. Polymorphisms in oxidative stress and inflammation pathway genes, low-dose ionizing radiation, and the risk of breast cancer among US radiologic technologists. Cancer Causes Control 2010, 21, 1857–1866. [Google Scholar] [CrossRef]
- Møller, A.P.; Mousseau, T.A. Strong effects of ionizing radiation from Chernobyl on mutation rates. Sci. Rep. 2015, 5, 8363. [Google Scholar] [CrossRef] [PubMed]
- Belli, M.; Tabocchini, M.A. Ionizing Radiation-Induced Epigenetic Modifications and Their Relevance to Radiation Protection. Int. J. Mol. Sci. 2020, 21, 5993. [Google Scholar] [CrossRef]
- Su, S.; Jin, Y.; Zhang, W.; Yang, L.; Shen, Y.; Cao, Y.; Tong, J. Aberrant Promoter Methylation of p16 INK4a and O 6 -Methylguanine-DNA Methyltransferase Genes in Workers at a Chinese Uranium Mine. J. Occup. Health 2006, 48, 261–266. [Google Scholar] [CrossRef]
- Romanenko, A.; Morell-Quadreny, L.; Lopez-Guerrero, J.A.; Pellin, A.; Nepomnyaschy, V.; Vozianov, A.; Llombart-Bosch, A. The INK4a/ARF locus: Role in cell cycle control for renal cell epithelial tumor growth after the Chernobyl accident. Virchows Arch. 2004, 445, 298–304. [Google Scholar] [CrossRef]
- Szumiel, I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: The pivotal role of mitochondria. Int. J. Radiat. Biol. 2015, 91, 1–12. [Google Scholar] [CrossRef]
- Zheng, F.; Gonçalves, F.M.; Abiko, Y.; Li, H.; Kumagai, Y.; Aschner, M. Redox toxicology of environmental chemicals causing oxidative stress. Redox Biol. 2020, 34, 101475. [Google Scholar] [CrossRef]
- Sun, H.; He, F.; Choi, W. Production of Reactive Oxygen Species by the Reaction of Periodate and Hydroxylamine for Rapid Removal of Organic Pollutants and Waterborne Bacteria. Environ. Sci. Technol. 2020, 54, 6427–6437. [Google Scholar] [CrossRef]
- Lelieveld, S.; Wilson, J.; Dovrou, E.; Mishra, A.; Lakey, P.S.J.; Shiraiwa, M.; Pöschl, U.; Berkemeier, T. Hydroxyl Radical Production by Air Pollutants in Epithelial Lining Fluid Governed by Interconversion and Scavenging of Reactive Oxygen Species. Environ. Sci. Technol. 2021, 55, 14069–14079. [Google Scholar] [CrossRef]
- Paithankar, J.G.; Saini, S.; Dwivedi, S.; Sharma, A.; Chowdhuri, D.K. Heavy metal associated health hazards: An interplay of oxidative stress and signal transduction. Chemosphere 2021, 262, 128350. [Google Scholar] [CrossRef]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic Mechanisms of Five Heavy Metals: Mercury, Lead, Chromium, Cadmium, and Arsenic. Front. Pharmacol. 2021, 12, 643972. [Google Scholar] [CrossRef]
- Exley, C. Aluminum in Biological Systems. In Encyclopedia of Metalloproteins; Kretsinger, R.H., Uversky, V.N., Permyakov, E.A., Eds.; Springer New York: New York, NY, USA, 2013; pp. 33–34. ISBN 978-1-4614-1532-9. [Google Scholar]
- Hossain, M.A.; Piyatida, P.; da Silva, J.A.T.; Fujita, M. Molecular Mechanism of Heavy Metal Toxicity and Tolerance in Plants: Central Role of Glutathione in Detoxification of Reactive Oxygen Species and Methylglyoxal and in Heavy Metal Chelation. J. Bot. 2012, 2012, 1–37. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Luo, N.; Chen, Z.; Wu, K.; Yin, G. Redox inactive metal ion triggered N-dealkylation by an iron catalyst with dioxygen activation: A lesson from lipoxygenases. Dalton Trans. 2015, 44, 9847–9859. [Google Scholar] [CrossRef]
- Niu, P.Y.; Niu, Q.; Zhang, Q.L.; Wang, L.P.; He, S.C.; Wu, T.C.; Conti, P.; Di Gioacchino, M.; Boscolo, P. Aluminum Impairs Rat Neural Cell Mitochondria In Vitro. Int. J. Immunopathol. Pharmacol. 2005, 18, 683–689. [Google Scholar] [CrossRef]
- Wei, W.; Smith, N.; Wu, X.; Kim, H.; Seravalli, J.; Khalimonchuk, O.; Lee, J. YCF1-Mediated Cadmium Resistance in Yeast Is Dependent on Copper Metabolism and Antioxidant Enzymes. Antioxid. Redox Signal. 2014, 21, 1475–1489. [Google Scholar] [CrossRef]
- Saito, K.; Nakagawa, M.; Mandal, M.; Ishikita, H. Role of redox-inactive metals in controlling the redox potential of heterometallic manganese–oxido clusters. Photosynth. Res. 2021, 148, 153–159. [Google Scholar] [CrossRef]
- Jayaraj, R.; Megha, P.; Sreedev, P. Review Article. Organochlorine pesticides, their toxic effects on living organisms and their fate in the environment. Interdiscip. Toxicol. 2016, 9, 90–100. [Google Scholar] [CrossRef]
- Georgiadis, N.; Tsarouhas, K.; Tsitsimpikou, C.; Vardavas, A.; Rezaee, R.; Germanakis, I.; Tsatsakis, A.; Stagos, D.; Kouretas, D. Pesticides and cardiotoxicity. Where do we stand? Toxicol. Appl. Pharmacol. 2018, 353, 1–14. [Google Scholar] [CrossRef]
- Litchfield, M.H. Estimates of Acute Pesticide Poisoning in Agricultural Workers in Less Developed Countries: Toxicol. Rev. 2005, 24, 271–278. [Google Scholar] [CrossRef]
- Dou, T.; Yan, M.; Wang, X.; Lu, W.; Zhao, L.; Lou, D.; Wu, C.; Chang, X.; Zhou, Z. Nrf2/ARE Pathway Involved in Oxidative Stress Induced by Paraquat in Human Neural Progenitor Cells. Oxid. Med. Cell Longev. 2016, 2016, 8923860. [Google Scholar] [CrossRef]
- Ranjbar, A.; Pasalar, P.; Sedighi, A.; Abdollahi, M. Induction of oxidative stress in paraquat formulating workers. Toxicol. Lett. 2002, 131, 191–194. [Google Scholar] [CrossRef]
- Abass, K.; Lämsä, V.; Reponen, P.; Küblbeck, J.; Honkakoski, P.; Mattila, S.; Pelkonen, O.; Hakkola, J. Characterization of human cytochrome P450 induction by pesticides. Toxicology 2012, 294, 17–26. [Google Scholar] [CrossRef]
- Delescluse, C.; Ledirac, N.; Li, R.; Piechocki, M.P.; Hines, R.N.; Gidrol, X.; Rahmani, R. Induction of cytochrome P450 1A1 gene expression, oxidative stress, and genotoxicity by carbaryl and thiabendazole in transfected human HepG2 and lymphoblastoid cells. Biochem. Pharmacol. 2001, 61, 399–407. [Google Scholar] [CrossRef]
- Puntarulo, S.; Cederbaum, A.I. Production of Reactive Oxygen Species by Microsomes Enriched in Specific Human Cytochrome P450 Enzymes. Free Radic. Biol. Med. 1998, 24, 1324–1330. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Prakasam, A.; Sethupathy, S.; Lalitha, S. Plasma and RBCs antioxidant status in occupational male pesticide sprayers. Clin. Chim. Acta 2001, 310, 107–112. [Google Scholar] [CrossRef]
- Sule, R.O.; Condon, L.; Gomes, A.V. A Common Feature of Pesticides: Oxidative Stress—The Role of Oxidative Stress in Pesticide-Induced Toxicity. Oxid. Med. Cell. Longev. 2022, 2022, 5563759. [Google Scholar] [CrossRef] [PubMed]
- Carloni, M.; Nasuti, C.; Fedeli, D.; Montani, M.; Vadhana, M.S.D.; Amici, A.; Gabbianelli, R. Early life permethrin exposure induces long-term brain changes in Nurr1, NF-kB and Nrf-2. Brain Res. 2013, 1515, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Yamamoto, N.; Matsushima, S.; Yamamoto, T.; Takada-Takatori, Y.; Akaike, A.; Kume, T. Compensatory role of the Nrf2–ARE pathway against paraquat toxicity: Relevance of 26S proteasome activity. J. Pharmacol. Sci. 2015, 129, 150–159. [Google Scholar] [CrossRef]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell Biol. 2015, 36, 271–284. [Google Scholar] [CrossRef]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758.e4. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Gangwar, R.S.; Bevan, G.H.; Palanivel, R.; Das, L.; Rajagopalan, S. Oxidative stress pathways of air pollution mediated toxicity: Recent insights. Redox Biol. 2020, 34, 101545. [Google Scholar] [CrossRef]
- Mukherjee, A.; Agrawal, M. World air particulate matter: Sources, distribution and health effects. Environ. Chem. Lett. 2017, 15, 283–309. [Google Scholar] [CrossRef]
- Li, N.; Sioutas, C.; Cho, A.; Schmitz, D.; Misra, C.; Sempf, J.; Wang, M.; Oberley, T.; Froines, J.; Nel, A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ. Health Perspect. 2003, 111, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Mazuryk, O.; Stochel, G.; Brindell, M. Variations in Reactive Oxygen Species Generation by Urban Airborne Particulate Matter in Lung Epithelial Cells—Impact of Inorganic Fraction. Front. Chem. 2020, 8, 581752. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, L.S.; Fleck, A.d.S.; Zanchi, A.C.; Saldiva, P.H.N.; Rhoden, C.R. Direct contact with particulate matter increases oxidative stress in different brain structures. Inhal. Toxicol. 2015, 27, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Lawal, A.O. Air particulate matter induced oxidative stress and inflammation in cardiovascular disease and atherosclerosis: The role of Nrf2 and AhR-mediated pathways. Toxicol. Lett. 2017, 270, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Dimova, E.Y.; Petry, A.; Martínez-Ruiz, A.; Hernansanz-Agustín, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, T.; Li, M.; Shi, D.; Tan, X.; Qiu, F. Lycopene attenuates d -galactose-induced insulin signaling impairment by enhancing mitochondrial function and suppressing the oxidative stress/inflammatory response in mouse kidneys and livers. Food Funct. 2022, 13, 7720–7729. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef]
- Motamed, M.; Nargesi, A.; Heidari, B.; Mirmiranpour, H.; Esteghamati, A.; Nakhjavani, M. Oxidized Low-Density Lipoprotein (ox-LDL) to LDL Ratio (ox-LDL/LDL) and ox-LDL to High-Density Lipoprotein Ratio (ox-LDL/HDL). Clin. Lab. 2016, 62, 1609–1617. [Google Scholar] [CrossRef]
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.-Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and TNF-α on appetite-regulating NPY neurons. Int. J. Obes. 2017, 41, 149–158. [Google Scholar] [CrossRef]
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Greenamyre, J.T. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell Mol. Life Sci. 2017, 74, 2851–2874. [Google Scholar] [CrossRef]
- Jiang, S.; Liu, H.; Li, C. Dietary Regulation of Oxidative Stress in Chronic Metabolic Diseases. Foods 2021, 10, 1854. [Google Scholar] [CrossRef] [PubMed]
- Dguzeh, U.; Haddad, N.; Smith, K.; Johnson, J.; Doye, A.; Gwathmey, J.; Haddad, G. Alcoholism: A Multi-Systemic Cellular Insult to Organs. Int. J. Environ. Res. Public. Health 2018, 15, 1083. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Liu, Y.; Chang, X.; Gou, W.; Zhou, X.; Liu, Z.; Li, Z.; Wu, Y.; Zuo, D. Acetaldehyde Induces Neurotoxicity In Vitro via Oxidative Stress- and Ca 2+ Imbalance-Mediated Endoplasmic Reticulum Stress. Oxid. Med. Cell Longev. 2019, 2019, 2593742. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cederbaum, A.I. Alcohol, Oxidative Stress, and Free Radical Damage. Alcohol Res. Health 2003, 27, 277–284. [Google Scholar] [PubMed]
- Baker, R.R.; Massey, E.D.; Smith, G. An overview of the effects of tobacco ingredients on smoke chemistry and toxicity. Food Chem. Toxicol. 2004, 42, 53–83. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.P.; Reddy, A.T.; Kodidhela, L.D.; Varadacharyulu, N.C. Epigallocatechin gallate diminishes cigarette smoke-induced oxidative stress, lipid peroxidation, and inflammation in human bronchial epithelial cells. Life Sci. 2020, 259, 118260. [Google Scholar] [CrossRef] [PubMed]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-Induced Oxidative Stress and Toxicity. J. Toxicol. 2012, 2012, 645460. [Google Scholar] [CrossRef] [PubMed]
- Chignell, C.F.; Motten, A.G.; Buettner, G.R. Photoinduced free radicals from chlorpromazine and related phenothiazines: Relationship to phenothiazine-induced photosensitization. Environ. Health Perspect. 1985, 64, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Micallef, I.; Baron, B. Doxorubicin: An Overview of the Anti-Cancer and Chemoresistance Mechanisms. Ann. Clin. Toxicol. 2020, 3, 12. [Google Scholar]
- Miyake, M.; Nakai, Y.; Hori, S.; Morizawa, Y.; Hirao, Y.; Fujimoto, K. Transient liver toxicity as a result of the oral administration of 5-aminolevulinic acid for photodynamic diagnosis in patients with bladder cancer. Int. J. Urol. 2019, 26, 315–317. [Google Scholar] [CrossRef]
- Sánchez-Borges, M.; Capriles-Hulett, A.; Caballero-Fonseca, F. Risk of skin reactions when using ibuprofen-based medicines. Expert Opin. Drug Saf. 2005, 4, 837–848. [Google Scholar] [CrossRef]
- Bagheri, H.; Lhiaubet, V.; Montastruc, J.L.; Chouini-Lalanne, N. Photosensitivity to Ketoprofen: Mechanisms and Pharmacoepidemiological Data. Drug Saf. 2000, 22, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, I.; Persson, E.; Ekebergh, A.; Mårtensson, J.; Börje, A. Ketoprofen-Induced Formation of Amino Acid Photoadducts: Possible Explanation for Photocontact Allergy to Ketoprofen. Chem. Res. Toxicol. 2014, 27, 1294–1303. [Google Scholar] [CrossRef]
- Onoue, S.; Igarashi, N.; Yamada, S.; Tsuda, Y. High-throughput reactive oxygen species (ROS) assay: An enabling technology for screening the phototoxic potential of pharmaceutical substances. J. Pharm. Biomed. Anal. 2008, 46, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Akat, P. Severe photosensitivity reaction induced by topical diclofenac. Indian J. Pharmacol. 2013, 45, 408. [Google Scholar] [CrossRef]
- Lhiaubet, V.; Paillous, N.; Chouini-Lalanne, N. Comparison of DNA Damage Photoinduced by Ketoprofen, Fenofibric Acid and Benzophenone via Electron and Energy Transfer. Photochem. Photobiol. 2007, 74, 670–678. [Google Scholar] [CrossRef]
- Kowalska, J.; Rok, J.; Rzepka, Z.; Wrześniok, D. Drug-Induced Photosensitivity—From Light and Chemistry to Biological Reactions and Clinical Symptoms. Pharmaceuticals 2021, 14, 723. [Google Scholar] [CrossRef] [PubMed]
- Köroğlu, K.M.; Çevik, Ö.; Şener, G.; Ercan, F. Apocynin alleviates cisplatin-induced testicular cytotoxicity by regulating oxidative stress and apoptosis in rats. Andrologia 2019, 51, e13227. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yan, M.-H.; Liu, Y.; Liu, Z.; Wang, Z.; Chen, C.; Zhang, J.; Sun, Y.-S. Ginsenoside Rg5 Ameliorates Cisplatin-Induced Nephrotoxicity in Mice through Inhibition of Inflammation, Oxidative Stress, and Apoptosis. Nutrients 2016, 8, 566. [Google Scholar] [CrossRef] [PubMed]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxid. Med. Cell Longev. 2018, 2018, 7582730. [Google Scholar] [CrossRef]
- Ghosh, R.; Hwang, S.M.; Cui, Z.; Gilda, J.E.; Gomes, A.V. Different effects of the nonsteroidal anti-inflammatory drugs meclofenamate sodium and naproxen sodium on proteasome activity in cardiac cells. J. Mol. Cell Cardiol. 2016, 94, 131–144. [Google Scholar] [CrossRef]
- Щекіна, К.Г.; Belik, G.; Semeniv, D.; Ulanova, V. A comparative study of the hepatotrophic properties of non-steroidal anti-inflammatory drugs. Ukr. Biopharm. J. 2021, 66, 36–41. [Google Scholar] [CrossRef]
- Martens, M.E.; Lee, C.-P. Reye’s syndrome: Salicylates and mitochondrial functions. Biochem. Pharmacol. 1984, 33, 2869–2876. [Google Scholar] [CrossRef]
- Jiang, J.; Briedé, J.J.; Jennen, D.G.J.; Van Summeren, A.; Saritas-Brauers, K.; Schaart, G.; Kleinjans, J.C.S.; de Kok, T.M.C.M. Increased mitochondrial ROS formation by acetaminophen in human hepatic cells is associated with gene expression changes suggesting disruption of the mitochondrial electron transport chain. Toxicol. Lett. 2015, 234, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R. The epidemiology of acute renal failure: 1975 versus 2005: Curr. Opin. Crit. Care 2006, 12, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Ghane Shahrbaf, F. Drug-induced renal disorders. Drug-Induc. Ren. Disord. 2015, 4, 57–60. [Google Scholar] [CrossRef]
- Perneger, T.V.; Whelton, P.K.; Klag, M.J. Risk of Kidney Failure Associated with the Use of Acetaminophen, Aspirin, and Nonsteroidal Antiinflammatory Drugs. N. Engl. J. Med. 1994, 331, 1675–1679. [Google Scholar] [CrossRef]
- Huang, C.; Chen, J.; Kuo, C.; Pai, P.; Ho, T.; Chen, T.; Tsai, F.; Padma, V.V.; Kuo, W.; Huang, C. Mitochondrial ROS-induced ERK1/2 activation and HSF2-mediated AT 1 R upregulation are required for doxorubicin-induced cardiotoxicity. J. Cell Physiol. 2018, 233, 463–475. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.-R.; Rossner, P.; Haghdoost, S.; Jeng, H.A.; Hu, C.-W. Nucleic Acid Oxidation in Human Health and Disease. Oxid. Med. Cell Longev. 2013, 2013, 368651. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Van Remmen, H.; Hamilton, M.L.; Richardson, A. Oxidative Damage to DNA and Aging. Exerc. Sport Sci. Rev. 2003, 31, 149–153. [Google Scholar] [CrossRef]
- Bruic, M.; Grujic-Milanovic, J.; Miloradovic, Z.; Jovovic, D.; Zivkovic, L.; Mihailovic-Stanojevic, N.; Karanovic, D.; Spremo-Potparevic, B. DNA, protein and lipid oxidative damage in tissues of spontaneously hypertensive versus normotensive rats. Int. J. Biochem. Cell Biol. 2021, 141, 106088. [Google Scholar] [CrossRef]
- Besaratinia, A.; Caliri, A.W.; Tommasi, S. Hydroxychloroquine induces oxidative DNA damage and mutation in mammalian cells. DNA Repair 2021, 106, 103180. [Google Scholar] [CrossRef]
- Fu, Z.; Xi, S. The effects of heavy metals on human metabolism. Toxicol. Mech. Methods 2020, 30, 167–176. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef]
- Gianazza, E.; Brioschi, M.; Martinez Fernandez, A.; Casalnuovo, F.; Altomare, A.; Aldini, G.; Banfi, C. Lipid Peroxidation in Atherosclerotic Cardiovascular Diseases. Antioxid. Redox Signal. 2021, 34, 49–98. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, H.; Nair, J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: Role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Spickett, C.M. The lipid peroxidation product 4-hydroxy-2-nonenal: Advances in chemistry and analysis. Redox Biol. 2013, 1, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Dyall, S.C.; Balas, L.; Bazan, N.G.; Brenna, J.T.; Chiang, N.; da Costa Souza, F.; Dalli, J.; Durand, T.; Galano, J.-M.; Lein, P.J.; et al. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: Recent advances in the understanding of their biosynthesis, structures, and functions. Prog. Lipid Res. 2022, 86, 101165. [Google Scholar] [CrossRef] [PubMed]
- Vangaveti, V.; Baune, B.T.; Kennedy, R.L. Hydroxyoctadecadienoic acids: Novel regulators of macrophage differentiation and atherogenesis. Ther. Adv. Endocrinol. Metab. 2010, 1, 51–60. [Google Scholar] [CrossRef]
- Schuster, S.; Johnson, C.D.; Hennebelle, M.; Holtmann, T.; Taha, A.Y.; Kirpich, I.A.; Eguchi, A.; Ramsden, C.E.; Papouchado, B.G.; McClain, C.J.; et al. Oxidized linoleic acid metabolites induce liver mitochondrial dysfunction, apoptosis, and NLRP3 activation in mice. J. Lipid Res. 2018, 59, 1597–1609. [Google Scholar] [CrossRef]
- Ramsden, C.E.; Ringel, A.; Feldstein, A.E.; Taha, A.Y.; MacIntosh, B.A.; Hibbeln, J.R.; Majchrzak-Hong, S.F.; Faurot, K.R.; Rapoport, S.I.; Cheon, Y.; et al. Lowering dietary linoleic acid reduces bioactive oxidized linoleic acid metabolites in humans. Prostaglandins Leukot. Essent. Fat. Acids 2012, 87, 135–141. [Google Scholar] [CrossRef]
- Shishehbor, M.H.; Zhang, R.; Medina, H.; Brennan, M.-L.; Brennan, D.M.; Ellis, S.G.; Topol, E.J.; Hazen, S.L. Systemic elevations of free radical oxidation products of arachidonic acid are associated with angiographic evidence of coronary artery disease. Free Radic. Biol. Med. 2006, 41, 1678–1683. [Google Scholar] [CrossRef]
- Mutemberezi, V.; Guillemot-Legris, O.; Muccioli, G.G. Oxysterols: From cholesterol metabolites to key mediators. Prog. Lipid Res. 2016, 64, 152–169. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Levy, D.; Bydlowski, S.P. Oxysterols and mesenchymal stem cell biology. In Vitamins and Hormones; Elsevier: Amsterdam, The Netherlands, 2021; Volume 116, pp. 409–436. ISBN 978-0-323-85550-1. [Google Scholar]
- Zhang, X.; Alhasani, R.H.; Zhou, X.; Reilly, J.; Zeng, Z.; Strang, N.; Shu, X. Oxysterols and retinal degeneration. Br. J. Pharmacol. 2021, 178, 3205–3219. [Google Scholar] [CrossRef]
- Samadi, A.; Sabuncuoglu, S.; Samadi, M.; Isikhan, S.Y.; Chirumbolo, S.; Peana, M.; Lay, I.; Yalcinkaya, A.; Bjørklund, G. A Comprehensive Review on Oxysterols and Related Diseases. Curr. Med. Chem. 2020, 28, 110–136. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2021, 41, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Jiang, W.; Wang, W.; Xiong, R.; Wu, X.; Geng, Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol. Res. 2021, 166, 105466. [Google Scholar] [CrossRef] [PubMed]
- Tudek, B.; Zdżalik-Bielecka, D.; Tudek, A.; Kosicki, K.; Fabisiewicz, A.; Speina, E. Lipid peroxidation in face of DNA damage, DNA repair and other cellular processes. Free Radic. Biol. Med. 2017, 107, 77–89. [Google Scholar] [CrossRef]
- Bayr, H. Reactive oxygen species. Crit. Care Med. 2005, 33, S498. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhu, X.; Sayre, L.M. Chemical nature of stochastic generation of protein-based carbonyls: Metal-catalyzed oxidation versus modification by products of lipid oxidation. Chem. Res. Toxicol. 2007, 20, 129–139. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Xie, H.; Griffin, T.J.; Bernlohr, D.A. Oxidative Stress and Covalent Modification of Protein with Bioactive Aldehydes. J. Biol. Chem. 2008, 283, 21837–21841. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Generation and propagation of radical reactions on proteins. Biochim. Biophys. Acta 2001, 1504, 196–219. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef]
- Fuentes-Lemus, E.; Hägglund, P.; López-Alarcón, C.; Davies, M.J. Oxidative Crosslinking of Peptides and Proteins: Mechanisms of Formation, Detection, Characterization and Quantification. Molecules 2021, 27, 15. [Google Scholar] [CrossRef]
- Angelova, P.R. Sources and triggers of oxidative damage in neurodegeneration. Free Radic. Biol. Med. 2021, 173, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Starosta, V.; Griese, M. Oxidative damage to surfactant protein D in pulmonary diseases. Free Radic. Res. 2006, 40, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.; Portero-Otin, M.; Pamplona, R.; Ferrer, I. Protein Targets of Oxidative Damage in Human Neurodegenerative Diseases with Abnormal Protein Aggregates. Brain Pathol. 2009, 20, 281–297. [Google Scholar] [CrossRef]
- Krisko, A.; Radman, M. Protein damage, ageing and age-related diseases. Open Biol. 2019, 9, 180249. [Google Scholar] [CrossRef] [PubMed]
- Godic, A.; Poljšak, B.; Adamic, M.; Dahmane, R. The role of antioxidants in skin cancer prevention and treatment. Oxid. Med. Cell Longev. 2014, 2014, 860479. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gregorio, A.; Manzo-Merino, J.; Lizano, M. Cellular redox, cancer and human papillomavirus. Virus Res. 2018, 246, 35–45. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.-F.; Teixeira, M.; Valentine, J.S. Superoxide dismutases and superoxide reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [CrossRef]
- Robbins, D.; Zhao, Y. Manganese superoxide dismutase in cancer prevention. Antioxid. Redox Signal. 2014, 20, 1628–1645. [Google Scholar] [CrossRef]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Castellano, I.; Cecere, F.; De Vendittis, A.; Cotugno, R.; Chambery, A.; Di Maro, A.; Michniewicz, A.; Parlato, G.; Masullo, M.; Avvedimento, E.V.; et al. Rat mitochondrial manganese superoxide dismutase: Amino acid positions involved in covalent modifications, activity, and heat stability. Biopolymers 2009, 91, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Crow, J.P.; Kerby, J.D.; Beckman, J.S.; Thompson, J.A. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc. Natl. Acad. Sci. USA 1996, 93, 11853–11858. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Cruthirds, D.L.; Ahki, K.M.; Sanders, P.W.; Thompson, J.A. Mitochondrial tyrosine nitration precedes chronic allograft nephropathy. Free Radic. Biol. Med. 2001, 31, 1603–1608. [Google Scholar] [CrossRef]
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-Mediated Inactivation of Manganese Superoxide Dismutase Involves Nitration and Oxidation of Critical Tyrosine Residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Aebi, H. Catalase In Vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Glorieux, C.; Sandoval, J.M.; Fattaccioli, A.; Dejeans, N.; Garbe, J.C.; Dieu, M.; Verrax, J.; Renard, P.; Huang, P.; Calderon, P.B. Chromatin remodeling regulates catalase expression during cancer cells adaptation to chronic oxidative stress. Free Radic. Biol. Med. 2016, 99, 436–450. [Google Scholar] [CrossRef]
- Glorieux, C.; Sandoval, J.M.; Dejeans, N.; Nonckreman, S.; Bahloula, K.; Poirel, H.A.; Calderon, P.B. Evaluation of Potential Mechanisms Controlling the Catalase Expression in Breast Cancer Cells. Oxid. Med. Cell Longev. 2018, 2018, 5351967. [Google Scholar] [CrossRef]
- Flor, S.; Oliva, C.R.; Ali, M.Y.; Coleman, K.L.; Greenlee, J.D.; Jones, K.A.; Monga, V.; Griguer, C.E. Catalase Overexpression Drives an Aggressive Phenotype in Glioblastoma. Antioxid. Basel Switz. 2021, 10, 1988. [Google Scholar] [CrossRef]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas-Rodríguez, N.; Pedraza-Chaverri, J. Especies reactivas de oxígeno y sistemas antioxidantes: Aspectos básicos. Profr. Al Día Biomed. 2005, 17, 164–173. [Google Scholar]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef]
- Zhu, H.; Santo, A.; Li, Y. The antioxidant enzyme peroxiredoxin and its protective role in neurological disorders. Exp. Biol. Med. Maywood NJ 2012, 237, 143–149. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Flohé, L.; Brigelius-Flohé, R. Selenoproteins of the Glutathione Peroxidase Family. In Selenium; Springer: New York, NY, USA, 2011; pp. 167–180. [Google Scholar]
- Rohr-Udilova, N.; Bauer, E.; Timelthaler, G.; Eferl, R.; Stolze, K.; Pinter, M.; Seif, M.; Hayden, H.; Reiberger, T.; Schulte-Hermann, R.; et al. Impact of glutathione peroxidase 4 on cell proliferation, angiogenesis and cytokine production in hepatocellular carcinoma. Oncotarget 2018, 9, 10054–10068. [Google Scholar] [CrossRef]
- Espinoza, S.E.; Guo, H.; Fedarko, N.; DeZern, A.; Fried, L.P.; Xue, Q.-L.; Leng, S.; Beamer, B.; Walston, J.D. Glutathione peroxidase enzyme activity in aging. J. Gerontol. A. Biol. Sci. Med. Sci. 2008, 63, 505–509. [Google Scholar] [CrossRef]
- Sarıkaya, E.; Doğan, S. Glutathione Peroxidase in Health and Diseases; IntechOpen: London, UK, 2020; ISBN 978-1-83880-126-7. [Google Scholar]
- Zhou, Y.; Lin, W.; Rao, T.; Zheng, J.; Zhang, T.; Zhang, M.; Lin, Z. Ferroptosis and Its Potential Role in the Nervous System Diseases. J. Inflamm. Res. 2022, 15, 1555–1574. [Google Scholar] [CrossRef]
- Saunders, N.; Walshe, J.; Serewko-Auret, M.; Chu, F.-F.; Esworthy, S.; Burcham, P.; Reeve, V. Glutathione peroxidase deficiency is a predisposing factor for UV-induced SCC in humans. Cancer Res. 2007, 67, 2632. [Google Scholar]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.-H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappalà, A.; Li Volti, G. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef]
- Deshane, J.; Wright, M.; Agarwal, A. Heme oxygenase-1 expression in disease states. Acta Biochim. Pol. 2005, 52, 273–284. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta BBA-Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Gregg, X.T.; Prchal, J.T. Chapter 44—Red Blood Cell Enzymopathies. In Hematology, 7th ed.; Hoffman, R., Benz, E.J., Silberstein, L.E., Heslop, H.E., Weitz, J.I., Anastasi, J., Salama, M.E., Abutalib, S.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 616–625. ISBN 978-0-323-35762-3. [Google Scholar]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef]
- Yan, C.; Duan, L.; Fu, C.; Tian, C.; Zhang, B.; Shao, X.; Zhu, G. Association Between Glutathione S-Transferase (GST) Polymorphisms and Schizophrenia in a Chinese Han Population. Neuropsychiatr. Dis. Treat. 2020, 16, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, M.; Nisticò, S.; Noce, A.; Tarantino, A.; Marrone, G.; Costa, A.; Rovella, V.; Di Cola, G.; Campia, U.; Lauro, D.; et al. The possible role of glutathione-S-transferase activity in diabetic nephropathy. Int. J. Immunopathol. Pharmacol. 2015, 28, 129–133. [Google Scholar] [CrossRef]
- Singh, R.R.; Reindl, K.M. Glutathione S-Transferases in Cancer. Antioxidants 2021, 10, 701. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and Thioredoxin Target Proteins: From Molecular Mechanisms to Functional Significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.H.; Yang, X.; Choi, Y.E.; Jones, D.P.; Kehrer, J.P. Thioredoxin and its role in toxicology. Toxicol. Sci. Off. J. Soc. Toxicol. 2004, 78, 3–14. [Google Scholar] [CrossRef]
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system1 1This review is based on the licentiate thesis “Thioredoxin reductase—Interactions with the redox active compounds 1-chloro-2,4-dinitrobenzene and lipoic acid” by Jonas Nordberg. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Mahmood, D.F.D.; Abderrazak, A.; El Hadri, K.; Simmet, T.; Rouis, M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid. Redox Signal. 2013, 19, 1266–1303. [Google Scholar] [CrossRef]
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346 Pt 1, 1–8. [Google Scholar] [CrossRef]
- Patwardhan, R.S.; Sharma, D.; Sandur, S.K. Thioredoxin reductase: An emerging pharmacologic target for radiosensitization of cancer. Transl. Oncol. 2022, 17, 101341. [Google Scholar] [CrossRef]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous non-enzymatic antioxidants in the human body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Moussa, Z.; Judeh, Z.M.A.; Ahmed, S.A. Nonenzymatic Exogenous and Endogenous Antioxidants. In Free Radical Medicine and Biology; Das, K., Das, S., Shivanagouda Biradar, M., Bobbarala, V., Subba Tata, S., Eds.; IntechOpen: London, UK, 2020; ISBN 978-1-78985-143-4. [Google Scholar]
- Janciauskiene, S. The Beneficial Effects of Antioxidants in Health and Diseases. Chronic Obstr. Pulm. Dis. J. COPD Found. 2020, 7, 182–202. [Google Scholar] [CrossRef] [PubMed]
- Noctor, G.; Queval, G.; Mhamdi, A.; Chaouch, S.; Foyer, C.H. Glutathione. Arab. Book 2011, 9, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Moinova, H.R.; Mulcahy, R.T. Up-Regulation of the Human γ-Glutamylcysteine Synthetase Regulatory Subunit Gene Involves Binding of Nrf-2 to an Electrophile Responsive Element. Biochem. Biophys. Res. Commun. 1999, 261, 661–668. [Google Scholar] [CrossRef]
- Giustarini, D.; Colombo, G.; Garavaglia, M.L.; Astori, E.; Portinaro, N.M.; Reggiani, F.; Badalamenti, S.; Aloisi, A.M.; Santucci, A.; Rossi, R.; et al. Assessment of glutathione/glutathione disulphide ratio and S-glutathionylated proteins in human blood, solid tissues, and cultured cells. Free Radic. Biol. Med. 2017, 112, 360–375. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. bchm 2009, 390, 191–214. [Google Scholar] [CrossRef]
- Iskusnykh, I.Y.; Zakharova, A.A.; Pathak, D. Glutathione in Brain Disorders and Aging. Molecules 2022, 27, 324. [Google Scholar] [CrossRef]
- Korge, P.; Calmettes, G.; Weiss, J.N. Increased reactive oxygen species production during reductive stress: The roles of mitochondrial glutathione and thioredoxin reductases. Biochim. Biophys. Acta BBA-Bioenerg. 2015, 1847, 514–525. [Google Scholar] [CrossRef]
- Alli, J.A.; Kehinde, A.O.; Kosoko, A.M.; Ademowo, O.G. Oxidative Stress and Reduced Vitamins C and E Levels Are Associated with Multi-Drug Resistant Tuberculosis. J. Tuberc. Res. 2014, 02, 52–58. [Google Scholar] [CrossRef]
- Chatterjee, A. Reduced Glutathione: A Radioprotector or a Modulator of DNA-Repair Activity? Nutrients 2013, 5, 525–542. [Google Scholar] [CrossRef]
- Tram, N.K.; McLean, R.M.; Swindle-Reilly, K.E. Glutathione Improves the Antioxidant Activity of Vitamin C in Human Lens and Retinal Epithelial Cells: Implications for Vitreous Substitutes. Curr. Eye Res. 2021, 46, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed]
- Christ-Hazelhof, E.; Nugteren, D.H. Purification and characterisation of prostaglandin endoperoxide D-isomerase, a cytoplasmic, glutathione-requiring enzymE. Biochim. Biophys. Acta BBA-Lipids Lipid Metab. 1979, 572, 43–51. [Google Scholar] [CrossRef]
- Layrisse, M.; Martínez-Torres, C.; Leets, I.; Taylor, P.; Ramírez, J. Effect of Histidine, Cysteine, Glutathione or Beef on Iron Absorption in Humans. J. Nutr. 1984, 114, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders: Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P. Role of glutathione in immunity and inflammation in the lung. Int. J. Gen. Med. 2011, 4, 105–113. [Google Scholar] [CrossRef]
- Quinlan, G.J.; Martin, G.S.; Evans, T.W. Albumin: Biochemical properties and therapeutic potential. Hepatology 2005, 41, 1211–1219. [Google Scholar] [CrossRef]
- Ronit, A.; Kirkegaard-Klitbo, D.M.; Dohlmann, T.L.; Lundgren, J.; Sabin, C.A.; Phillips, A.N.; Nordestgaard, B.G.; Afzal, S. Plasma Albumin and Incident Cardiovascular Disease: Results From the CGPS and an Updated Meta-Analysis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 473–482. [Google Scholar] [CrossRef]
- Health, Aging and Body Composition Study; Visser, M.; Kritchevsky, S.B.; Newman, A.B.; Goodpaster, B.H.; Tylavsky, F.A.; Nevitt, M.C.; Harris, T.B. Lower serum albumin concentration and change in muscle mass: The Health, Aging and Body Composition Study. Am. J. Clin. Nutr. 2005, 82, 531–537. [Google Scholar] [CrossRef]
- Gomi, I.; Fukushima, H.; Shiraki, M.; Miwa, Y.; Ando, T.; Takai, K.; Moriwaki, H. Relationship between Serum Albumin Level and Aging in Community-Dwelling Self-Supported Elderly Population. J. Nutr. Sci. Vitaminol. 2007, 53, 37–42. [Google Scholar] [CrossRef]
- Lloyd, D.R.; Phillips, D.H. Oxidative DNA damage mediated by copper(II), iron(II) and nickel(II) Fenton reactions: Evidence for site-specific mechanisms in the formation of double-strand breaks, 8-hydroxydeoxyguanosine and putative intrastrand cross-links. Mutat. Res. Mol. Mech. Mutagen. 1999, 424, 23–36. [Google Scholar] [CrossRef]
- Di Simplicio, P.; Cheeseman, K.H.; Slater, T.F. The Reactivity of the Sh Group of Bovine Serum Albumin with Free Radicals. Free Radic. Res. Commun. 1991, 14, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Otagiri, M.; Chuang, V.T.G. Pharmaceutically Important Pre- and Posttranslational Modifications on Human Serum Albumin. Biol. Pharm. Bull. 2009, 32, 527–534. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; di Masi, A.; Ascenzi, P. Serum Albumin: A Multifaced Enzyme. Int. J. Mol. Sci. 2021, 22, 10086. [Google Scholar] [CrossRef]
- Taverna, M.; Marie, A.-L.; Mira, J.-P.; Guidet, B. Specific antioxidant properties of human serum albumin. Ann. Intensive Care 2013, 3, 4. [Google Scholar] [CrossRef]
- Roche, M.; Rondeau, P.; Singh, N.R.; Tarnus, E.; Bourdon, E. The antioxidant properties of serum albumin. FEBS Lett. 2008, 582, 1783–1787. [Google Scholar] [CrossRef]
- Rondeau, P.; Bourdon, E. The glycation of albumin: Structural and functional impacts. Biochimie 2011, 93, 645–658. [Google Scholar] [CrossRef]
- McGuinness, A.; Sapey, E. Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. J. Clin. Med. 2017, 6, 21. [Google Scholar] [CrossRef]
- Bomholt, T.; Adrian, T.; Nørgaard, K.; Ranjan, A.G.; Almdal, T.; Larsson, A.; Vadstrup, M.; Rix, M.; Feldt-Rasmussen, B.; Hornum, M. The Use of HbA1c, Glycated Albumin and Continuous Glucose Monitoring to Assess Glucose Control in the Chronic Kidney Disease Population Including Dialysis. Nephron 2021, 145, 14–19. [Google Scholar] [CrossRef]
- Peacock, T.P.; Shihabi, Z.K.; Bleyer, A.J.; Dolbare, E.L.; Byers, J.R.; Knovich, M.A.; Calles-Escandon, J.; Russell, G.B.; Freedman, B.I. Comparison of glycated albumin and hemoglobin A1c levels in diabetic subjects on hemodialysis. Kidney Int. 2008, 73, 1062–1068. [Google Scholar] [CrossRef]
- Nocentini, A.; Bonardi, A.; Pratesi, S.; Gratteri, P.; Dani, C.; Supuran, C.T. Pharmaceutical strategies for preventing toxicity and promoting antioxidant and anti-inflammatory actions of bilirubin. J. Enzym. Inhib. Med. Chem. 2022, 37, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Sticova, E. New insights in bilirubin metabolism and their clinical implications. World J. Gastroenterol. 2013, 19, 6398. [Google Scholar] [CrossRef]
- Bhutani, V.K.; Wong, R.J.; Stevenson, D.K. Hyperbilirubinemia in Preterm Neonates. Clin. Perinatol. 2016, 43, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wei, X.; Bales, K.R.; Paul, A.B.C.; Ma, Z.; Yan, G.; Paul, S.M.; Du, Y. Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced cerebellar hypoplasia in the Gunn rat. Eur. J. Neurosci. 2005, 22, 21–27. [Google Scholar] [CrossRef]
- Zhou, Z.-X.; Chen, J.-K.; Hong, Y.-Y.; Zhou, R.; Zhou, D.-M.; Sun, L.-Y.; Qin, W.-L.; Wang, T.-C. Relationship Between the Serum Total Bilirubin and Inflammation in Patients With Psoriasis Vulgaris: Relationship Between the Serum Total Bilirubin and Inflammation. J. Clin. Lab. Anal. 2016, 30, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Ziberna, L.; Martelanc, M.; Franko, M.; Passamonti, S. Bilirubin is an Endogenous Antioxidant in Human Vascular Endothelial Cells. Sci. Rep. 2016, 6, 29240. [Google Scholar] [CrossRef]
- Liu, X.-T. Relationship between bilirubin free radical and formation of pigment gallstone. World J. Gastroenterol. 2002, 8, 413. [Google Scholar] [CrossRef] [PubMed]
- DiNicolantonio, J.J.; McCarty, M.F.; O’Keefe, J.H. Antioxidant bilirubin works in multiple ways to reduce risk for obesity and its health complications. Open Heart 2018, 5, e000914. [Google Scholar] [CrossRef]
- Hinds, T.D.; Stec, D.E. Bilirubin Safeguards Cardiorenal and Metabolic Diseases: A Protective Role in Health. Curr. Hypertens. Rep. 2019, 21, 87. [Google Scholar] [CrossRef]
- Lacko, M.; Roelofs, H.M.J.; te Morsche, R.H.M.; Voogd, A.C.; Ophuis, M.B.O.; Peters, W.H.M.; Manni, J.J. Genetic polymorphism in the conjugating enzyme UGT1A1 and the risk of head and neck cancer. Int. J. Cancer 2010, 127, 2815–2821. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Liao, Y.; Chen, X.; Mai, N.; Ouyang, C.; Chen, B.; Zhang, M.; Peng, Q.; Liang, W.; Zhang, W.; et al. Abnormal Serum Bilirubin/Albumin Concentrations in Dementia Patients With Aβ Deposition and the Benefit of Intravenous Albumin Infusion for Alzheimer’s Disease Treatment. Front. Neurosci. 2020, 14, 859. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Serafini, M.; Colic Baric, I.; Hazen, S.L.; Klein, S. Effect of Plasma Uric Acid on Antioxidant Capacity, Oxidative Stress, and Insulin Sensitivity in Obese Subjects. Diabetes 2014, 63, 976–981. [Google Scholar] [CrossRef]
- Klein, B.E.K.; Klein, R.; Lee, K.E. Components of the Metabolic Syndrome and Risk of Cardiovascular Disease and Diabetes in Beaver Dam. Diabetes Care 2002, 25, 1790–1794. [Google Scholar] [CrossRef]
- Zurlo, A.; Veronese, N.; Giantin, V.; Maselli, M.; Zambon, S.; Maggi, S.; Musacchio, E.; Toffanello, E.D.; Sartori, L.; Perissinotto, E.; et al. High serum uric acid levels increase the risk of metabolic syndrome in elderly women: The PRO.V.A study. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Dhaun, N.; Vachiery, J.-L.; Benza, R.L.; Naeije, R.; Hwang, L.-J.; Liu, X.; Teal, S.; Webb, D.J. Endothelin antagonism and uric acid levels in pulmonary arterial hypertension: Clinical associations. J. Heart Lung Transplant. 2014, 33, 521–527. [Google Scholar] [CrossRef]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Montagnana, M.; Franchini, M.; Favaloro, E.J.; Targher, G. The paradoxical relationship between serum uric acid and cardiovascular disease. Clin. Chim. Acta 2008, 392, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stinefelt, B.; Leonard, S.S.; Blemings, K.P.; Shi, X.; Klandorf, H. Free Radical Scavenging, DNA Protection, and Inhibition of Lipid Peroxidation Mediated by Uric Acid. Ann. Clin. Lab. Sci. 2005, 35, 37–45. [Google Scholar]
- Bowman, G.L.; Shannon, J.; Frei, B.; Kaye, J.A.; Quinn, J.F. Uric Acid as a CNS Antioxidant. J. Alzheimers Dis. 2010, 19, 1331–1336. [Google Scholar] [CrossRef]
- Reyes, A.J. The increase in serum uric acid concentration caused by diuretics might be beneficial in heart failure. Eur. J. Heart Fail. 2005, 7, 461–467. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol.-Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef]
- Hink, H.U.; Santanam, N.; Dikalov, S.; McCann, L.; Nguyen, A.D.; Parthasarathy, S.; Harrison, D.G.; Fukai, T. Peroxidase Properties of Extracellular Superoxide Dismutase: Role of Uric Acid in Modulating In Vivo Activity. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1402–1408. [Google Scholar] [CrossRef]
- Mahajan, M.; Kaur, S.; Mahajan, S.; Kant, R. Uric acid a better scavenger of free radicals than vitamin C in rheumatoid arthritis. Indian J. Clin. Biochem. 2009, 24, 205–207. [Google Scholar] [CrossRef][Green Version]
- Laudon, M.; Frydman-Marom, A. Therapeutic Effects of Melatonin Receptor Agonists on Sleep and Comorbid Disorders. Int. J. Mol. Sci. 2014, 15, 15924–15950. [Google Scholar] [CrossRef]
- Reiter, R.; Tan, D.; Rosales-Corral, S.; Galano, A.; Zhou, X.; Xu, B. Mitochondria: Central Organelles for Melatonin′s Antioxidant and Anti-Aging Actions. Molecules 2018, 23, 509. [Google Scholar] [CrossRef]
- Ding, M.; Ning, J.; Feng, N.; Li, Z.; Liu, Z.; Wang, Y.; Wang, Y.; Li, X.; Huo, C.; Jia, X.; et al. Dynamin-related protein 1-mediated mitochondrial fission contributes to post-traumatic cardiac dysfunction in rats and the protective effect of melatonin. J. Pineal Res. 2018, 64, e12447. [Google Scholar] [CrossRef] [PubMed]
- Quera Salva, M.A.; Hartley, S.; Barbot, F.; Alvarez, J.C.; Lofaso, F.; Guilleminault, C. Circadian Rhythms, Melatonin and Depression. Curr. Pharm. Des. 2011, 17, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Pandiperumal, S.; Trakht, I.; Srinivasan, V.; Spence, D.; Maestroni, G.; Zisapel, N.; Cardinali, D. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 2008, 85, 335–353. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.-X.; Mayo, J.C.; Sainz, R.M.; Leon, J.; Czarnocki, Z. Melatonin as an antioxidant: Biochemical mechanisms and pathophysiological implications in humans. Acta Biochim. Pol. 2003, 50, 1129–1146. [Google Scholar] [CrossRef]
- Miller, E.; Mrowicka, M.; Malinowska, K.; Mrowicki, J.; Saluk-Juszczak, J.; Kędziora, J. The effects of whole-body cryotherapy on oxidative stress in multiple sclerosis patients. J. Therm. Biol. 2010, 35, 406–410. [Google Scholar] [CrossRef]
- Othman, A.I.; El-Missiry, M.A.; Amer, M.A.; Arafa, M. Melatonin controls oxidative stress and modulates iron, ferritin, and transferrin levels in adriamycin treated rats. Life Sci. 2008, 83, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, Classification, and Pharmacology of G Protein-Coupled Melatonin Receptors. Pharmacol. Rev. 2010, 62, 343–380. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sirianni, A.; Pei, Z.; Cormier, K.; Smith, K.; Jiang, J.; Zhou, S.; Wang, H.; Zhao, R.; Yano, H.; et al. The Melatonin MT1 Receptor Axis Modulates Mutant Huntingtin-Mediated Toxicity. J. Neurosci. 2011, 31, 14496–14507. [Google Scholar] [CrossRef] [PubMed]
- Back, K.; Tan, D.-X.; Reiter, R.J. Melatonin biosynthesis in plants: Multiple pathways catalyze tryptophan to melatonin in the cytoplasm or chloroplasts. J. Pineal Res. 2016, 61, 426–437. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.; Qin, L.; Reiter, R. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef]
- Zheng, X.; Tan, D.X.; Allan, A.C.; Zuo, B.; Zhao, Y.; Reiter, R.J.; Wang, L.; Wang, Z.; Guo, Y.; Zhou, J.; et al. Chloroplastic biosynthesis of melatonin and its involvement in protection of plants from salt stress. Sci. Rep. 2017, 7, 41236. [Google Scholar] [CrossRef]
- Bonnefont-Rousselot, D.; Collin, F. Melatonin: Action as antioxidant and potential applications in human disease and aging. Toxicology 2010, 278, 55–67. [Google Scholar] [CrossRef]
- Devore, E.E.; Harrison, S.L.; Stone, K.L.; Holton, K.F.; Barrett-Connor, E.; Ancoli-Israel, S.; Yaffe, K.; Ensrud, K.; Cawthon, P.M.; Redline, S.; et al. Association of urinary melatonin levels and aging-related outcomes in older men. Sleep Med. 2016, 23, 73–80. [Google Scholar] [CrossRef]
- Navarro-Alarcon, M.; Ruiz-Ojeda, F.J.; Blanca-Herrera, R.M.; Agil, A. Antioxidant activity of melatonin in diabetes in relation to the regulation and levels of plasma Cu, Zn, Fe, Mn, and Se in Zucker diabetic fatty rats. Nutrition 2013, 29, 785–789. [Google Scholar] [CrossRef]
- López-Lluch, G.; Rodríguez-Aguilera, J.C.; Santos-Ocaña, C.; Navas, P. Is coenzyme Q a key factor in aging? Mech. Ageing Dev. 2010, 131, 225–235. [Google Scholar] [CrossRef]
- Jankowski, J.; Korzeniowska, K.; Cieślewicz, A.; Jabłecka, A. Coenzyme Q10—A new player in the treatment of heart failure? Pharmacol. Rep. 2016, 68, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Petrova, E.V.; Korotkova, E.I.; Kratochvil, B.; Voronova, O.A.; Dorozhko, E.V.; Bulycheva, E.V. Investigation of Coenzyme Q10 by Voltammetry. Procedia Chem. 2014, 10, 173–178. [Google Scholar] [CrossRef]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—Biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef]
- Acosta, M.J.; Vazquez Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochim. Biophys. Acta BBA-Bioenerg. 2016, 1857, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Navas, P.; Villalba, J.M.; de Cabo, R. The importance of plasma membrane coenzyme Q in aging and stress responses. Mitochondrion 2007, 7, S34–S40. [Google Scholar] [CrossRef]
- Rodick, T.C.; Seibels, D.R.; Babu, J.R.; Huggins, K.W.; Ren, G.; Mathews, S.T. Potential role of coenzyme Q10 in health and disease conditions. Nutr. Diet. Suppl. 2018, 10, 1325. [Google Scholar] [CrossRef]
- Potgieter, M.; Pretorius, E.; Pepper, M.S. Primary and secondary coenzyme Q10 deficiency: The role of therapeutic supplementation. Nutr. Rev. 2013, 71, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta BBA-Biomembr. 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Donnino, M.W.; Cocchi, M.N.; Salciccioli, J.D.; Kim, D.; Naini, A.B.; Buettner, C.; Akuthota, P. Coenzyme Q10 levels are low and may be associated with the inflammatory cascade in septic shock. Crit. Care 2011, 15, R189. [Google Scholar] [CrossRef]
- Noh, Y.H.; Kim, K.-Y.; Shim, M.S.; Choi, S.-H.; Choi, S.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A.; Ju, W.-K. Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis. 2013, 4, e820. [Google Scholar] [CrossRef] [PubMed]
- Cooney, R.V.; Dai, Q.; Gao, Y.-T.; Chow, W.-H.; Shu, X.-O.; Li, H.; Ji, B.; Cai, Q.; Franke, A.A.; Zheng, W. Abstract 874: Low plasma coenzyme Q10 levels and breast cancer risk in Chinese women: Influence of menopausal status. Cancer Res. 2010, 70, 874. [Google Scholar] [CrossRef]
- Blatt, T.; Littarru, G.P. Biochemical rationale and experimental data on the antiaging properties of CoQ10 at skin level. BioFactors 2011, 37, 381–385. [Google Scholar] [CrossRef]
- Lee, B.-J.; Huang, Y.-C.; Chen, S.-J.; Lin, P.-T. Coenzyme Q10 supplementation reduces oxidative stress and increases antioxidant enzyme activity in patients with coronary artery disease. Nutrition 2012, 28, 250–255. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Predoi, G.; Serban, A.I. Oxidative stress mitigation by antioxidants—An overview on their chemistry and influences on health status. Eur. J. Med. Chem. 2021, 209, 112891. [Google Scholar] [CrossRef]
- Neha, K.; Haider, M.R.; Pathak, A.; Yar, M.S. Medicinal prospects of antioxidants: A review. Eur. J. Med. Chem. 2019, 178, 687–704. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Jahns, L.; Conrad, Z.; Johnson, L.K.; Whigham, L.D.; Wu, D.; Claycombe-Larson, K.J. A diet high in carotenoid-rich vegetables and fruits favorably impacts inflammation status by increasing plasma concentrations of IFN-α2 and decreasing MIP-1β and TNF-α in healthy individuals during a controlled feeding trial. Nutr. Res. 2018, 52, 98–104. [Google Scholar] [CrossRef]
- Silva, S.; Costa, E.M.; Veiga, M.; Morais, R.M.; Calhau, C.; Pintado, M. Health promoting properties of blueberries: A review. Crit. Rev. Food Sci. Nutr. 2020, 60, 181–200. [Google Scholar] [CrossRef]
- Forman, H.J.; Davies, K.J.A.; Ursini, F. How Do Nutritional Antioxidants Really Work: Nucleophilic Tone and Para-Hormesis Versus Free Radical Scavenging In Vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef]
- Njus, D.; Kelley, P.M.; Tu, Y.-J.; Schlegel, H.B. Ascorbic acid: The chemistry underlying its antioxidant properties. Free Radic. Biol. Med. 2020, 159, 37–43. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Michels, A.J.; Frei, B. Vitamin C. Adv. Nutr. 2014, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Valdés, F. Vitamina C. Actas Dermo-Sifiliográficas 2006, 97, 557–568. [Google Scholar] [CrossRef]
- Carr, A.C.; Vissers, M.C.M. Synthetic or Food-Derived Vitamin C—Are They Equally Bioavailable? Nutrients 2013, 5, 4284–4304. [Google Scholar] [CrossRef]
- Ishizuka, K.; Ohira, Y. Scurvy. QJM Int. J. Med. 2022, 115, 475. [Google Scholar] [CrossRef]
- Guo, H.; Ding, J.; Liu, Q.; Li, Y.; Liang, J.; Zhang, Y. Vitamin C and Metabolic Syndrome: A Meta-Analysis of Observational Studies. Front. Nutr. 2021, 8, 728880. [Google Scholar] [CrossRef]
- Michels, A.J.; Hagen, T.M.; Frei, B. Human Genetic Variation Influences Vitamin C Homeostasis by Altering Vitamin C Transport and Antioxidant Enzyme Function. Annu. Rev. Nutr. 2013, 33, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The Pharmacokinetics of Vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.A.; Diab, A.S.; Mohammed, A.A. Antioxidant Categories and Mode of Action; IntechOpen: London, UK, 2019; ISBN 978-1-78923-920-1. [Google Scholar]
- Kaźmierczak-Barańska, J.; Boguszewska, K.; Adamus-Grabicka, A.; Karwowski, B.T. Two Faces of Vitamin C—Antioxidative and Pro-Oxidative Agent. Nutrients 2020, 12, 1501. [Google Scholar] [CrossRef]
- Chakraborthy, A.; Ramani, P.; Sherlin, H.J.; Premkumar, P.; Natesan, A. Antioxidant and pro-oxidant activity of Vitamin C in oral environment. Indian J. Dent. Res. 2014, 25, 499. [Google Scholar] [CrossRef] [PubMed]
- Bravo, L. Polyphenols: Chemistry, dietary sources, metabolism, and nutritional significance. Nutr. Rev. 1998, 56, 317–333. [Google Scholar] [CrossRef]
- Zeb, A. Concept, mechanism, and applications of phenolic antioxidants in foods. J. Food Biochem. 2020, 44, e13394. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; González-Paramás, A.M.; Oludemi, T.; Ayuda-Durán, B.; González-Manzano, S. Chapter Four—Plant phenolics as functional food ingredients. In Advances in Food and Nutrition Research; Ferreira, I.C.F.R., Barros, L., Eds.; Functional Food Ingredients from Plants; Academic Press: Cambridge, MA, USA, 2019; Volume 90, pp. 183–257. [Google Scholar]
- Nijveldt, R.J.; van Nood, E.; van Hoorn, D.E.; Boelens, P.G.; van Norren, K.; van Leeuwen, P.A. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Orallo, F.; Álvarez, E.; Camiña, M.; Leiro, J.M.; Gómez, E.; Fernández, P. The Possible Implication of trans-Resveratrol in the Cardioprotective Effects of Long-Term Moderate Wine Consumption. Mol. Pharmacol. 2002, 61, 294–302. [Google Scholar] [CrossRef]
- Shoskes, D.A. Effect of Bioflavonoids Quercetin and Curcumin on Ischemic Renal Injury: A New Class of Renoprotective Agents: 1. Transplantation 1998, 66, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Jin, B. Plant flavonoids: Classification, distribution, biosynthesis, and antioxidant activity. Food Chem. 2022, 383, 132531. [Google Scholar] [CrossRef] [PubMed]
- Porras-Loaiza, A.P.; López-Malo, A. Importancia de los grupos fenólicos en los alimentos. Temas Sel. Ing. Aliment. 2009, 31, 121–134. [Google Scholar]
- Singh, R.B.; Fedacko, J.; Fatima, G.; Magomedova, A.; Watanabe, S.; Elkilany, G. Why and How the Indo-Mediterranean Diet May Be Superior to Other Diets: The Role of Antioxidants in the Diet. Nutrients 2022, 14, 898. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef]
- Di Lorenzo, C.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef]
- Carlsen, M.H.; Halvorsen, B.L.; Holte, K.; Bøhn, S.K.; Dragland, S.; Sampson, L.; Willey, C.; Senoo, H.; Umezono, Y.; Sanada, C.; et al. The total antioxidant content of more than 3100 foods, beverages, spices, herbs and supplements used worldwide. Nutr. J. 2010, 9, 3. [Google Scholar] [CrossRef]
- Liu, J.; Hefni, M.E.; Witthöft, C.M. Characterization of Flavonoid Compounds in Common Swedish Berry Species. Foods 2020, 9, 358. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.L. Wine Phenolics. Ann. N. Y. Acad. Sci. 2002, 957, 21–36. [Google Scholar] [CrossRef]
- Miller, K.B.; Hurst, W.J.; Flannigan, N.; Ou, B.; Lee, C.Y.; Smith, N.; Stuart, D.A. Survey of Commercially Available Chocolate- and Cocoa-Containing Products in the United States. 2. Comparison of Flavan-3-ol Content with Nonfat Cocoa Solids, Total Polyphenols, and Percent Cacao. J. Agric. Food Chem. 2009, 57, 9169–9180. [Google Scholar] [CrossRef]
- Cabrera, C.; Artacho, R.; Giménez, R. Beneficial Effects of Green Tea—A Review. J. Am. Coll. Nutr. 2006, 25, 79–99. [Google Scholar] [CrossRef] [PubMed]
- McKay, D.L.; Blumberg, J.B. The Role of Tea in Human Health: An Update. J. Am. Coll. Nutr. 2002, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Galiniak, S.; Aebisher, D.; Bartusik-Aebisher, D. Health benefits of resveratrol administration. Acta Biochim. Pol. 2019, 66, 13–21. [Google Scholar] [CrossRef]
- Ungvari, Z.; Orosz, Z.; Rivera, A.; Labinskyy, N.; Xiangmin, Z.; Olson, S.; Podlutsky, A.; Csiszar, A. Resveratrol increases vascular oxidative stress resistance. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H2417–H2424. [Google Scholar] [CrossRef]
- Leonard, S.S.; Xia, C.; Jiang, B.-H.; Stinefelt, B.; Klandorf, H.; Harris, G.K.; Shi, X. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochem. Biophys. Res. Commun. 2003, 309, 1017–1026. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef]
- Ungvari, Z.; Labinskyy, N.; Mukhopadhyay, P.; Pinto, J.T.; Bagi, Z.; Ballabh, P.; Zhang, C.; Pacher, P.; Csiszar, A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H1876–H1881. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Singh, R.; Verma, S.S.; Rai, V.; Kaschula, C.H.; Maiti, P.; Gupta, S.C. Health benefits of resveratrol: Evidence from clinical studies. Med. Res. Rev. 2019, 39, 1851–1891. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, R.; Rocic, P. The metabolic syndrome, oxidative stress, environment, and cardiovascular disease: The great exploration. Exp. Diabetes Res. 2012, 2012, 271028. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Hallan, S.; Sharma, K. The Role of Mitochondria in Diabetic Kidney Disease. Curr. Diab. Rep. 2016, 16, 61. [Google Scholar] [CrossRef]
- Medema, R.H.; Kops, G.J.P.L.; Bos, J.L.; Burgering, B.M.T. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27(kip1). Nature 2000, 404, 782–787. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal Growth Factor (EGF)-induced Generation of Hydrogen Peroxide: Role in EGF Receptor-Mediated Tyrosine Phosphorylation. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar] [CrossRef]
- Daroui, P.; Desai, S.D.; Li, T.-K.; Liu, A.A.; Liu, L.F. Hydrogen Peroxide Induces Topoisomerase I-mediated DNA Damage and Cell Death. J. Biol. Chem. 2004, 279, 14587–14594. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Arancibia-Hernández, Y.L.; Hernández-Cruz, E.Y.; Pedraza-Chaverri, J. RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen 2022, 2, 437-478. https://doi.org/10.3390/oxygen2040030
Aranda-Rivera AK, Cruz-Gregorio A, Arancibia-Hernández YL, Hernández-Cruz EY, Pedraza-Chaverri J. RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen. 2022; 2(4):437-478. https://doi.org/10.3390/oxygen2040030
Chicago/Turabian StyleAranda-Rivera, Ana Karina, Alfredo Cruz-Gregorio, Yalith Lyzet Arancibia-Hernández, Estefani Yaquelin Hernández-Cruz, and José Pedraza-Chaverri. 2022. "RONS and Oxidative Stress: An Overview of Basic Concepts" Oxygen 2, no. 4: 437-478. https://doi.org/10.3390/oxygen2040030
APA StyleAranda-Rivera, A. K., Cruz-Gregorio, A., Arancibia-Hernández, Y. L., Hernández-Cruz, E. Y., & Pedraza-Chaverri, J. (2022). RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen, 2(4), 437-478. https://doi.org/10.3390/oxygen2040030