
Quantitative Analysis of NDMA in Drug Products: A Proposed High-Throughput Approach Using Headspace–SIFT-MS


Abstract
1. Introduction
2. Materials and Methods
2.1. Samples, Chemicals, and Standards
2.1.1. Drug Product Samples
2.1.2. Chemicals and Standards
2.2. SIFT-MS
2.2.1. SIFT-MS Analysis
2.2.2. Automated Headspace–SIFT-MS Analysis
2.3. Automated LC-MS/MS Analysis
3. Results
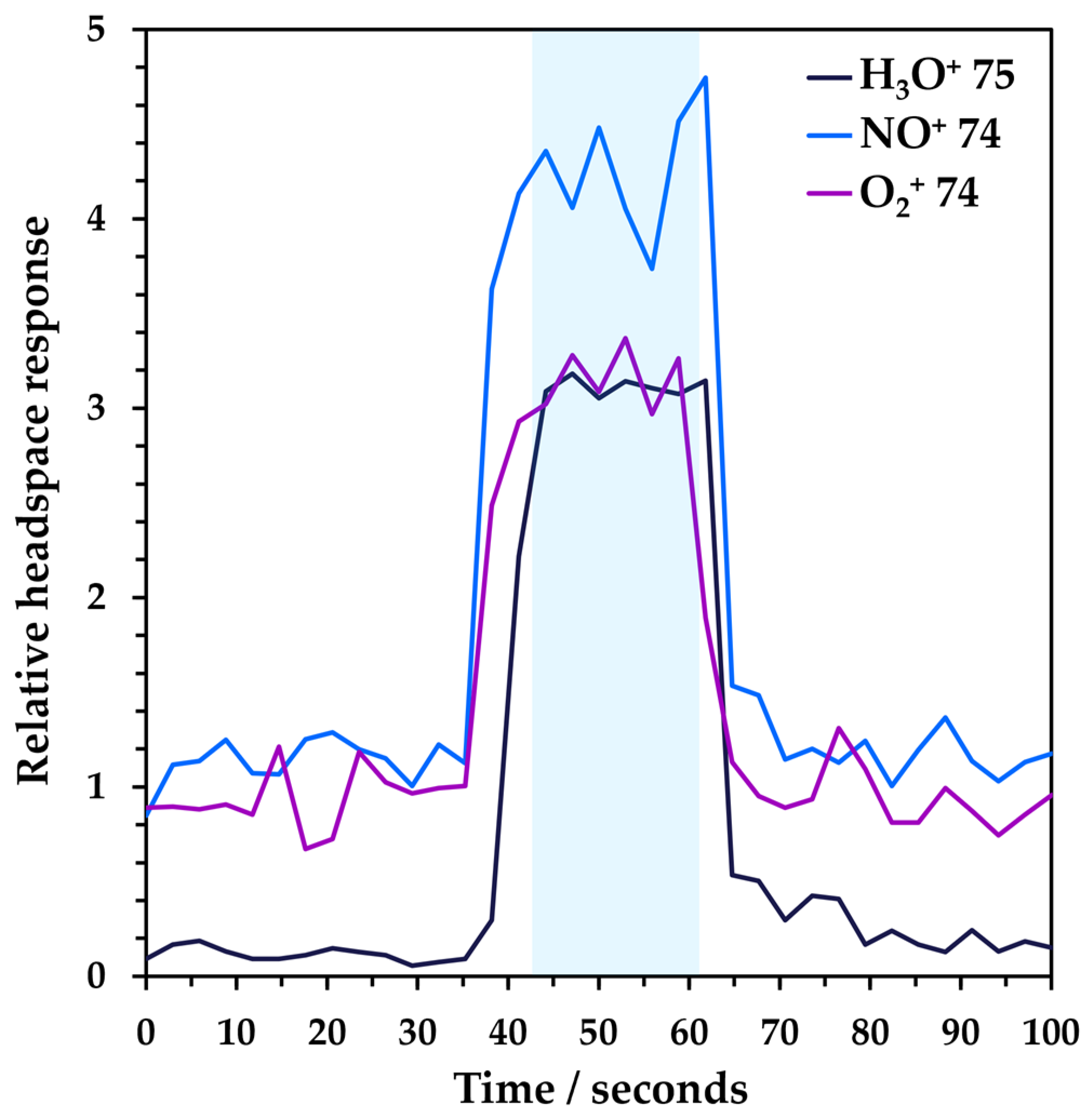
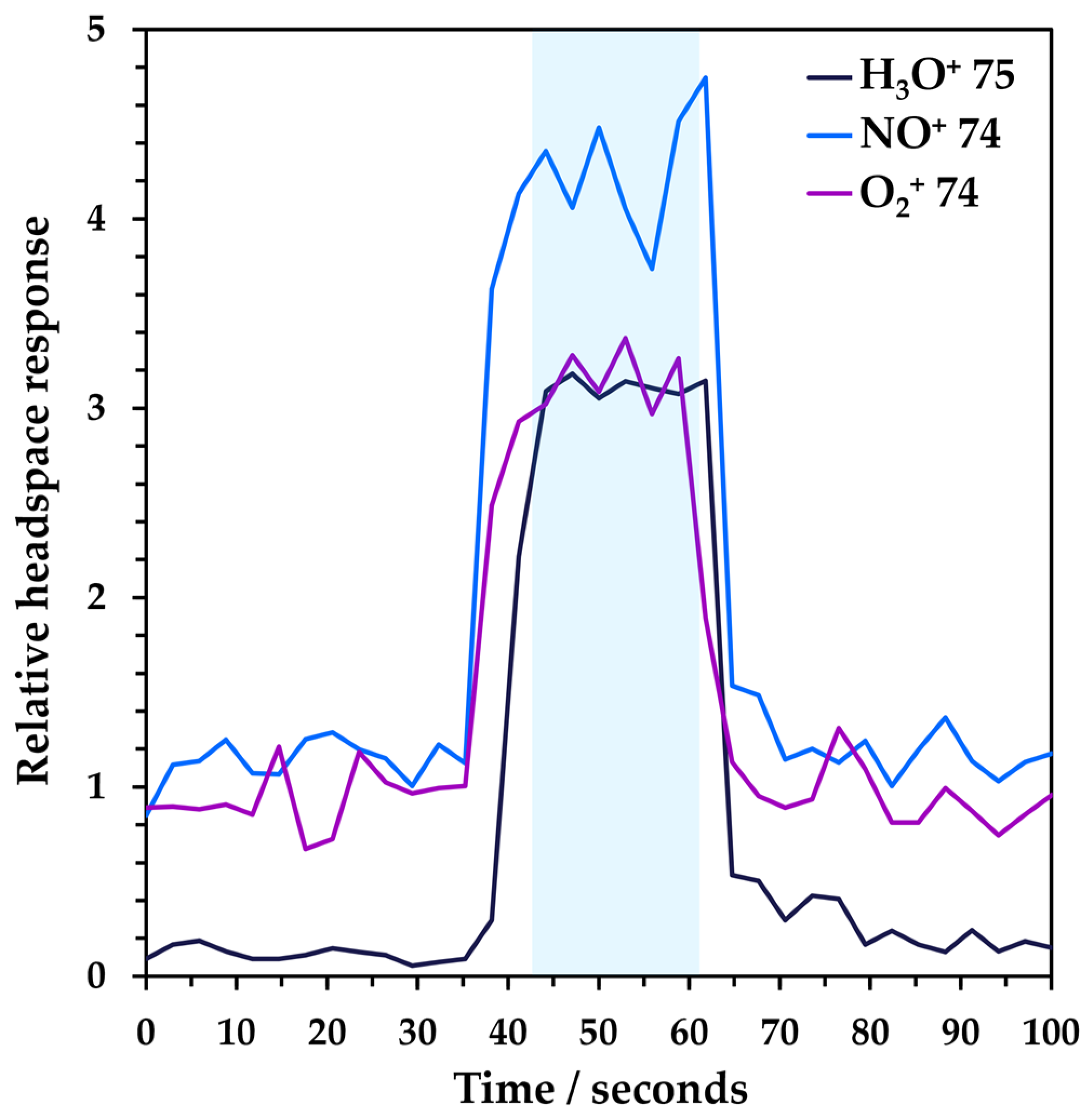
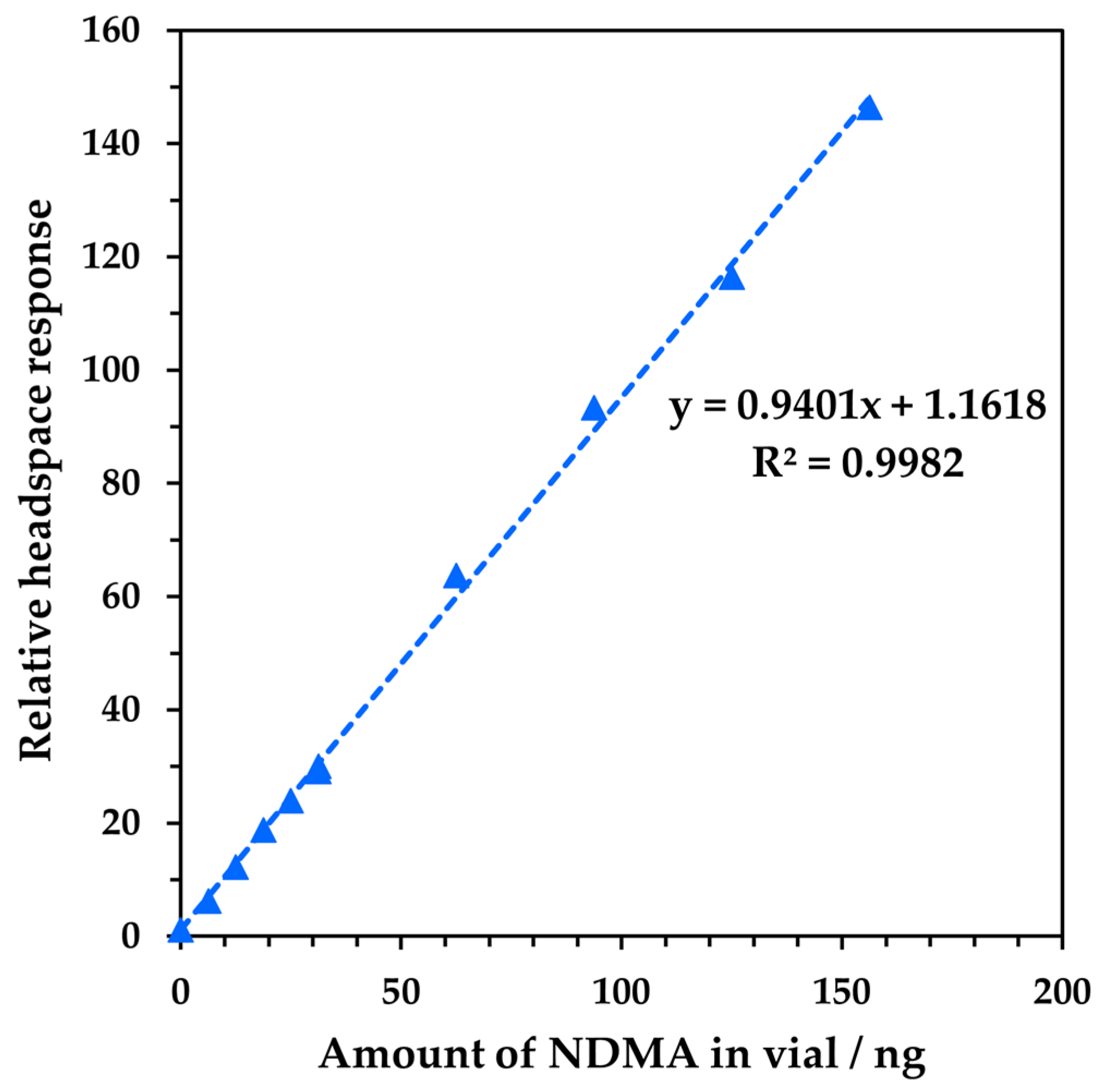
3.1. Analytical Performance of Headspace–SIFT-MS Analysis of NDMA
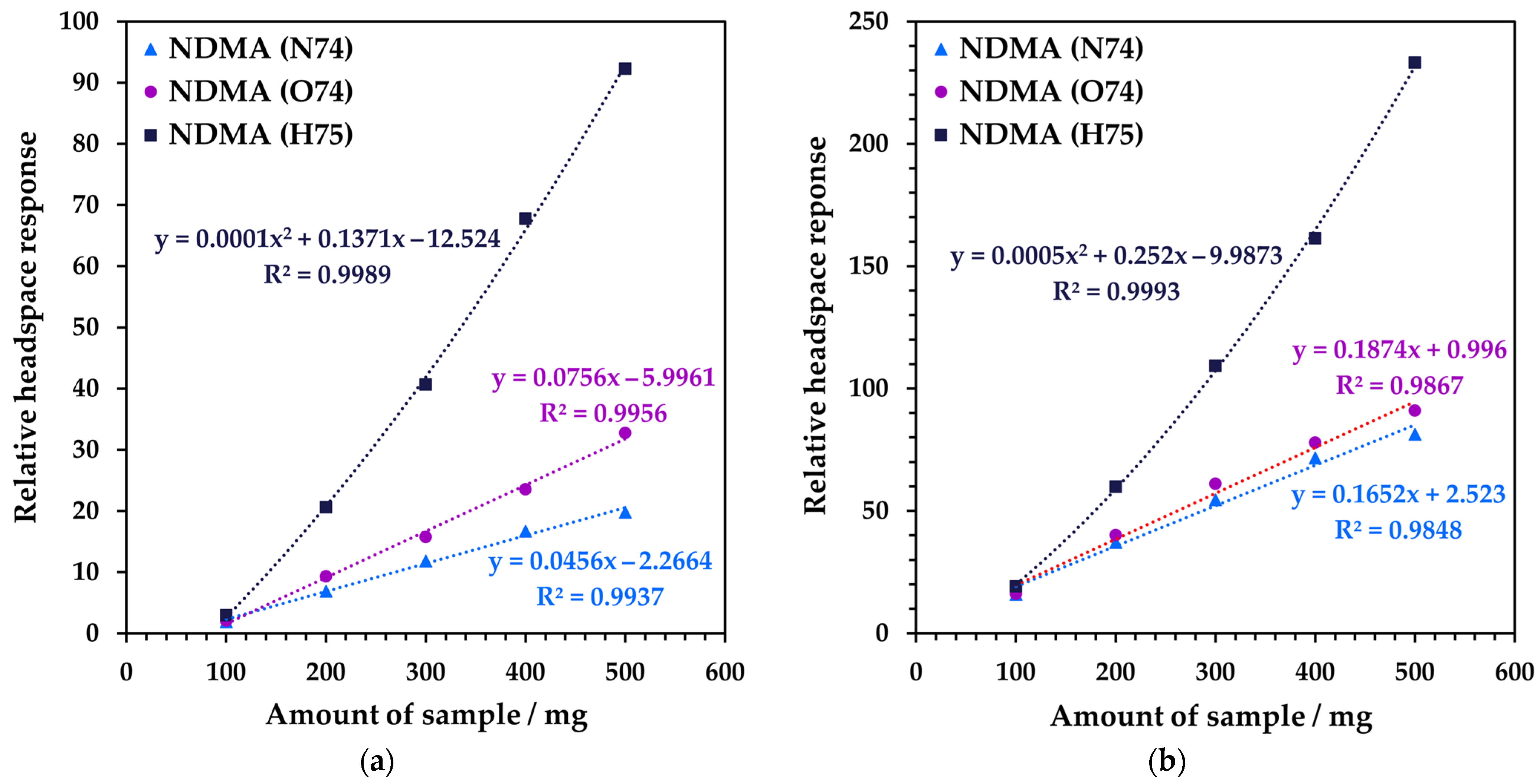
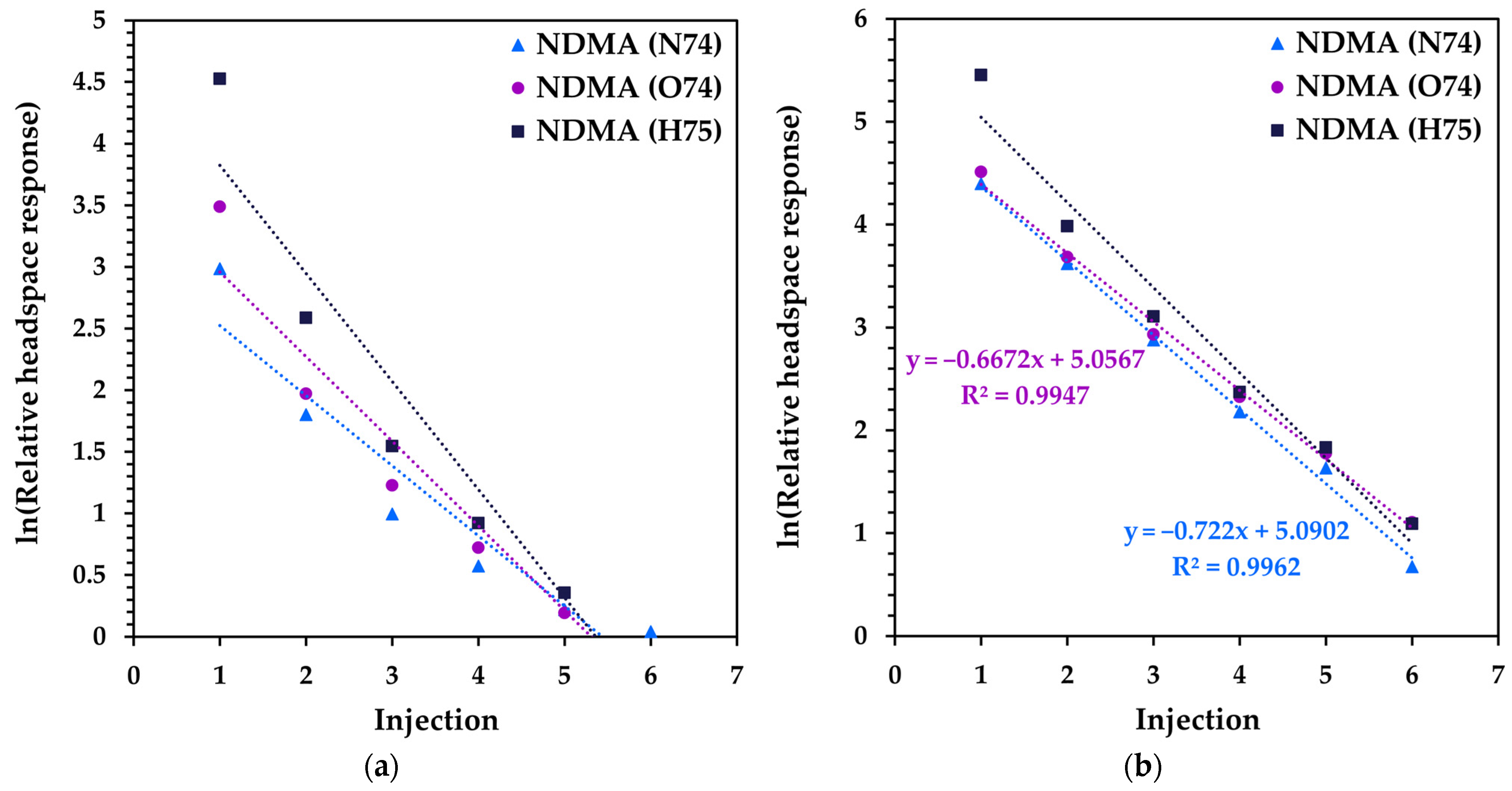
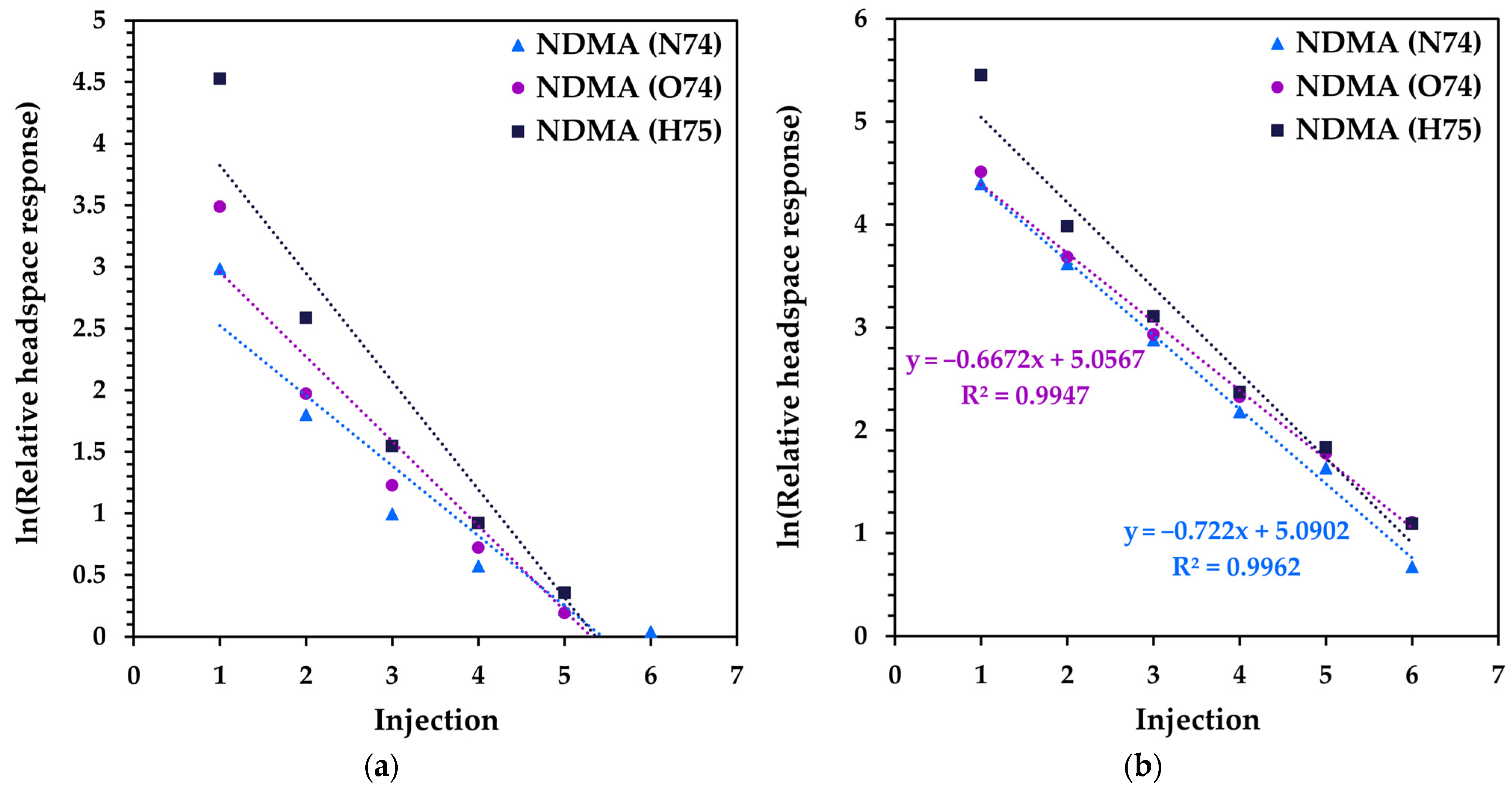
3.2. Method Development for Quantitative SIFT-MS Analysis of Tablet Formulations
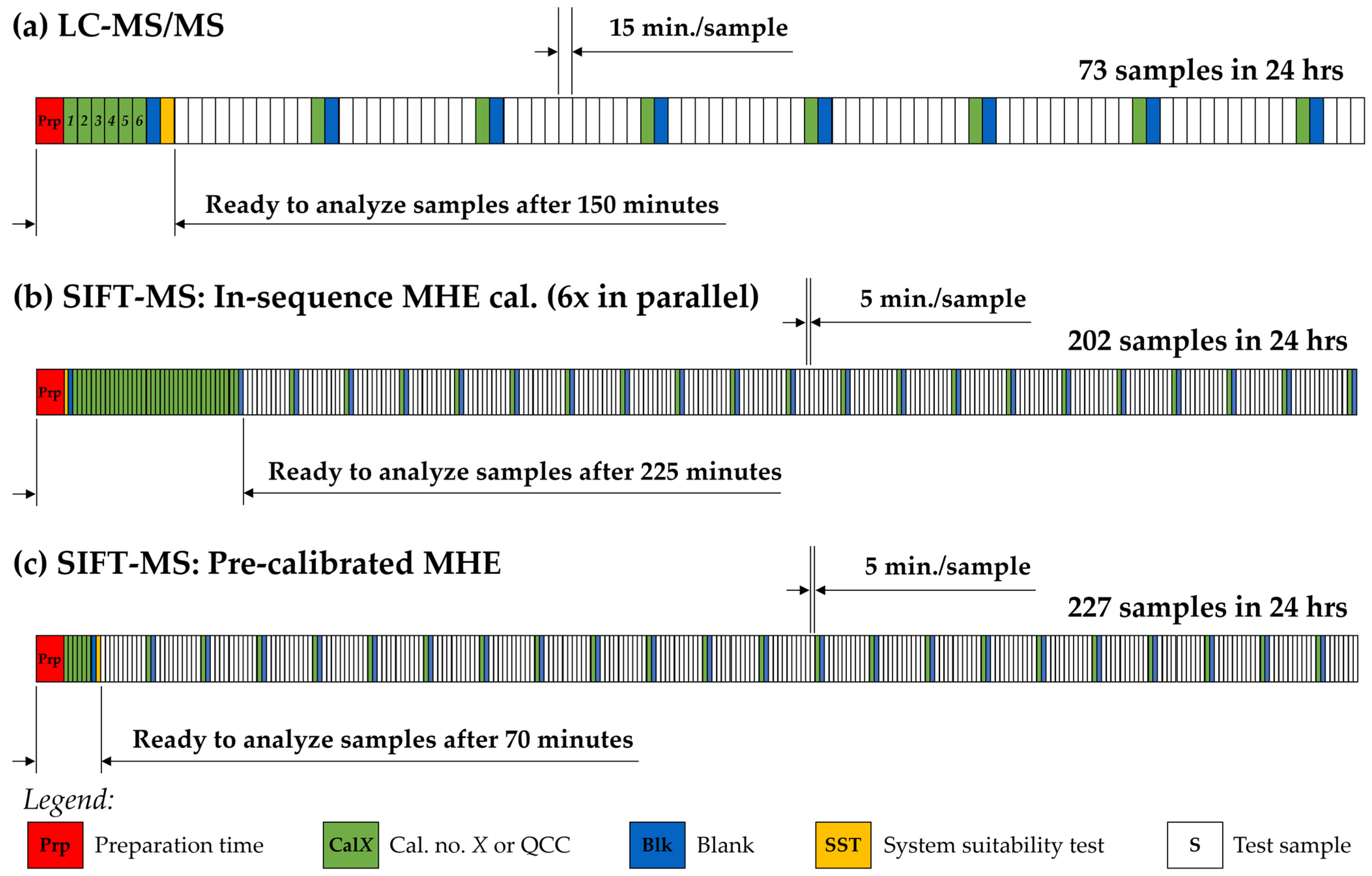
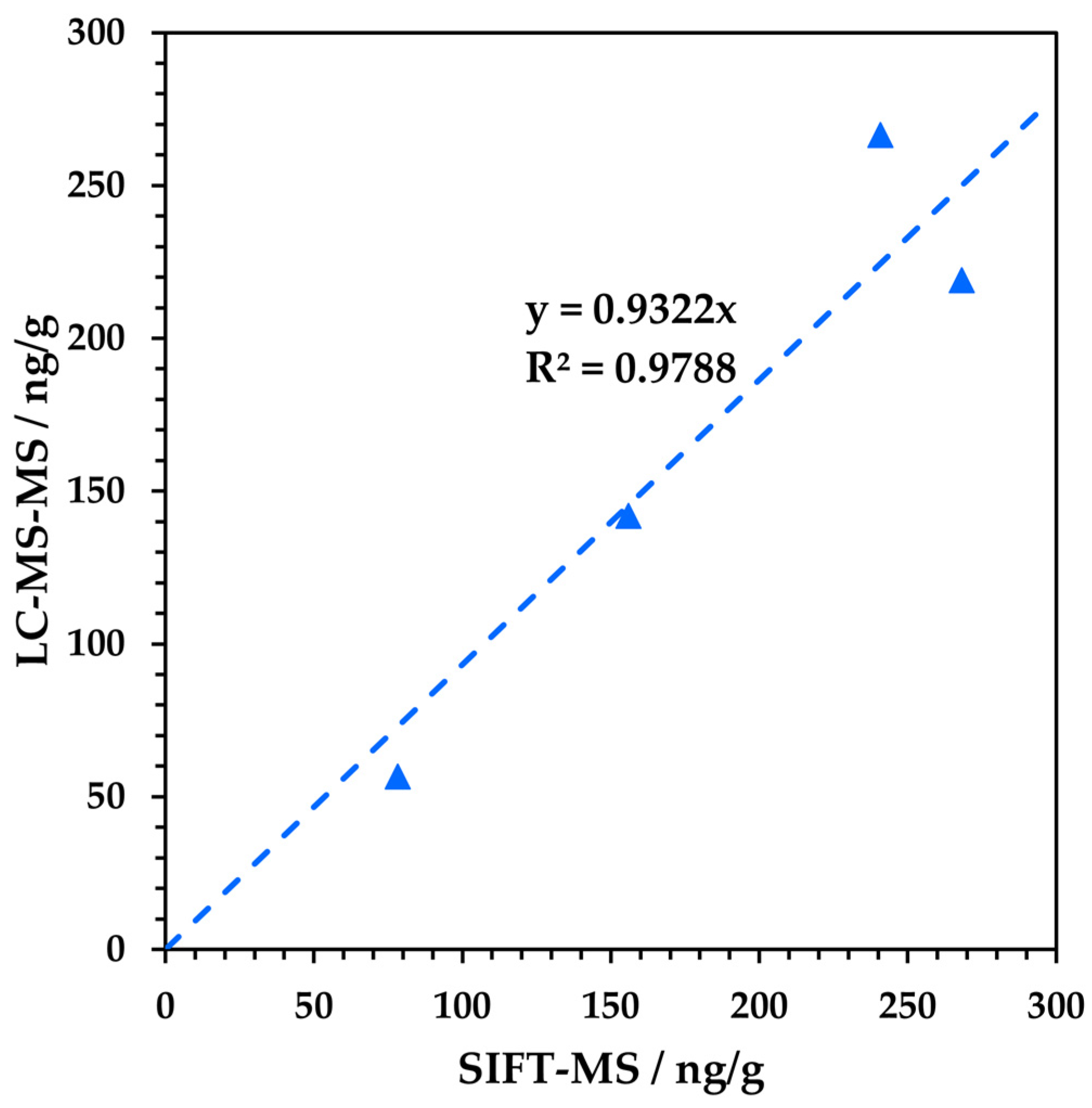
3.3. Head-to-Head Comparison with LC-MS/MS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halden, R.U.; Gushgari, A.J. Critical review of major sources of human exposure to N-nitrosamines. Chemosphere 2018, 210, 1124–1136. [Google Scholar] [CrossRef]
- Akkaraju, H.; Tatia, R.; Mane, S.S.; Khade, A.B.; Dengale, S.J. A comprehensive review of sources of nitrosamine contamination of pharmaceutical substances and products. Regul. Toxicol. Pharmacol. 2023, 139, 105355. [Google Scholar] [CrossRef]
- World Health Organization. N-Nitrosodimethylamine in Drinking Water. 2008. Available online: https://cdn.who.int/media/docs/default-source/wash-documents/wash-chemicals/ndma-2add-feb2008.pdf?sfvrsn=122aa3d3_4 (accessed on 5 February 2024).
- McWeeny, D.J. Nitrosamines in beverages. Food Chem. 1983, 11, 273–287. [Google Scholar] [CrossRef]
- Park, J.; Seo, J.; Lee, J.; Kwon, H. Distribution of seven N-nitrosamines in food. Toxicol. Res. 2015, 31, 279–288. [Google Scholar] [CrossRef]
- Tricker, A.R.; Preussmann, R. Carcinogenic N-nitrosamines in the diet: Occurrence, formation, mechanisms and carcinogenic potential. Mutat. Res. 1991, 259, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Boltres, B. Evaluating nitrosamines from elastomers in pharmaceutical primary packaging. PDA J. Pharm. Sci. Tech. 2022, 76, 136–150. [Google Scholar] [CrossRef] [PubMed]
- United States Food and Drug Administration (US FDA). FDA Updates on Valsartan Recalls. 2018. Available online: https://www.fda.gov/news-events/press-announcements/fda-announces-voluntary-recall-several-medicines-containing-valsartan-following-detection-impurity (accessed on 5 February 2024).
- Golob, N.; Grahek, R.; Ross, M.; Roskar, R. Nitrocellulose blister material as a source of N-nitrosamine contamination of pharmaceutical drug products. Int. J. Pharmaceutics 2022, 618, 121687. [Google Scholar] [CrossRef] [PubMed]
- Schlingemann, J.; Burns, M.J.; Ponting, D.J.; Avila, C.M.; Romero, N.E.; Jaywant, M.A.; Smith, G.F.; Ashworth, I.W.; Simon, S.; Saal, C.; et al. The landscape of potential small and drug substance related nitrosamines in pharmaceuticals. J. Pharm. Sci. 2023, 112, 1287–1304. [Google Scholar] [CrossRef] [PubMed]
- Thresher, A.; Foster, R.; Ponting, D.J.; Stalford, S.A.; Tennant, R.E.; Thomas, R. Are all nitrosamines concerning? A review of mutagenicity and carcinogenicity data. Regul. Toxicol. Pharmacol. 2020, 116, 104749. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). Questions and Answers for Marketing Authorisation Holders/Applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 Referral on Nitrosamine Impurities in Human Medicinal Products, EMA/409815/2020. 2021. Available online: https://www.ema.europa.eu/en/documents/referral/nitrosamines-emea-h-a53-1490-questions-answers-marketing-authorisation-holders/applicants-chmp-opinion-article-53-regulation-ec-no-726/2004-referral-nitrosamine-impurities-human-medicinal-products_en.pdf (accessed on 1 February 2024).
- United States Food and Drug Administration (US FDA). Control of Nitrosamine Impurities in Human Drugs: Guidance for Industry. 2021. Available online: https://www.fda.gov/media/141720/download (accessed on 1 February 2024).
- United States Pharmacopeia. Nitrosamine Impurities <1469>; United States Pharmacopeia: Rockville, MD, USA, 2021. [Google Scholar]
- European Pharmacopeia. N-Nitrosamines in Active Substances, 11th ed.; European Pharmacopeia: Strasbourg, France, 2022; Chapter 2.5.42. [Google Scholar]
- Zhang, J.; Selaya, S.D.; Shakleya, D.; Mohammad, A.; Faustino, P.J. Rapid quantitation of four nitrosamine impurities in angiotensin receptor blocker drug substances. J. Pharm. Sci. 2023, 112, 1246–1254. [Google Scholar] [CrossRef]
- Smith, D.; Španěl, P. Selected ion flow tube mass spectrometry (SIFT-MS) for on-line trace gas analysis. Mass Spec. Rev. 2005, 24, 661–700. [Google Scholar] [CrossRef]
- Smith, D.; Španěl, P.; Demarais, N.; Langford, V.S.; McEwan, M.J. Recent developments and applications of selected ion flow tube mass spectrometry (SIFT-MS). Mass Spec. Rev. 2023, e21835. [Google Scholar] [CrossRef] [PubMed]
- Langford, V.S.; Gray, J.D.C.; Maclagan, R.G.A.R.; Milligan, D.B.; McEwan, M.J. Real-time measurement of nitrosamines in air. Int. J. Mass Spectrom. 2015, 377, 490–495. [Google Scholar] [CrossRef]
- Perkins, M.J.; Silva, L.P.; Langford, V.S. Evaluation of solvent compatibilities for headspace-SIFT-MS analysis of pharmaceutical products. Analytica 2023, 4, 313–335. [Google Scholar] [CrossRef]
- Kolb, B.; Ettre, L.S. Static Headspace-Gas Chromatography—Theory and Practice, 2nd ed.; Wiley–Interscience: New York, NY, USA, 2006. [Google Scholar]
- Perkins, M.J.; Langford, V.S. Multiple headspace extraction-selected ion flow tube mass spectrometry (MHE-SIFT-MS). Part 1: A protocol for method development and transfer to routine analysis. Rev. Sep. Sci. 2022, 4, e22001. [Google Scholar] [CrossRef]
- Perkins, M.J.; Langford, V.S. Improved MHE-SIFT-MS Workflow: Even Faster Quantitation of Formaldehyde in Gelucire Excipient. Syft Technologies Application Note. 2023. Available online: http://bit.ly/431xTp2 (accessed on 1 February 2024).
- United States Food and Drug Administration (US FDA). FDA Requests Removal of All Ranitidine Products (Zantac) from the Market. 2020. Available online: https://www.fda.gov/news-events/press-announcements/fda-requests-removal-all-ranitidine-products-zantac-market (accessed on 5 February 2024).
- United States Food and Drug Administration (US FDA). FDA Alerts Patients and Health Care Professionals to Nitrosamine Impurity Findings in Certain Metformin Extended-Release Products. 2020. Available online: https://www.fda.gov/news-events/press-announcements/fda-alerts-patients-and-health-care-professionals-nitrosamine-impurity-findings-certain-metformin (accessed on 5 February 2024).
- Langford, V.S.; Dryahina, K.; Španěl, P. Robust automated SIFT-MS quantitation of volatile compounds in air using a multicomponent gas standard. J. Am. Soc. Mass Spectrom. 2023, 34, 2630–2645. [Google Scholar] [CrossRef] [PubMed]
- McEwan, M.J. Direct analysis mass spectrometry. In Ion Molecule Attachment Reactions: Mass Spectrometry; Fujii, T., Ed.; Springer: New York, NY, USA, 2015; pp. 263–317. [Google Scholar]
- United States Pharmacopeia. Residual Solvents <467>; United States Pharmacopeia: Rockville, MD, USA, 2007. [Google Scholar]
- Parr, M.K.; Joseph, J.F. NDMA impurity in valsartan and other pharmaceutical products: Analytical methods for the determination of N-nitrosamines. J. Pharm. Biomed. Anal. 2019, 164, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.J.; Langford, V.S. Standard validation protocol for selected ion flow tube mass spectrometry methods applied to direct headspace analysis of aqueous volatile organic compounds. Anal. Chem. 2021, 93, 8386–8392. [Google Scholar] [CrossRef] [PubMed]
- Liscio, C. Full Method Validation of an Automated Solution for Nitrosamines in Irbesartan and Metformin by LC-MS-MS Analysis. Anatune Limited Application Note. 2020. Available online: https://bit.ly/3Sut1Ee (accessed on 5 February 2024).
- Španěl, P.; Smith, D. SIFT studies of the reactions of H3O+, NO+, and O2+• with a series of alcohols. Int. J. Mass Spectrom. Ion Proc. 1997, 167/168, 375–388. [Google Scholar] [CrossRef]
- Španěl, P.; Ji, Y.; Smith, D. SIFT studies of the reactions of H3O+, NO+, and O2+• with a series of aldehydes and ketones. Int. J. Mass Spectrom. Ion Proc. 1997, 165/166, 25–37. [Google Scholar] [CrossRef]
- United States Food and Drug Administration (US FDA). Laboratory Tests|Ranitidine. 2019. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/laboratory-tests-ranitidine (accessed on 1 February 2024).
- Perkins, M.J.; Langford, V.S. Application of routine analysis procedures to a direct mass spectrometry technique: Selected ion flow tube mass spectrometry (SIFT-MS). Rev. Sep. Sci. 2021, 3, e21003. [Google Scholar] [CrossRef]
- United States Food and Drug Administration (US FDA). Prinston Pharmaceutical Inc Issues Voluntary Nationwide Recall of Valsartan and Valsartan HCTZ Tablets Due to Detection of a Trace Amount of Unexpected Impurity, N-nitrosodimethylamine (NDMA) in the Products. 2018. Available online: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/prinston-pharmaceutical-inc-issues-voluntary-nationwide-recall-valsartan-and-valsartan-hctz-tablets (accessed on 1 February 2024).
- United States Food and Drug Administration (US FDA). Laboratory Tests|Metformin. 2020. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/laboratory-tests-metformin (accessed on 5 February 2024).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Pharmaceutical Ingredient (API) | Manufacturer | US National Drug Code (NDC) | Lot Number | Expiration Date 1 | Measured Date 1,2 | Sample Code | |
|---|---|---|---|---|---|---|---|
| Name | Quantity (mg) | ||||||
| Ranitidine | 150 | Medreich 3 | Unknown | 970043 | 01/2022 | 9/2022 | R1 |
| 150 | Sanofi 4 | Unknown | 19A430U | 01/2021 | 9/2022 | R2 | |
| Valsartan | 160 | Prinston Pharm./Solco Healthcare 5 | 43547-369-09 | 342B17022 | 10/2019 | 10/2022 | V1 |
| Metformin 6 | 500 | Amneal 7 | 53746-178-01 | HM02918A | 01/2021 | 11/2023 | M1 |
| 500 | Amneal 7 | 53746-178-90 | HD03319A | 04/2021 | 11/2023 | M2 | |
| 500 | Apotex 8 | 60505-0260-1 | NE5801 | 04/2021 | 11/2023 | M3 | |
| 500 | Amneal 7 | 65162-178-10 | AM190107AA | 12/2020 | 11/2023 | M4 | |
| 750 | Amneal 7 | 65162-179-10 | AM180770A | 05/2020 | 11/2023 | M5 | |
| 500 | Lupin 9 | 68180-336-07 | G901203 | 12/2020 | 11/2023 | M6 | |
| NDMA Reagent Ion—Product Ion Pair | Signal-to-Noise Ratio (S/N) | Theoretical LOD Based on 3:1 S/N (ng) | Theoretical LOQ Based on 10:1 S/N (ng) |
|---|---|---|---|
| H3O+ 75 | 23.8 | 0.33 | 1.1 |
| NO+ 74 | 15.3 | 0.51 | 1.7 |
| O2+• 74 | 5.9 | 1.3 | 4.4 |
| Metformin Sample | NDMA Concentration (ng g−1) | |
|---|---|---|
| SIFT-MS | LC-MS/MS | |
| M1 | 268 | 219 |
| M2 | 241 | 267 |
| M3 | 78.2 | 57 |
| M4 | 156 | 142 |
| M5 | 131 | * |
| M6 | 55.1 | * |
| Metformin Sample and Statistical Parameters | Ratio of First MHE Injection to Full MHE * | Slope of Linear Fit to Six MHE Injections |
|---|---|---|
| M1 | 0.1232 | −0.2276 |
| M2 | 0.1245 | −0.2301 |
| M3 | 0.0924 | −0.1630 |
| M4 | 0.1281 | −0.2365 |
| M5 | 0.1119 | −0.2030 |
| M6 | 0.1070 | −0.1894 |
| Mean | 0.1145 | −0.2083 |
| Standard Dev. | 0.0135 | 0.0286 |
| RSD (%) | 11.8% | 13.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perkins, M.J.; Hastie, C.J.; Langford, V.S. Quantitative Analysis of NDMA in Drug Products: A Proposed High-Throughput Approach Using Headspace–SIFT-MS. AppliedChem 2024, 4, 107-121. https://doi.org/10.3390/appliedchem4010008
Perkins MJ, Hastie CJ, Langford VS. Quantitative Analysis of NDMA in Drug Products: A Proposed High-Throughput Approach Using Headspace–SIFT-MS. AppliedChem. 2024; 4(1):107-121. https://doi.org/10.3390/appliedchem4010008
Chicago/Turabian StylePerkins, Mark J., Colin J. Hastie, and Vaughan S. Langford. 2024. "Quantitative Analysis of NDMA in Drug Products: A Proposed High-Throughput Approach Using Headspace–SIFT-MS" AppliedChem 4, no. 1: 107-121. https://doi.org/10.3390/appliedchem4010008
APA StylePerkins, M. J., Hastie, C. J., & Langford, V. S. (2024). Quantitative Analysis of NDMA in Drug Products: A Proposed High-Throughput Approach Using Headspace–SIFT-MS. AppliedChem, 4(1), 107-121. https://doi.org/10.3390/appliedchem4010008