
Application of a Universal Calibration Method for True Molar Mass Determination of Fluoro-Derivatized Technical Lignins by Size-Exclusion Chromatography

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Technical Lignins
2.2. Derivatizations
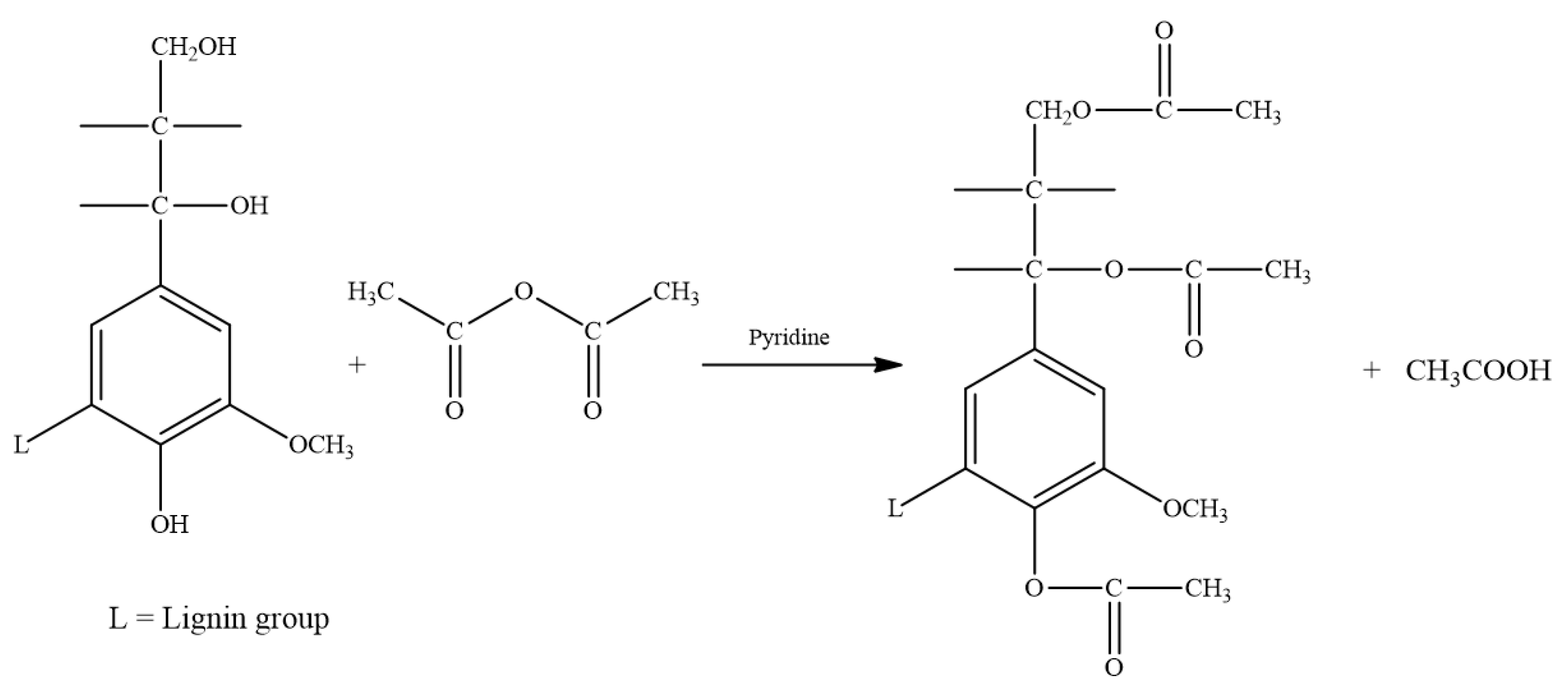
2.2.1. Acetylation
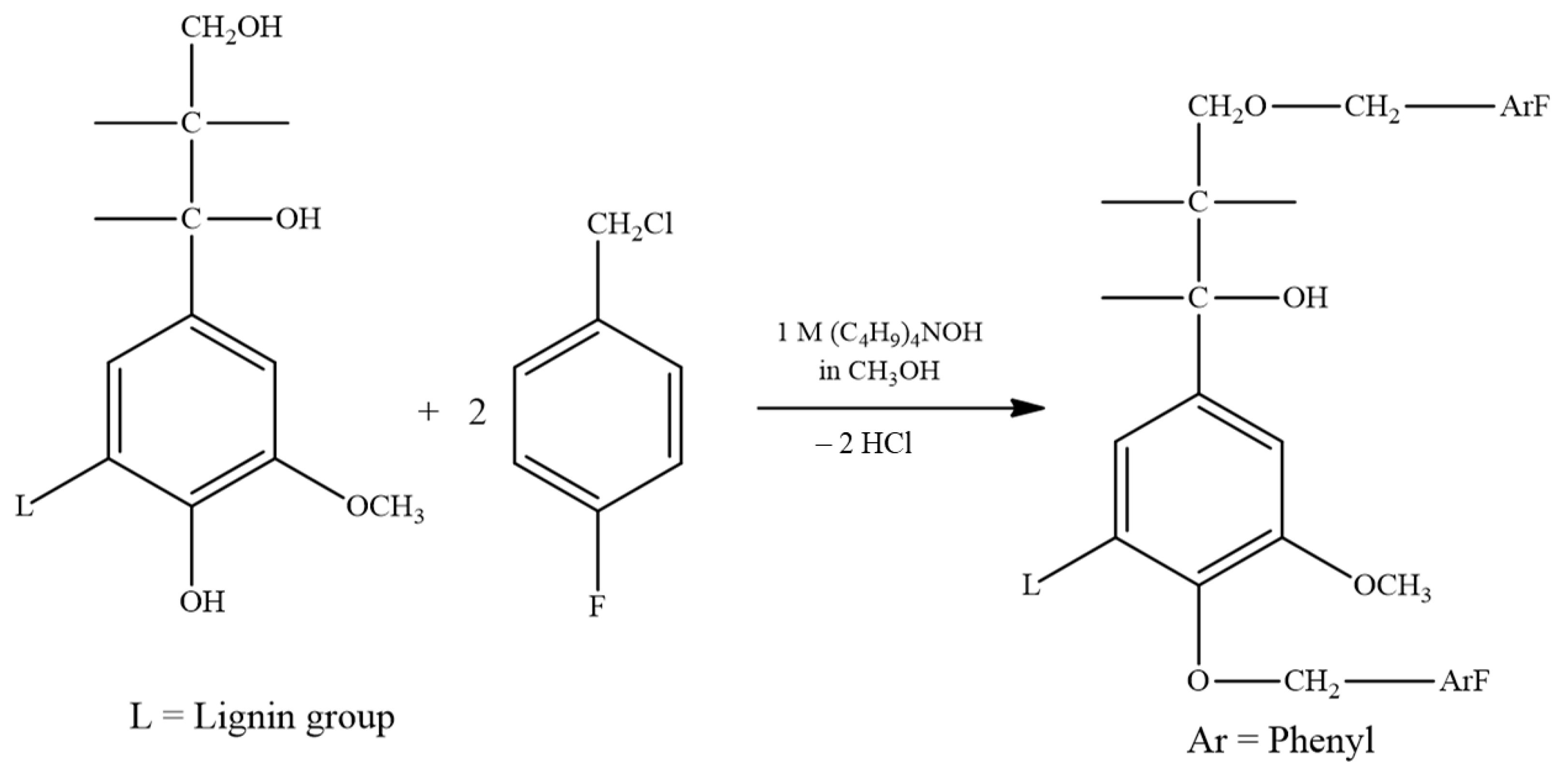
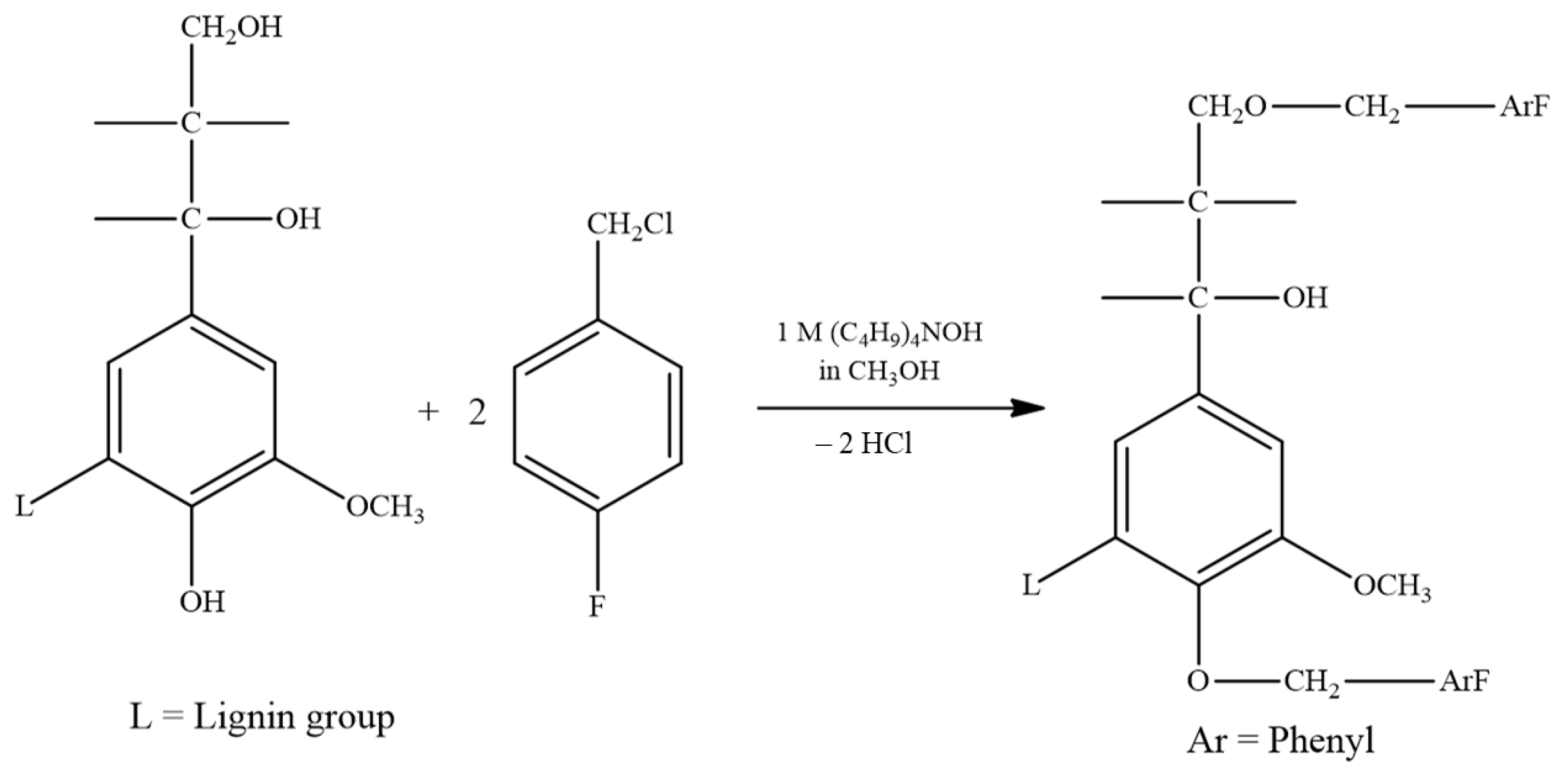
2.2.2. Fluorobenzylation
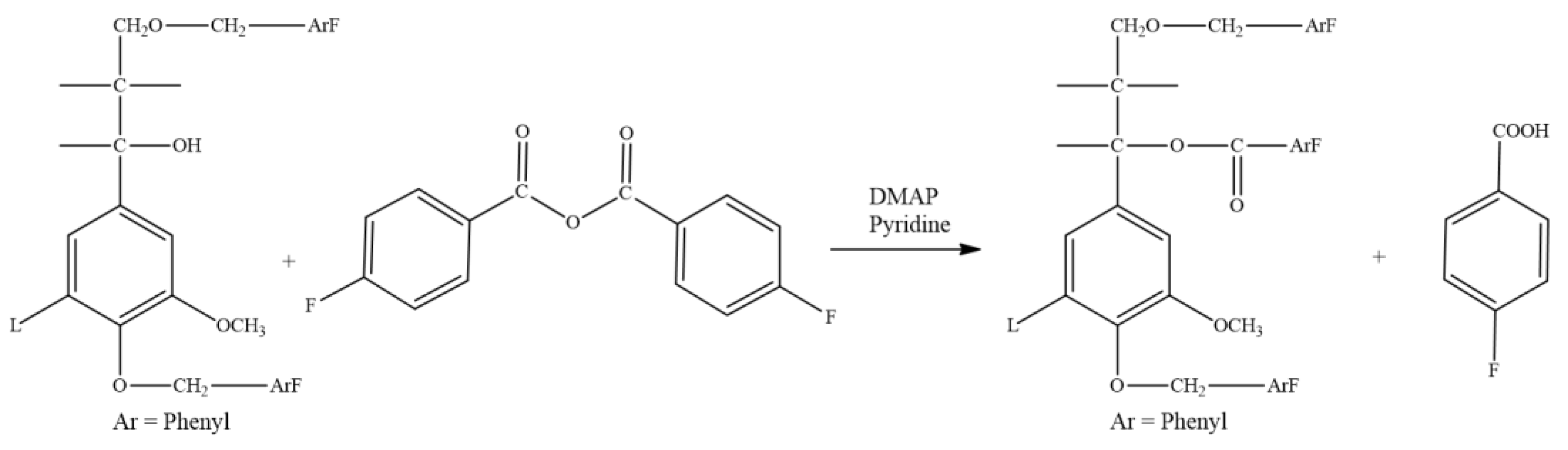
2.2.3. Fluorobenzoylation
2.3. MMD–SEC Conditions
3. Results and Discussion
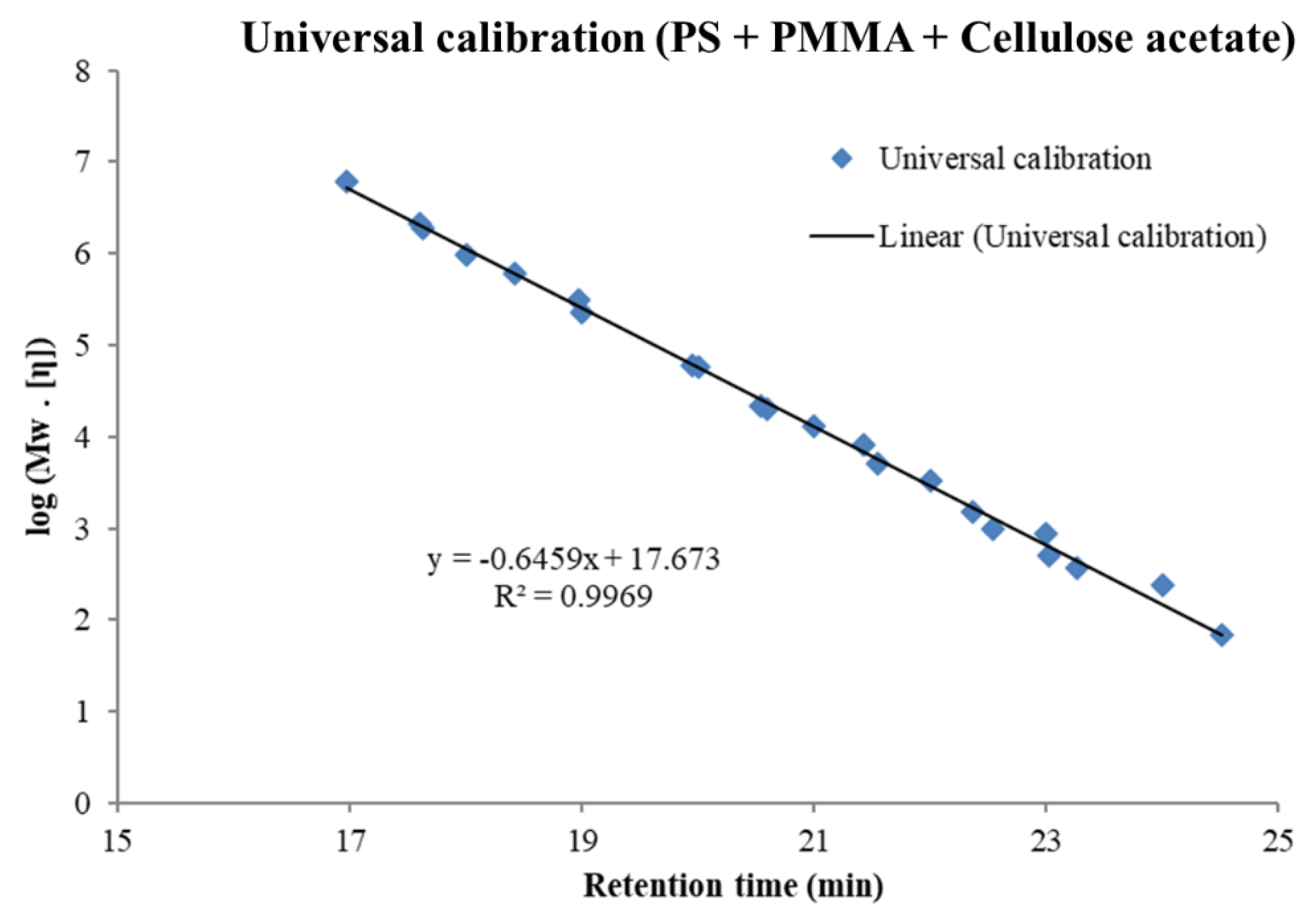
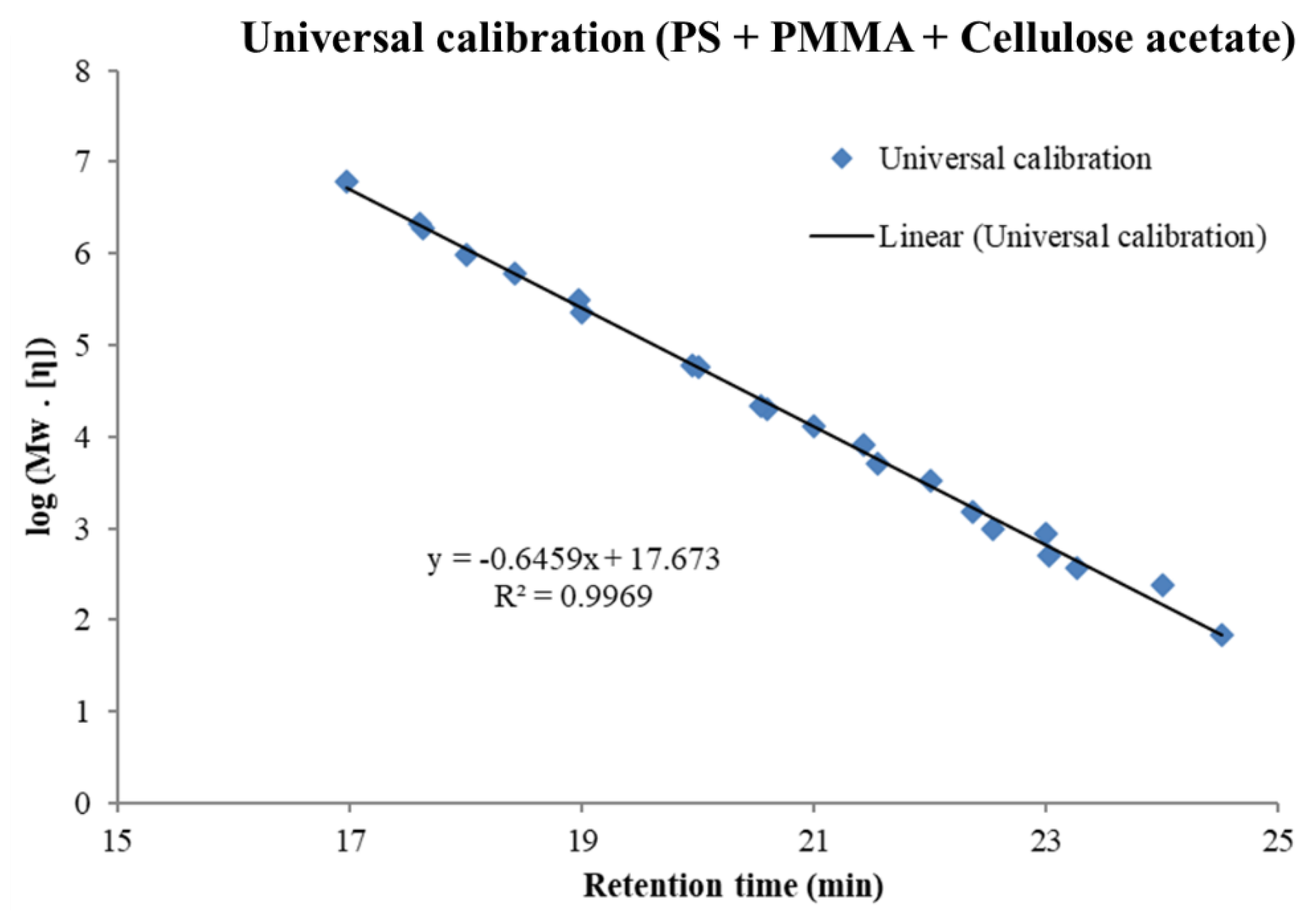
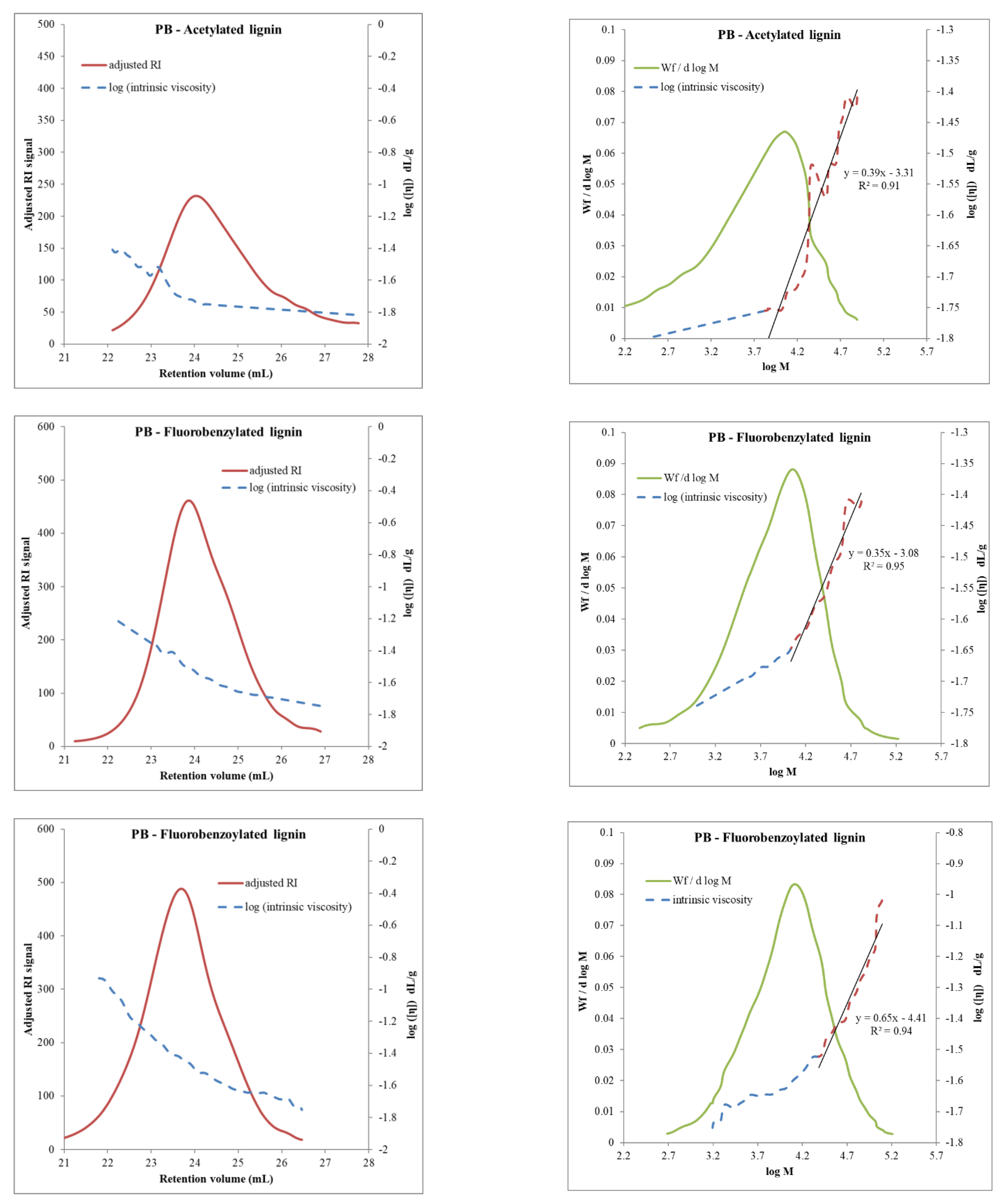
3.1. Molar Mass Determination Using the Universal Calibration
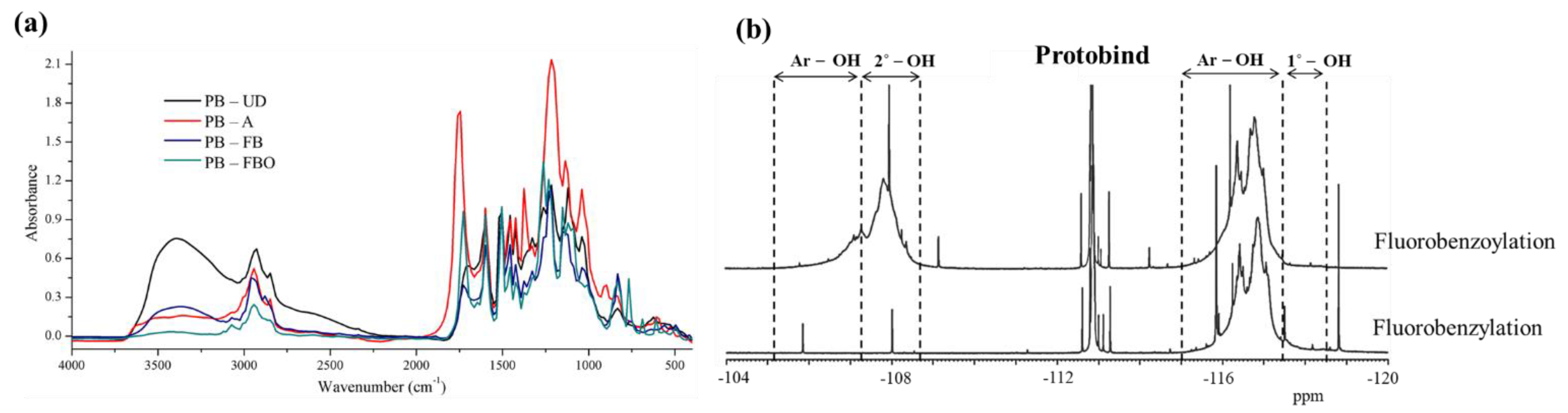
3.2. FTIR and 19F NMR Spectra of Different Lignin and Its Derivatives
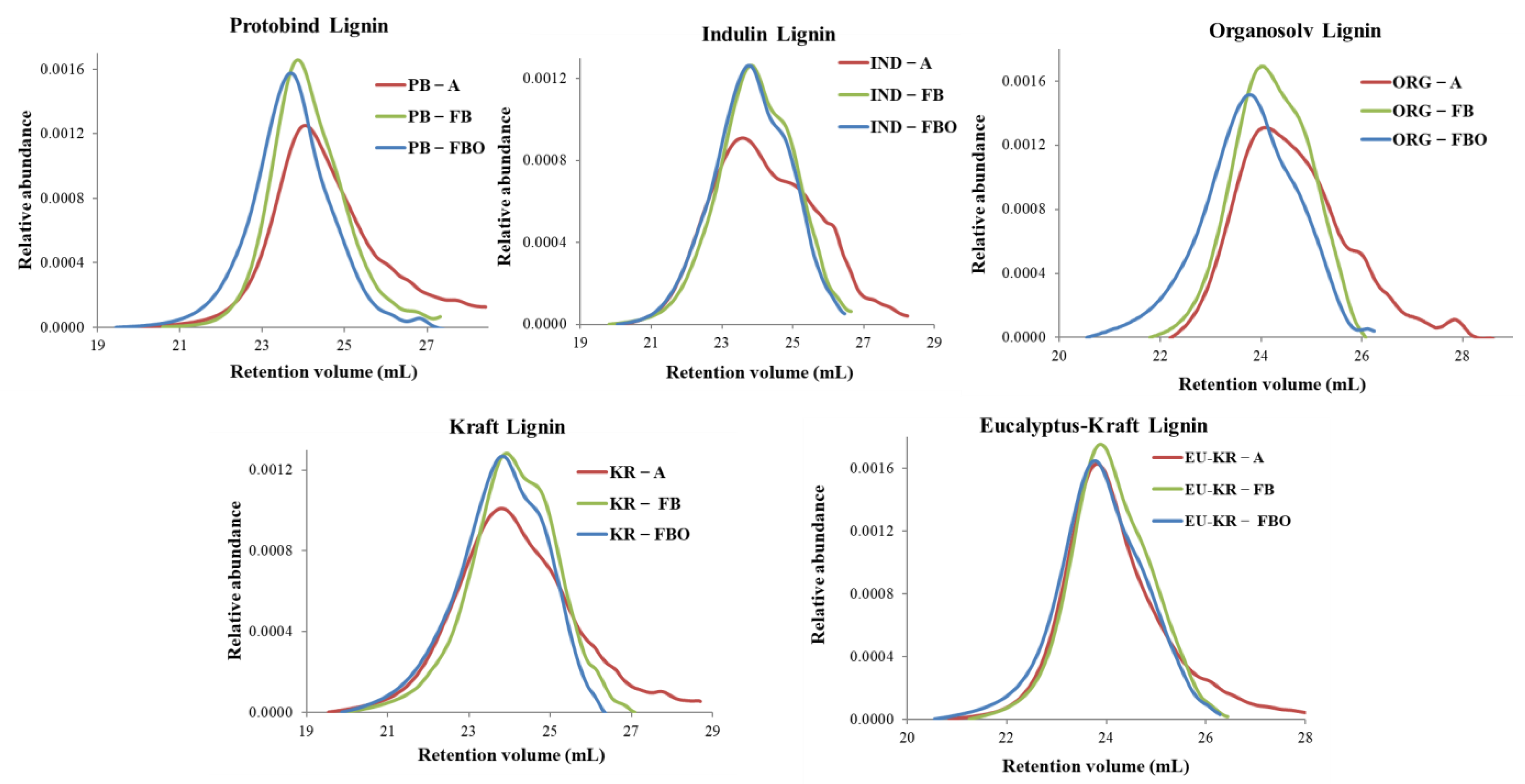
3.3. Molar Mass Distribution Curves of Different Derivatized Lignin Samples in SEC-THF System
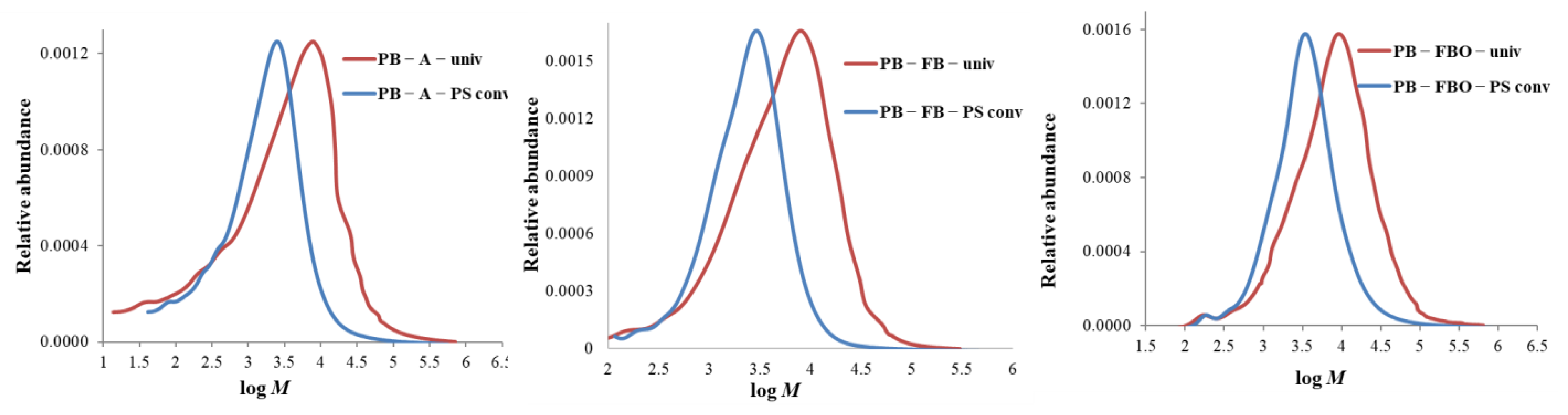
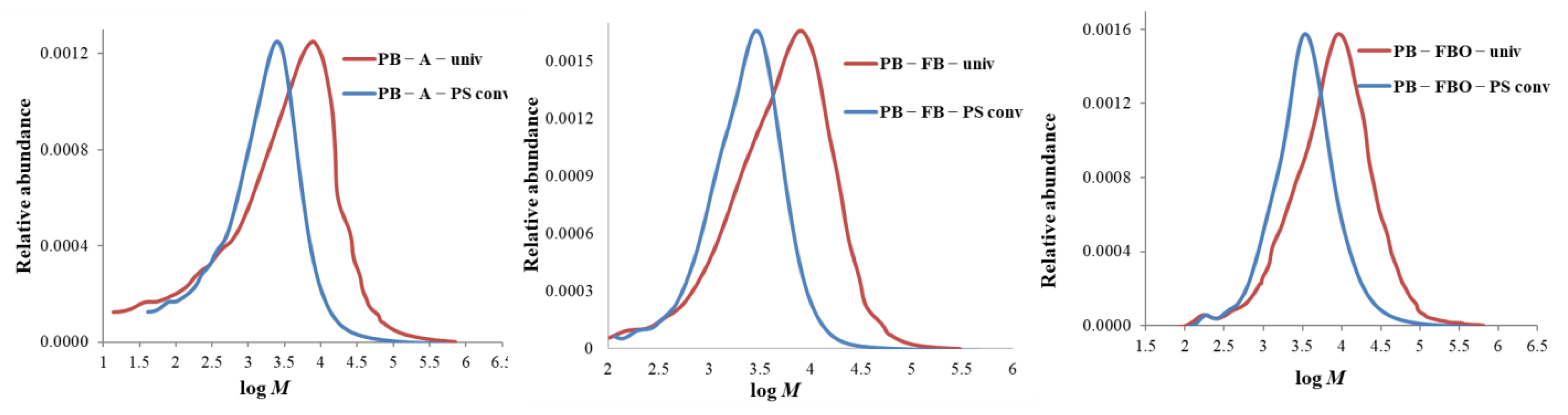
3.3.1. Molar Mass Distribution Curves: Comparison between the Standard and the Universal Calibration
- In all cases of universal calibration, average molar mass values using PS and PMMA as standards were rather close but more distant to the values using CA as standard.
- The difference between universal and conventional calibration is significant: M-averages and dispersity values obtained by conventional calibration appear 3 to 5 times lower than those obtained by universal calibration.
- In the case of PB lignin in Table 3, dispersity values of fluorobenzylated and fluorobenzoylated lignins were significantly lower than the acetylated sample.
- Similarly, in Table S4, acetylated IND showed Mw values closer to fluorobenzoylated IND than to fluorobenzylated IND. This is due to the width of the acetylated lignin MMD which extends in the high mass region, increasing Mw.
- As Mn values for acetylated lignin appear generally much lower than for fluoro-derivatives, this explains the large differences in dispersity index. It is likely that compared to other derivatives, acetylated lignins contain a larger number of small molecules which contribute to the decrease of Mn. Because of the size selection process during the precipitation procedure, fluoro-derivatives contain bigger molecular fractions, thus contributing to an increase of Mn and decrease of dispersity index.
- It is likely that in the case of ORG lignin, the solubility of the fluorobenzoylated lignin was better than for the two other derivatives and a major part of the higher-molar-mass lignin could be analysed, which increased the Mw value (Table S5). In the case of the ORG FB curve, the molecular weight was calculated until log M = 4.8 and after log M > 4.8, erratic measurement of [η] was observed which was represented as a dashed line (shown in Figure S4). Therefore, M-average value calculations were limited to the integration of the full-line portion of the curve.
- In the case of KR lignin, the acetylated lignin using universal calibration showed the highest MMD compared to fluoro-derivatives (Table S6). However, the Mn value was very low and it greatly influenced the dispersity value, compared to fluoro-derivatives. Fluorobenzoylated lignin dispersity value was lower than fluorobenzylated and acetylated derivatives and the higher molar mass addition increased significantly the average Mw and Mn in this case.
- Similarly, as for PB lignin, a small shift (compared to other lignins) towards higher molar mass was observed for EU-KR lignin. Average-M values in Table S7 indicate that standard calibration yields about 2–2.5 times lower average mass than universal calibration. With universal calibration, acetylated and fluorobenzylated lignins showed similar Mw values, but as for other lignins, the acetylated lignin Mn value was lower than for the fluorobenzylated sample, leading to a higher dispersity value.
3.3.2. Degree of Polymerization of Different Derivatives Using Universal Calibration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demirbaş, A. Relationships between Lignin Contents and Heating Values of Biomass. Energy Convers. Manag. 2001, 42, 183–188. [Google Scholar] [CrossRef]
- Hamelinck, C.N.; van Hooijdonk, G.; Faaij, A.P. Ethanol from Lignocellulosic Biomass: Techno-Economic Performance in Short-, Middle- and Long-Term. Biomass Bioenergy 2005, 28, 384–410. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Hatakeyama, T. Lignin Structure, Properties, and Applications. In Biopolymers: Lignin, Proteins, Bioactive Nanocomposites; Advances in Polymer Science; Abe, A., Dusek, K., Kobayashi, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1–63. ISBN 978-3-642-13630-6. [Google Scholar]
- Fengel, D.; Wegener, G. Wood: Chemistry, Ultrastructure, Reactions; Walter de Gruyter: Berlin, Germany, 2011; ISBN 978-3-11-083965-4. [Google Scholar]
- Pellinen, J.; Salkinoja-Salonen, M. High-Performance Size-Exclusion Chromatography of Lignin and Its Derivatives. J. Chromatogr. A 1985, 328, 299–308. [Google Scholar] [CrossRef]
- Baumberger, S.; Abaecherli, A.; Fasching, M.; Gellerstedt, G.; Gosselink, R.; Hortling, B.; Li, J.; Saake, B.; de Jong, E. Molar Mass Determination of Lignins by Size-Exclusion Chromatography: Towards Standardisation of the Method. Holzforschung 2007, 61, 459–468. [Google Scholar] [CrossRef]
- Mansouri, N.-E.E.; Salvadó, J. Structural Characterization of Technical Lignins for the Production of Adhesives: Application to Lignosulfonate, Kraft, Soda-Anthraquinone, Organosolv and Ethanol Process Lignins. Ind. Crop. Prod. 2006, 24, 8–16. [Google Scholar] [CrossRef]
- Sulaeva, I.; Zinovyev, G.; Plankeele, J.-M.; Sumerskii, I.; Rosenau, T.; Potthast, A. Fast Track to Molar-Mass Distributions of Technical Lignins. ChemSusChem 2017, 10, 629–635. [Google Scholar] [CrossRef]
- Ringena, O.; Lebioda, S.; Lehnen, R.; Saake, B. Size-Exclusion Chromatography of Technical Lignins in Dimethyl Sulfoxide/Water and Dimethylacetamide. J. Chromatogr. A 2006, 1102, 154–163. [Google Scholar] [CrossRef]
- Araújo, L.C.P.; Yamaji, F.M.; Lima, V.H.; Botaro, V.R. Kraft Lignin Fractionation by Organic Solvents: Correlation between Molar Mass and Higher Heating Value. Bioresour. Technol. 2020, 314, 123757. [Google Scholar] [CrossRef]
- Schure, M.R.; Moran, R.E. Size Exclusion Chromatography with Superficially Porous Particles. J. Chromatogr. A 2017, 1480, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Cathala, B.; Saake, B.; Faix, O.; Monties, B. Association Behaviour of Lignins and Lignin Model Compounds Studied by Multidetector Size-Exclusion Chromatography. J. Chromatogr. A 2003, 1020, 229–239. [Google Scholar] [CrossRef]
- Lange, H.; Rulli, F.; Crestini, C. Gel Permeation Chromatography in Determining Molecular Weights of Lignins: Critical Aspects Revisited for Improved Utility in the Development of Novel Materials. ACS Sustain. Chem. Eng. 2016, 4, 5167–5180. [Google Scholar] [CrossRef]
- Wang, L.; Shigetomi, K.; Koda, K.; Gele, A.; Uraki, Y. A Branched Structure Provides Kraft Lignins a Denser Morphology and a High Molar Mass for a given Hydrodynamic Radius. Holzforschung 2020, 74, 551–558. [Google Scholar] [CrossRef]
- Gosselink, R.J.A.; Abächerli, A.; Semke, H.; Malherbe, R.; Käuper, P.; Nadif, A.; van Dam, J.E.G. Analytical Protocols for Characterisation of Sulphur-Free Lignin. Ind. Crop. Prod. 2004, 19, 271–281. [Google Scholar] [CrossRef]
- Asikkala, J.; Tamminen, T.; Argyropoulos, D.S. Accurate and Reproducible Determination of Lignin Molar Mass by Acetobromination. J. Agric. Food Chem. 2012, 60, 8968–8973. [Google Scholar] [CrossRef] [PubMed]
- Duval, A.; Avérous, L. Mild and Controlled Lignin Methylation with Trimethyl Phosphate: Towards a Precise Control of Lignin Functionality. Green Chem. 2020, 22, 1671–1680. [Google Scholar] [CrossRef]
- Buono, P.; Duval, A.; Verge, P.; Averous, L.; Habibi, Y. New Insights on the Chemical Modification of Lignin: Acetylation versus Silylation. ACS Sustain. Chem. Eng. 2016, 4, 5212–5222. [Google Scholar] [CrossRef]
- Glasser, W.G.; Jain, R.K. Lignin Derivatives. I. Alkanoates. Holzforschung 1993, 47, 225–233. [Google Scholar] [CrossRef]
- Glasser, W.G.; Davé, V.; Frazier, C.E. Molecular Weight Distribution of (Semi-) Commercial Lignin Derivatives. J. Wood Chem. Technol. 1993, 13, 545–559. [Google Scholar] [CrossRef]
- Dong, D.; Fricke, A.L. Intrinsic Viscosity and the Molecular Weight of Kraft Lignin. Polymer 1995, 36, 2075–2078. [Google Scholar] [CrossRef]
- Alzahrani, Q.E.; Adams, G.G.; Gillis, R.B.; Besong, T.M.D.; Kök, M.S.; Fong, E.; Harding, R.A.; van Dam, J.E.G.; Gosselink, R.J.A.; Rowe, A.J.; et al. Matrix-Free Hydrodynamic Study on the Size Distribution and Conformation of Three Technical Lignins from Wood and Non-Wood. Holzforschung 2016, 70, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Gidh, A.V.; Decker, S.R.; See, C.H.; Himmel, M.E.; Williford, C.W. Characterization of Lignin Using Multi-Angle Laser Light Scattering and Atomic Force Microscopy. Anal. Chim. Acta 2006, 555, 250–258. [Google Scholar] [CrossRef]
- Jacobs, A.; Dahlman, O. Absolute Molar Mass of Lignins by Size Exclusion Chromatography and MALDI-TOF Mass Spectroscopy. Nord. Pulp Pap. Res. J. 2000, 15, 120–127. [Google Scholar] [CrossRef]
- Richel, A.; Vanderghem, C.; Simon, M.; Wathelet, B.; Paquot, M. Evaluation of Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry for Second-Generation Lignin Analysis. Anal. Chem. Insights 2012, 7, ACI-S10799. [Google Scholar] [CrossRef] [PubMed]
- Podzimek, S. Molar Mass Distribution by Size Exclusion Chromatography: Comparison of Multi-Angle Light Scattering and Universal Calibration. J. Appl. Polym. Sci. 2019, 136, 47561. [Google Scholar] [CrossRef]
- Wang, L.; Uraki, Y.; Koda, K.; Gele, A.; Zhou, X.; Chen, F. Determination of the Absolute Molar Mass of Acetylated Eucalyptus Kraft Lignin by Two Types of Size-Exclusion Chromatography Combined with Multi-Angle Laser Light-Scattering Detectors. Holzforschung 2019, 73, 363–369. [Google Scholar] [CrossRef]
- Zinovyev, G.; Sulaeva, I.; Podzimek, S.; Rössner, D.; Kilpeläinen, I.; Sumerskii, I.; Rosenau, T.; Potthast, A. Getting Closer to Absolute Molar Masses of Technical Lignins. ChemSusChem 2018, 11, 3259–3268. [Google Scholar] [CrossRef] [Green Version]
- Brown, W. Solution Properties of Lignin. Thermodynamic Properties and Molecular Weight Determinations. J. Appl. Polym. Sci. 1967, 11, 2381–2396. [Google Scholar] [CrossRef]
- Faix, O.; Lange, W.; Salud, E.C. The Use of HPLC for the Determination of Average Molecular Weights and Molecular Weight Distributions of Milled Wood Lignins from Shorea Polysperma (Blco.). Holzforschung 1981, 35, 3–9. [Google Scholar] [CrossRef]
- Chum, H.L.; Johnson, D.K.; Tucker, M.P.; Himmel, M.E. Some Aspects of Lignin Characterization by High Performance Size Exclusion Chromatography Using Styrene Divinylbenzene Copolymer Gels. Holzforschung 1987, 41, 97–108. [Google Scholar] [CrossRef]
- Himmel, M.E.; Tatsumoto, K.; Oh, K.K.; Grohmann, K.; Johnson, D.K.; Chum, H.L. Molecular Weight Distribution of Aspen Lignins Estimated by Universal Calibration. In Lignin; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1989; Volume 397, pp. 82–99. ISBN 978-0-8412-1631-0. [Google Scholar]
- Clauss, M.M.; Weldin, D.L.; Frank, E.; Giebel, E.; Buchmeiser, M.R. Size-Exclusion Chromatography and Aggregation Studies of Acetylated Lignins in N,N-Dimethylacetamide in the Presence of Salts. Macromol. Chem. Phys. 2015, 216, 2012–2019. [Google Scholar] [CrossRef]
- Sulaeva, I.; Vejdovszky, P.; Henniges, U.; Mahler, A.K.; Rosenau, T.; Potthast, A. Molar Mass Characterization of Crude Lignosulfonates by Asymmetric Flow Field-Flow Fractionation. ACS Sustain. Chem. Eng. 2019, 7, 216–223. [Google Scholar] [CrossRef]
- Faix, O. Fourier Transform Infrared Spectroscopy. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; Volume 10, pp. 83–109. [Google Scholar]
- Rönnols, J.; Jacobs, A.; Aldaeus, F. Consecutive Determination of Softwood Kraft Lignin Structure and Molar Mass from NMR Measurements. Holzforschung 2017, 71, 563–570. [Google Scholar] [CrossRef]
- Serrano, L.; Esakkimuthu, E.S.; Marlin, N.; Brochier-Salon, M.-C.; Mortha, G.; Bertaud, F. Fast, Easy, and Economical Quantification of Lignin Phenolic Hydroxyl Groups: Comparison with Classical Techniques. Energy Fuels 2018, 32, 5969–5977. [Google Scholar] [CrossRef]
- Esakkimuthu, E.S. Study of New Chemical Derivatization Techniques for Lignin Analysis by Size Exclusion Chromatography. Ph.D. Thesis, Université Grenoble Alpes, Grenoble, France, 2020. [Google Scholar]
- Esakkimuthu, E.S.; Marlin, N.; Brochier-Salon, M.-C.; Mortha, G. Study of the Reactivity of Lignin Model Compounds to Fluorobenzylation Using 13C and 19F NMR: Application to Lignin Phenolic Hydroxyl Group Quantification by 19F NMR. Molecules 2020, 25, 3211. [Google Scholar] [CrossRef]
- Barrelle, M.; Fernandes, J.C.; Froment, P.; Lachenal, D. An Approach to the Determination of Functional Groups in Oxidized Lignins by 19F Nmr. J. Wood Chem. Technol. 1992, 12, 413–424. [Google Scholar] [CrossRef]
- Barrelle, M. Improvements in the Structural Investigation of Lignins by 19F NMR Spectroscopy. J. Wood Chem. Technol. 1995, 15, 179–188. [Google Scholar] [CrossRef]
- Benoit, H.; Grubisic, Z.; Rempp, P.; Decker, D.; Zilliox, J.-G. Étude par chromatographie en phase liquide de polystyrènes linéaires et ramifiés de structures connues. J. Chim. Phys. 1966, 63, 1507–1514. [Google Scholar] [CrossRef]
- Ahvazi, B.C.; Crestini, C.; Argyropoulos, D.S. 19F Nuclear Magnetic Resonance Spectroscopy for the Quantitative Detection and Classification of Carbonyl Groups in Lignins. J. Agric. Food Chem. 1999, 47, 190–201. [Google Scholar] [CrossRef]
- Zoia, L.; King, A.W.T.; Argyropoulos, D.S. Molecular Weight Distributions and Linkages in Lignocellulosic Materials Derivatized from Ionic Liquid Media. J. Agric. Food Chem. 2011, 59, 829–838. [Google Scholar] [CrossRef]
- Faix, O.; Lange, W.; Beinhoff, O. Molecular weights and molecular weight distributions of milled wood lignins of some wood and bambusoideae species. Holzforschung 1980, 34, 174–176. [Google Scholar] [CrossRef]
- Nascimento, E.A.; Morais, S.A.; Machado, A.E.; Veloso, D.P. Studies of Eucalyptus Grandis Lignin. Part II: High-Performance Size-Exclusion Chromatography of Milled Wood Lignin, Kraft and Organosolv Lignins. J. Braz. Chem. Soc. 1992, 3, 61–64. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lignin | Fluorobenzylated | Total OHCalculated mmol/g Derivatized Lignin | |
|---|---|---|---|
| Ph–OH | 1°–OH | ||
| PB | 1.785 | 0.106 | 1.891 |
| ORG | 1.708 | 0.120 | 1.828 |
| IND | 2.451 | 0.078 | 2.529 |
| KR | 2.252 | 0.089 | 2.341 |
| EU-KR | 2.322 | 0.059 | 2.381 |
| Lignin | Molar Mass of Monomer | |
|---|---|---|
| FB | FBO | |
| PB | 251 | 308 |
| ORG | 249 | 275 |
| IND | 275 | 328 |
| KR | 268 | 310 |
| EU-KR | 269 | 291 |
| Universal Calibration (Viscometric + RI Detectors) | Standard Calibration (RI Detector) | |||||||
|---|---|---|---|---|---|---|---|---|
| PB–Acetylated | Mn | Mw | Mw/Mn | Mpeak | Mn | Mw | Mw/Mn | Mpeak |
| Fit with Global curve a | 380 | 10,060 | 26.5 | 7780 | - | - | - | - |
| PS | 330 | 9030 | 27.4 | 6910 | 560 | 2890 | 5.2 | 2500 |
| PMMA | 240 | 8260 | 34.4 | 5981 | 725 | 3640 | 5.0 | 3150 |
| Cellulose acetate | 700 | 13,260 | 18.9 | 11,090 | 856 | 2720 | 3.2 | 2620 |
| PB–Fluorobenzylated | ||||||||
| Fit with Global curve a | 2000 | 9560 | 4.8 | 7980 | - | - | - | - |
| PS | 1760 | 8570 | 4.9 | 7110 | 1450 | 3180 | 2.2 | 2930 |
| PMMA | 1400 | 7700 | 5.5 | 6200 | 1840 | 4000 | 2.2 | 3690 |
| Cellulose acetate | 3220 | 12,890 | 4.0 | 11,230 | 1770 | 3060 | 1.7 | 2990 |
| PB–Fluorobenzoylated | ||||||||
| Fit with Global curve a | 3690 | 16,140 | 4.4 | 9240 | - | - | - | - |
| PS | 3230 | 14,550 | 4.5 | 8250 | 2140 | 5390 | 2.5 | 3430 |
| PMMA | 2640 | 13,510 | 5.1 | 7250 | 2710 | 6740 | 2.5 | 4320 |
| Cellulose acetate | 5640 | 20,680 | 3.7 | 12,850 | 2450 | 4660 | 1.9 | 3420 |
| Universal Calibration in THF | |||||
|---|---|---|---|---|---|
| Molar Mass of Monomer (g/mol) | DPn | DPw | Dispersity | ||
| PB | Acetylated | 240 | 2 | 42 | 26.2 |
| FB | 251 * | 8 | 38 | 4.8 | |
| FBO | 308 * | 12 | 52 | 4.4 | |
| IND | Acetylated | 240 | 5 | 83 | 18.3 |
| FB | 275 * | 9 | 56 | 6.3 | |
| FBO | 328 * | 11 | 68 | 5.9 | |
| ORG | Acetylated | 240 | 12 | 79 | 6.5 |
| FB | 249 * | -- | -- | -- | |
| FBO | 275 * | 31 | 125 | 4 | |
| KR | Acetylated | 240 | 2 | 129 | 57.5 |
| FB | 268 * | 8 | 51 | 6.2 | |
| FBO | 310 * | 10 | 59 | 5.7 | |
| EU-KR | Acetylated | 240 | 5 | 35 | 6.7 |
| FB | 269 * | 13 | 32 | 2.5 | |
| FBO | 291 * | 12 | 45 | 3.6 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esakkimuthu, E.S.; Marlin, N.; Brochier-Salon, M.-C.; Mortha, G. Application of a Universal Calibration Method for True Molar Mass Determination of Fluoro-Derivatized Technical Lignins by Size-Exclusion Chromatography. AppliedChem 2022, 2, 30-47. https://doi.org/10.3390/appliedchem2010002
Esakkimuthu ES, Marlin N, Brochier-Salon M-C, Mortha G. Application of a Universal Calibration Method for True Molar Mass Determination of Fluoro-Derivatized Technical Lignins by Size-Exclusion Chromatography. AppliedChem. 2022; 2(1):30-47. https://doi.org/10.3390/appliedchem2010002
Chicago/Turabian StyleEsakkimuthu, Esakkiammal Sudha, Nathalie Marlin, Marie-Christine Brochier-Salon, and Gérard Mortha. 2022. "Application of a Universal Calibration Method for True Molar Mass Determination of Fluoro-Derivatized Technical Lignins by Size-Exclusion Chromatography" AppliedChem 2, no. 1: 30-47. https://doi.org/10.3390/appliedchem2010002
APA StyleEsakkimuthu, E. S., Marlin, N., Brochier-Salon, M.-C., & Mortha, G. (2022). Application of a Universal Calibration Method for True Molar Mass Determination of Fluoro-Derivatized Technical Lignins by Size-Exclusion Chromatography. AppliedChem, 2(1), 30-47. https://doi.org/10.3390/appliedchem2010002