1. Introduction
Vascular endothelial growth factor A (VEGF) belongs to a superfamily of homodimeric proteins that modulate cell function from the extracellular environment through activation of cell surface receptors. VEGF, in particular, has been identified as a critical factor that drives vasculogenesis and angiogenesis [
1]. Angiogenesis, the growth of new blood vessels from existing ones, has been identified as a key step in the transition of cancer to malignancy and in promoting metastasis. Consequently, considerable effort has been directed at developing approaches to inhibit VEGF-mediated angiogenesis for the treatment of cancers and other diseases associated with excessive vascular growth [
2,
3]. VEGF receptor 2 has been identified as the key signaling receptor on endothelial cells responsible for VEGF-mediated angiogenesis. Thus, most therapeutic approaches have focused on blocking VEGF receptor 2 activation and/or signaling through the use of monoclonal antibodies, fusion protein ligand traps, tyrosine kinase inhibitors, and inhibitors of downstream signaling [
1]. However, VEGF also binds to and is modulated by other components present on the cell surface and within the extracellular matrix of endothelial cells where these interactions may alter the efficacy of anti-VEGF therapies [
4,
5]. Indeed, we have previously found that binding of VEGF to heparin and fibronectin can have dramatic effects on the binding of VEGF to a therapeutic anti-VEGF antibody, bevacizumab (known by the tradename Avastin) [
6].
Avastin was approved in 2004 by the FDA for treatment of colorectal cancer and has since been widely used to treat a variety of other cancers and ocular disorders [
7]. Avastin, also known as bevacizumab, is a recombinant humanized monoclonal antibody that binds to VEGF and inhibits its interaction with VEGF receptor 2 (VEGFR-2). While Avastin has proven to be an effective anti-angiogenic drug, factors such as its relatively short half-life, long adsorption time, and low bioavailability results in the need for repetitive administration of high doses. Moreover, a number of adverse side effects have also been noted including hemorrhage, retinal detachment, venous thrombosis, hypertension, proteinuria, stroke, and heart failure [
7,
8,
9]. Consequently, considerable effort has been directed at improving Avastin efficacy while reducing its negative side effects. Approaches have included incorporation of Avastin within various hydrogels and co-therapy with other drugs and proteins [
10,
11,
12,
13,
14]. These new approaches have shown promising results, but have not yet led to changes in the current state of Avastin therapy. Thus, the wide clinical use of Avastin continues to highlight the need for improvements that amplify the beneficial actions of this valuable pharmaceutical agent.
We have previously observed that the VEGF–Avastin interaction is dramatically destabilized at low pH [
6]. Reduced VEGF–Avastin binding may limit the effectiveness of Avastin within tumors and other undervascularized tissues that are characterized by low extracellular pH as a consequence of the local hypoxia. Heparin, a well-known binding partner of VEGF, has also been shown to modulate VEGF binding to VEGFR-2 [
4,
15,
16,
17,
18]. The effect of heparin on VEGF binding is complex. Heparin can both increase and decrease the binding of VEGF to VEGFR-2 and to the extracellular matrix protein fibronectin based on concentration and the specific experimental conditions [
4,
15,
19,
20,
21,
22,
23,
24]. We previously showed that heparin does not interfere with the binding of Avastin to VEGF, but instead it enhances this interaction [
6]. Importantly, heparin is also able to rescue the reduced binding of Avastin to VEGF at acidic pH. These results suggest that heparin might be used to enhance Avastin efficacy. Thus, here, we propose to conjugate Avastin and heparin to improve the VEGF binding properties of Avastin. Conjugation, rather than intravenous injections of the two drug mixtures, would ensure that both drugs are present within the same molecular space at the same time in order to form a high affinity ternary complex with VEGF. While Avastin has a half-life of 19.9 days [
25], heparin only has a half-life of 1.5 h [
26]; thus, conjugating the two molecules has the potential to coordinate the pharmacokinetics and pharmacodynamics of the two agents.
To establish the proof of concept of the proposed drug conjugate, biotin–heparin and biotin–Avastin were linked to streptavidin surfaces to bring the two molecules into close proximity. Immobilized heparin and Avastin were shown to have enhanced ability to bind VEGF. Increased VEGF binding was also noted at acidic pH, where Avastin alone showed decreased VEGF binding. Under conditions where Avastin and heparin showed enhanced VEGF binding, we observed reduced VEGF-induced migration of endothelial cells. These findings indicate design principles for a modified Avastin-based inhibitor of angiogenesis.
2. Materials and Methods
2.1. Materials
Streptavidin-coated 96-well plates (#15500) and protein A-coated plates (#15130) were from Pierce (Rockford, IL, USA). Avastin (bevacizumab) (#A2006) was from Selleck Chemicals (Houston, TX, USA). Heparin, from porcine intestinal mucosa (#H3393), human plasma fibronectin (FN) (#FC010), and biotin-heparin (#B9806) were from Millipore Sigma (Burlington, MA, USA). Cell Culture reagents: Dulbecco’s modified Eagle’s medium (DMEM) (#10014CV), penicillin–streptomycin solution, 50X (#30-001-CI), and L-glutamine (#25005CI) were purchased from Corning (Tewkesbury, MA, USA). Fetal bovine serum (FBS) (#S11550) and newborn calf serum (CS) (#S11250) were from Atlanta Biologics (Atlanta, GA, USA). Recombinant human vascular endothelial growth factor 165 (VEGF) (#293-VE) was purchased from R&D Systems (Minneapolis, MN, USA).
125 I-Bolton-Hunter’s Reagent was purchased from Perkin Elmer Life Science (Boston, MA, USA) and was used to label the VEGF as previously described [
19]. Casein (#37528) in phosphate buffered saline (PBS), the BCA assay kit (#23227), and the EZ-Link Sulfo-NHS-LC Biotinylation kit (#21935) were obtained from ThermoFisher Scientific (Waltham, MA, USA). The Streptavidin Conjuagation Kit (#ab102921) was obtained from Abcam Inc (Cambridge, MA, USA). Peroxidase-AffiniPure Donkey Anti-Human IgG, Fc (gamma) Fragment Specific (#709-035-098), and anti-rabbit horseradish peroxidase (HRP)-linked secondary antibody (#111-035-144), and streptavidin (#016-000-113) were obtained from Jackson Immunoresearch (West Grove, PA, USA).
2.2. Cell Culture
Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza (Walkersvile, MD, USA). Each preparation of cells was a batch of pooled cells from 4–6 individuals that were screened by the vendor for expression of PECAM-1, endoglin, and von Willebrand factor VIII. They were also shown to have acetylated low-density lipoprotein uptake. The cells were maintained in endothelial cell growth medium-2 containing 2% FBS, hydrocortisone, human fibroblast growth factor, 2 ng/mL vascular endothelial growth factor, insulin-like growth factor-1, ascorbic acid, human recombinant endothelial growth factor, GA-1000, and heparin in T-75 flasks that were coated with 0.1% porcine gelatin for 30 min at 37 °C, 5% CO2. The media was replaced every two days as per vendor’s instructions.
2.3. Avastin Biotinylation
Avastin was labeled with biotin in accordance with the manufacturer’s directions. The required volume for 200 µg Avastin (13.5 µL of 14.9 mg/mL) was diluted to a volume of 700 µL in PBS and mixed with 9 mM sulfo-NHS-LC-biotin (7.52 µL) to achieve a 50-fold molar excess of labeling reagent to the protein. The mixture was incubated for 1 h at RT. Zeba spin desalting columns were equilibrated by adding 1 mL of PBS and centrifuging at 1000× g for 2 min, and this was repeated 3 more times. The incubated biotin–Avastin labeling mixture was added to the column and allowed to absorb into the resin. The column was spun down at 1000× g for 2 min and the flow through containing the labeled protein was collected and saved for future experiments.
2.4. Streptavidin–Avastin Conjugation
Avastin was linked to streptavidin in accordance with the manufacturer’s instructions. A 100 µg quantity of Avastin (100 µL of 1 mg/mL) was mixed with a 1:10 volume of modifier and the mixture was used to reconstitute the lyophilized activated streptavidin mix. The mixture was allowed to incubate at RT for 3 h and then the quencher reagent was added at a ratio of 1:10 to the antibody volume. The mixture was incubated for 30 min at RT and then stored at 4 °C for future experiments. The concentration of streptavidin-conjugated Avastin was recalculated based on the additional volumes and the original Avastin quantity.
2.5. VEGF Binding Assays
VEGF binding assays were conducted with protein A-coated or streptavidin-coated 96-well plates as described in the legends to the figures. Protein A-coated plates were incubated with either Avastin or biotinylated-Avastin or streptavidin–Avastin in 0.1% BSA, 0.05% Tween-20 in PBS (PBS-B-T) for 2 h at RT on a shaker. The unbound protein was removed by washing each well three times with PBS-B-T, and then 125 I-VEGF was added to each well and incubated for 2 h on a shaker at RT. Biotinylated-Avastin, biotinylated-heparin, and heparin were incubated with streptavidin-coated plates in PBS overnight at 4 °C on a shaker (100 µL/well). Plates were washed to remove unbound molecules and 125 I-VEGF was added to each well in 100 µL PBS-B-T. The treatments were incubated for 2 h at 4 °C on a shaker, and then the unbound material was removed by washing each well three times with binding buffer. In both experimental conditions, bound 125 I-VEGF was extracted from the plate surface with 100 µL of 1 N NaOH for 5 min followed by an additional 100 µL of 1 N NaOH. The radioactivity in each sample was counted using a gamma counter.
2.6. Cell Migration
HUVEC migration was measured using a modified Boyden chamber assay system where cells were plated into the top chamber onto a porous membrane and VEGF was presented along with surface-immobilized Avastin, heparin, and both within the bottom chamber. HUVECs at 70–80% confluence (75 cm2 flasks) were washed twice with endothelial basal media (EBM) with 0.1% bovine serum albumin (BSA), then supplemented with EBM media (15 mL/flask) and placed in the incubator for 5 h at 37 °C, 5% CO2. The cells were then trypsinized and resuspended in trypsin neutralizing solution. The cells were collected by centrifugation (200× g, 5 min). The pellet was resuspended in EBM, containing 0.1% BSA. The cells were counted, and 50,000 cells were placed on the top compartment of 8 μM FluoroBlok inserts (Corning, Corning, NY, USA) that had been coated with 0.1% porcine gelatin for 30 min at 37 °C, 5% CO2.
The bottom chamber was prepared by coating 24-well falcon tissue culture plates with 300 µL/well of streptavidin (200 nM) in 1× PBS overnight at 4 °C. The following day, the wells were washed once with 1× PBS and then incubated with 300 µL of 1× PBS ± 1 nM biotinylated Avastin, ±50 nM biotinylated heparin overnight on the shaker at 4 °C. The wells were then incubated with 750 µL of EBM, containing 0.1% BSA for 2 h at 4 °C on the shaker. After VEGF was allowed to interact with the immobilized biotinylated Avastin and heparin, the inserts containing the cells were transferred to the plates containing the chemoattractant. The cells were placed in the incubator at 37 °C, 5% CO2, for 14–16 h overnight to allow sufficient time to migrate, but not enough time to undergo cell division. The following day, the unattached cells in the top chamber were removed, and then inserts were transferred to new wells containing 750 µL of 4 µg/mL Calcein AM (Thermo Fisher Scientific, Waltham, MA, USA). The plates were incubated for 1.5 h at 37 °C, 5% CO2 and then read on a Spectromax Fluorescence Plate Reader with excitation: 485 nm, emission: 530 nm and auto cutoff at 515 nm, with the bottom plate only read at 6 flashes/read.
2.7. Statistical Analysis
Student’s t-test (2-tailed) were used to evaluate the statistical significance when comparing two experimental groups. In experiments with more than two experimental groups, data were subjected to analysis of variance (ANOVA) followed by multi-comparison t-tests (Newman–Keuls method). Groups were considered to be statistically different from one another when p < 0.05.
3. Results
3.1. Co-Localizing Biotin–Avastin and Biotin–Heparin Leads to Enhanced Avastin–VEGF Binding
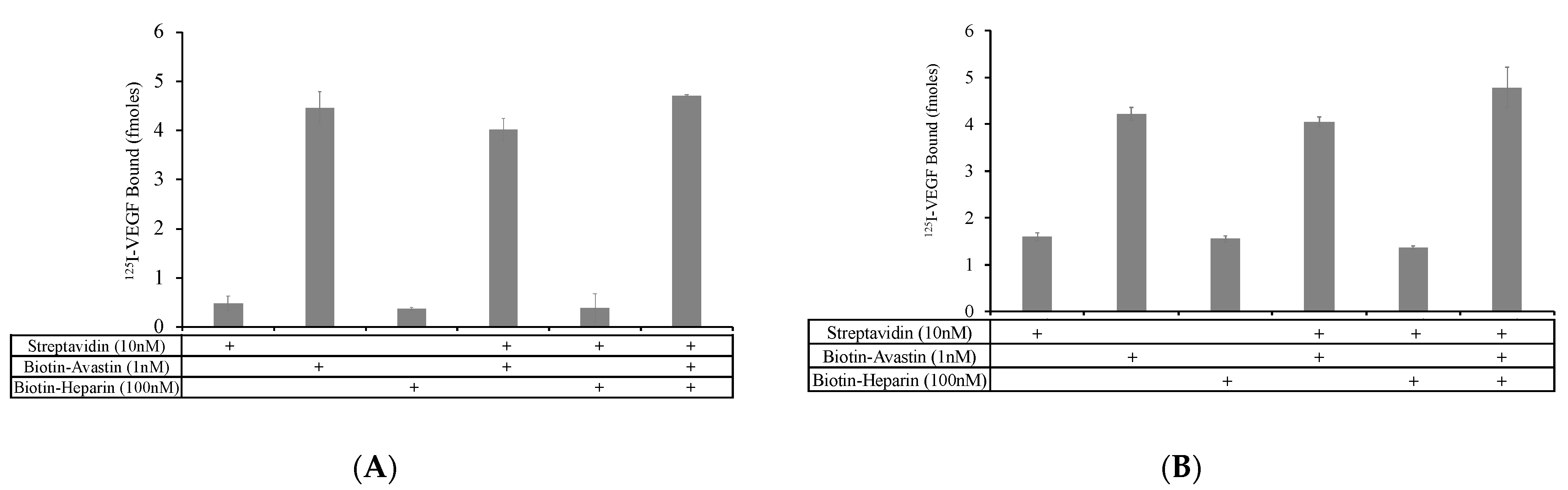
Avastin was conjugated to biotin so that complexes of biotin–Avastin and biotin–heparin could be linked via streptavidin. To confirm that biotin–Avastin was capable of binding to streptavidin, biotin–Avastin and Avastin (5 nM and 10 nM) were incubated with a streptavidin-coated plate (
Figure 1A). Binding of
125 I-VEGF to the streptavidin plate was measured. As expected, there was no detectable binding of
125 I-VEGF to the wells incubated with non-conjugated Avastin, whereas there was considerable binding to the wells incubated with the biotin–Avastin. These data indicate that the biotin-conjugation of Avastin did not block its VEGF binding site and allows the biotin–Avastin to be functionally immobilized onto streptavidin-coated surfaces. To more directly evaluate the relative binding of VEGF to Avastin with and without the biotin conjugation, a side-by-side comparison of VEGF binding to biotin–Avastin and unmodified Avastin was conducted by linking the two Avastin preparations to a protein A plate.
125 I-VEGF bound to both biotin–Avastin and unmodified Avastin, but the level of binding to biotin–Avastin was decreased by 45% compared to the unmodified version (
Figure 1B). The reduced binding of VEGF could reflect modification of the VEGF binding domain or the Fc regions on Avastin; thus, either destabilizing the Avastin–VEGF or Avastin–protein A interactions.
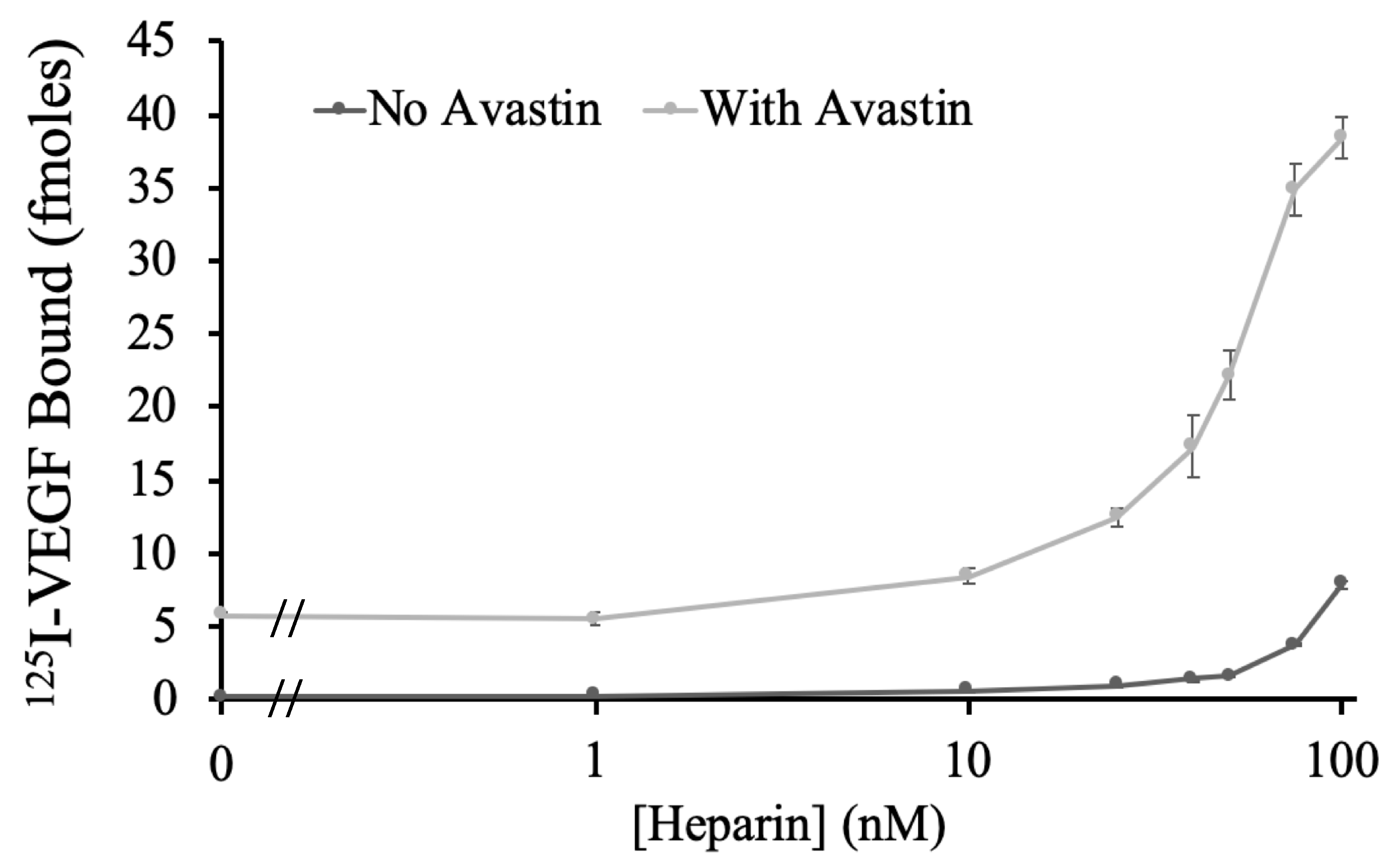
To examine if bringing heparin and Avastin into close proximity would enhance VEGF binding to Avastin, biotin–Avastin (1 nM) and a concentration range of biotin–heparin (0–100 nM) were bound onto a streptavidin plate and incubated with
125 I-VEGF. As shown in
Figure 2, biotin–heparin showed a dose-dependent increase in VEGF binding to biotin–Avastin, where the highest dose tested (100 nM) caused a greater than 6-fold increase. Since the combination of biotinylated Avastin and heparin showed enhanced VEGF binding, direct conjugation of these two molecules was attempted using streptavidin.
3.2. Creating Soluble Heparin–Avastin Complexes Using Biotin and Streptavidin
Streptavidin contains four binding sites for biotin; thus, in an effort to create soluble heparin–Avastin conjugates, we linked biotin–Avastin and biotin–heparin to streptavidin and then selectively isolated those complexes that contained Avastin using protein A.
Mixtures ± biotin–Avastin (1 nM) ± streptavidin (10 nM) were prepared and incubated overnight. By using a molar excess of streptavidin to Avastin, it was predicted that few of the streptavidin molecules would be occupied by multiple Avastin molecules such that biotin binding sites should remain available. However, it is also likely that each Avastin molecule contains multiple biotin molecules such that a single biotin–Avastin molecule might bind to multiple sites on streptavidin. In an attempt to link heparin and Avastin onto the same streptavidin molecules, the biotin–Avastin–streptavidin complexes were incubated with biotin–heparin (100 nM). The soluble complexes were allowed to bind to a protein A plate to selectively capture only those complexes containing Avastin. 125 I-VEGF binding was conducted with the various complexes.
No differences in VEGF binding to biotin–Avastin and streptavidin–biotin–Avastin were observed (
Figure 3A), indicating that linking of biotin–Avastin to streptavidin does not sterically interfere with VEGF binding to Avastin. The presence of biotin–heparin in combination with biotin–Avastin resulted in a modest, but not statistically significant, increase in VEGF binding compared to that observed with biotin–Avastin alone, with or without streptavidin. When the VEGF binding assays were conducted at pH 5.5, biotin–heparin caused a small statistically significant increase in VEGF binding to biotin–Avastin in the presence of streptavidin compared to that observed without biotin–heparin (
Figure 3B).
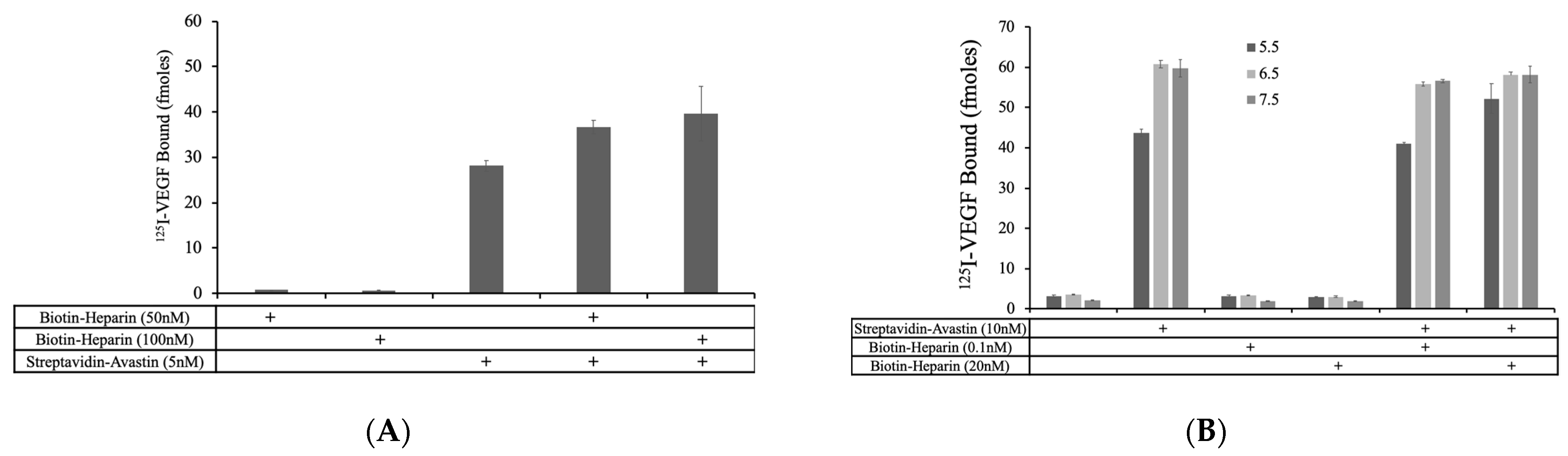
The fact that the addition of biotin–heparin had a relatively small effect on VEGF binding to biotin–Avastin when bound to soluble streptavidin suggest that it is unlikely that this approach led to a significant number of streptavidin molecules containing both biotin–heparin and biotin–Avastin so that the molecules could effectively synergize to bind VEGF. In an attempt to circumvent this limitation, Avastin was directly conjugated to streptavidin and then biotin–heparin was allowed to bind to this complex. Mixtures of streptavidin–Avastin (5 nM) ± biotin–heparin (50 nM and 100 nM) were prepared and coated onto protein A plates to selectively isolate complexes containing Avastin, and VEGF binding was measured.
The addition of biotin–heparin at 50 and 100 nM resulted in a 30.4% and 41.1% increase in VEGF binding, respectively, compared to that observed with streptavidin–Avastin alone (
Figure 4A). Additionally, when this experiment was performed with streptavidin–Avastin (10 nM) ± lower concentrations of biotin–heparin (0.1 nM and 20 nM) at pH 5.5, 6.5, and 7.5, VEGF binding showed a 19.4% increase in the presence of biotin–heparin at pH 5.5, with no change at pH 6.5 and 7.5 (
Figure 4B), confirming the ability of heparin to promote VEGF binding to Avastin at low pH under these conditions. These data indicate that it may be possible to link heparin and Avastin to enhance VEGF binding; therefore, we decided to characterize VEGF binding while these molecules were co-localized on the same surface to expand on these initial findings.
3.3. Biotin–Avastin and Biotin–Heparin Showed Enhanced VEGF Binding Dependent on Streptavidin Density and pH
Commercial streptavidin plates contain a saturated monolayer of streptavidin that leads to a high density of biotin binding sites, which, based on information provided by the manufacturer, is estimated to provide a binding site density of ~300 pmol/cm
2. Hence, when bound with biotin–Avastin (1, 5 and 10 nM in 100 µL; surface density ~0.3, 1.5 and 3 pmol/cm
2), only a small fraction of the binding sites on the streptavidin surface would be expected to be occupied with Avastin, such that the Avastin will be sparsely distributed across the surface. As such, it is predicted that high concentrations of biotin–heparin would be required in order for heparin and Avastin to be brought into close proximity to participate in forming a high affinity ternary complex with VEGF. Indeed, this is what was observed in
Figure 1, where concentrations of biotin–heparin between 10–100 nM (surface density of ~3–30 pmol/cm
2) were required to induce an increase in VEGF binding to biotin–Avastin. Moreover, the inability of biotin–heparin to replicate VEGF binding enhancement when bound to soluble streptavidin bound to biotin–Avastin suggest that biotin–heparin has a limited ability to bind to streptavidin that is already bound to biotin–Avastin. This could reflect issues associated with steric hindrance and/or the possibility that an individual biotin–Avastin molecule might bind to multiple biotin sites on streptavidin, thus blocking biotin–heparin binding. Based on these results, it is predicted that the ability of biotin–heparin to enhance VEGF binding to biotin–Avastin on a streptavidin surface would be closely linked to the density of streptavidin on the surface, such that sparsely distributed streptavidin would decrease the probability that bound heparin molecules would be close enough to Avastin to enhance VEGF binding.
To evaluate the hypothesis that the ability of heparin to enhance VEGF binding to Avastin requires that the two molecules be in close proximity, a range of streptavidin concentrations were coated onto the plate surface to produce a range of biotin binding site densities. If we assume that 100% of the streptavidin was adsorbed and all the biotin binding sites were available, the highest concentration tested (500 nM; ~156.25 pmol/cm2) would be estimated to result in a biotin binding site density of ~600 pmol/cm2 on the well surface. This is obviously a high estimate, thus, the highest streptavidin concentration tested might be within the range of the saturated commercial plate binding site 300 pmol/cm2. However, it is important to note that we do not know the details of the method used to prepare the commercial plates; thus, it is not likely to be very informative to directly compare results from the two plate types.
Biotin–Avastin (1 nM; 0.3 pmol/cm
2), biotin–heparin (50 nM; 15 pmol/cm
2), and a combined mixture were allowed to bind to the streptavidin surfaces, and then VEGF binding was measured (
Figure 5). VEGF binding to biotin–Avastin alone was nearly constant at streptavidin concentrations of 15.63–156.25 pmol/cm
2 suggesting that there were sufficient biotin sites available to bind all of the biotin–Avastin to the plate surface under these conditions. Interestingly, the ability of biotin–heparin to maximally enhance VEGF binding to biotin–Avastin required a higher density of streptavidin. Indeed, a concentration of 62.5 pmol/cm
2 (biotin binding site density of ~240 pmol/cm
2) was required to achieve a maximal heparin effect (~4-fold increase), whereas at concentrations of 15.63 and 31.25 pmol/cm
2 (equivalent to biotin binding site densities of 60 and 120 pmol/cm
2), where there should be excess sites available to bind the biotin–heparin, only a partial effect was observed. Thus, the streptavidin density dependence provides evidence that the ability of heparin to enhance VEGF binding to Avastin is dependent on the two molecules being brought into close proximity in order to act together in binding VEGF.
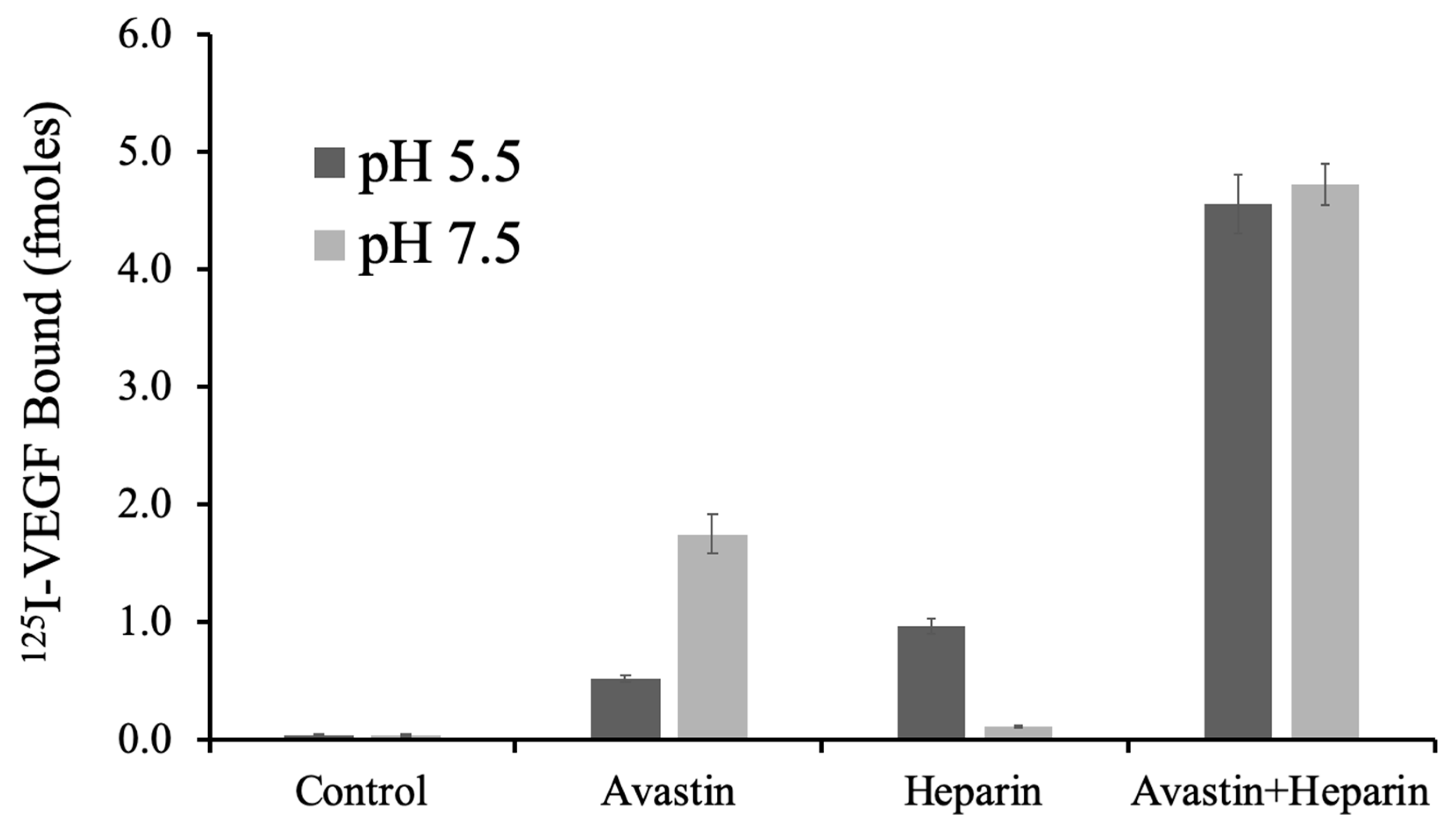
To determine if the pH sensitivity of VEGF binding to Avastin, and the function of heparin, are replicated under these conditions, VEGF binding was conducted with surface localized Avastin ± heparin at pH 5.5 and 7.5. A streptavidin surface density (93.75 pmol/cm
2) was selected to achieve colocalization of biotin–Avastin and biotin–heparin. VEGF binding with these conditions showed an intricate relationship that was consistent with pH dependent binding shown previously [
6].
Figure 6 shows that VEGF binding to Avastin was decreased at pH 5.5 compared to pH 7.5, whereas direct binding of VEGF to heparin increased at pH 5.5 compared to pH 7.5. The combination of Avastin and heparin showed similar VEGF binding at both pH 5.5 and 7.5 where each molecules appears to compensate for the other in establishing a high affinity ternary complex.
To further explore to concept that heparin and Avastin act through physical proximity, we directly compared conditions where heparin was allowed to freely diffuse in the binding buffer to that when it was bound to the plate surface along with Avastin. To do this, biotin–Avastin (1 nM) with and without biotin–heparin (80 nM), was bound to a streptavidin plate, and then soluble heparin (80 nM) was added into the VEGF binding reaction. As shown in
Figure 7A, soluble heparin was not able to enhance VEGF binding to immobilized Avastin at the concentration tested, whereas biotin–heparin induced an ~3-fold increase in VEGF binding to Avastin. This experiment was repeated with a concentration range of biotinylated and soluble heparin at pH 5.5 and 7.5 (
Figure 7B,C) and again, the biotin–heparin was more effective than soluble heparin at enhancing VEGF binding to Avastin at both pH 5.5 and 7.5.
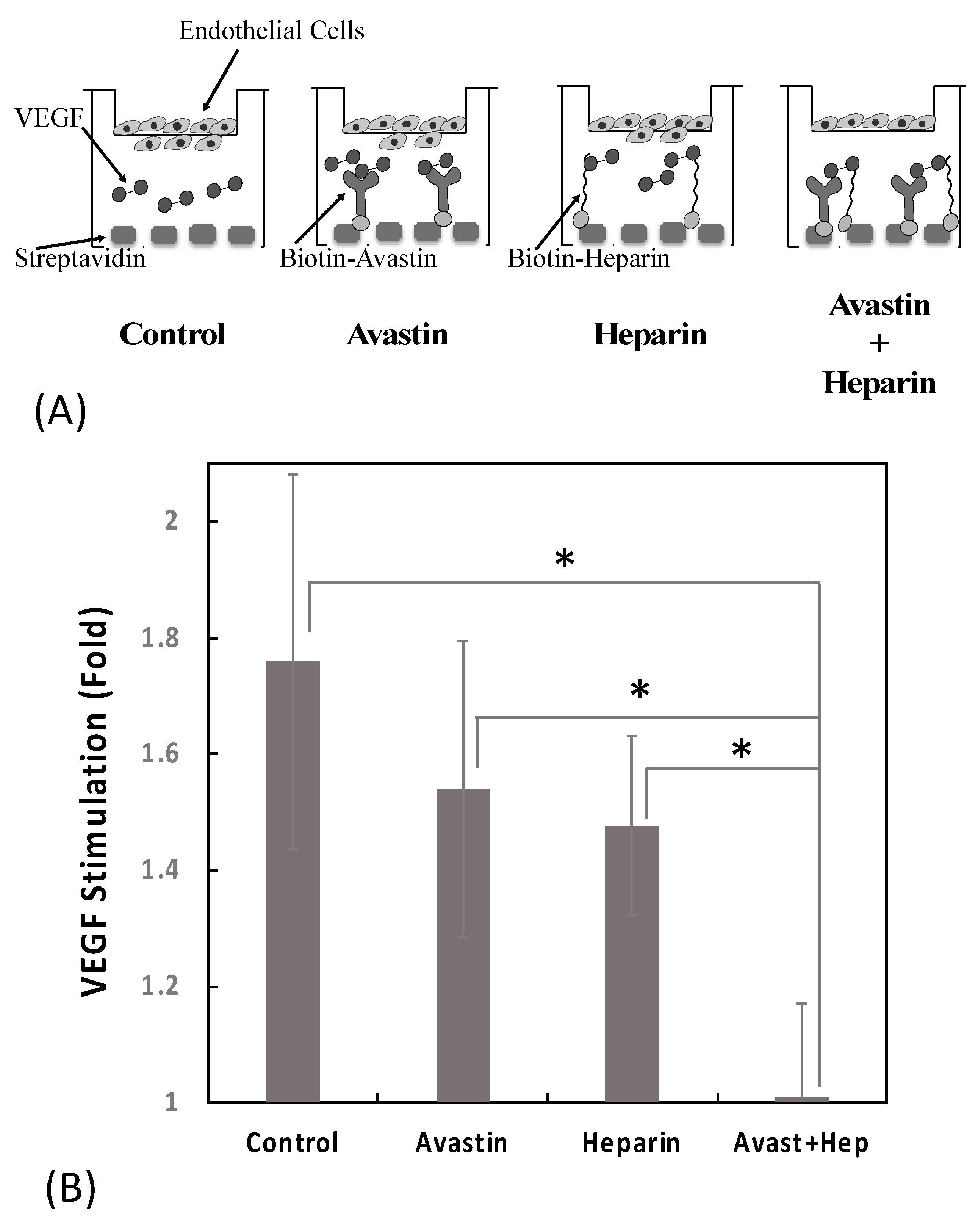
Colocalizing Avastin and Heparin Enhanced Inhibiton of VEGF-Induced Endothelial Cell Migration
One of the critical first steps in the angiogenic response is the activation of endothelial cell migration. Indeed, stimulation of cell migration is one of the most well characterized responses to VEGF. Thus, we evaluated the ability of heparin and Avastin to modulate VEGF-induced endothelial cell migration using primary HUVECs. In this experimental set up, a 24-well polystyrene plate was coated with streptavidin (200 nM) and then incubated ± biotin–Avastin (1 nM) ± biotin–heparin (50 nM). After modifying the surface with Avastin/heparin, VEGF (25 ng/mL) was added and then FluoroBlok inserts containing serum-starved HUVEC were placed into each well and the cells allowed to migrate for 14–16 h (
Figure 8A).
The number of cells that migrated through the membrane was measured and displayed as the fold change of VEGF-stimulated versus non-stimulated in 12 independent experiments. The presence of biotin–Avastin or biotin–heparin alone resulted in a modest 30% and 35% reduction in VEGF stimulation of cell migration, respectively (because of the high variability in this assay these changes were not statistically significant). The combined addition of biotin–Avastin with biotin–heparin resulted in complete inhibition of VEGF stimulation. These data indicate that the co-localization of Avastin and heparin via streptavidin enhanced the VEGF inhibitory activity compared to either molecule alone (
Figure 8B).
4. Discussion
The observation that heparin and heparan sulfate can potentiate the binding of VEGF to VEGFR-2 through the formation of a high affinity ternary complex suggests the concept that heparin might function similarly to enhance the activity of anti-VEGF drugs [
4,
5,
18,
27]. Indeed, our previous findings showing that heparin enhances VEGF binding to Avastin suggest that colocalizing heparin with Avastin might lead to enhanced VEGF inhibitory activity of Avastin [
6]. To explore this possibility, Avastin and heparin were conjugated using the biotin–streptavidin system. Biotin–Avastin was found to retain its ability to bind VEGF, protein A, and streptavidin (
Figure 1). Biotin–heparin was also found to facilitate VEGF binding to the modified Avastin, and this effect was enhanced when the two molecules were brought into close proximity via streptavidin (
Figure 2,
Figure 4,
Figure 5, and
Figure 7). However, it appeared that there were few available biotin binding sites on streptavidin that had bound to biotin–Avastin, such that it was difficult to generate effective soluble complexes containing both biotin–Avastin and biotin–heparin bound to the same streptavidin molecule (
Figure 3 and
Figure 4). Nevertheless, the results reported here demonstrate the proof of the concept that the combined action of heparin and Avastin can lead to enhanced VEGF inhibition compared to the action of either molecule alone, especially when the two molecules are co-localized (
Figure 7 and
Figure 8).
Avastin was first approved by the FDA in 2004 for treatment of metastatic colorectal cancer and has since been approved for treatment of a wide range of cancers in over 130 countries [
8]. Avastin and its related drug Lucentis are also used widely to treat age-related macular degeneration and other ocular diseases associated with excessive angiogensis [
28,
29]. While Avastin remains the most widely used anti-angiogenesis drug, negative side effects and the development of treatment resistance after long-term therapy have motivated the search for improvements [
11,
13,
14,
30]. Approaches have generally focused on encapsulating Avastin within various types of hydrogels to protect the protein and sustain its release within in vivo environments. Hydrogel-based delivery of Avastin from synthetic polymers, as well as natural polysaccharides, has shown promising results in vitro and in animal models in vivo. Indeed, Avastin delivered subcutaneously within a triblock copolymer of vitamin D-functionalized polycarbonate and poly (ethylene glycol) resulted in greater tumor suppression compared to intravenous or intraperitoneal administration using two separate HCT116 xenograft mouse models [
11]. Other approaches have considered combining Avastin with other therapeutic agents, as well as incorporation within liposomes to enhance bioavailability and efficacy [
10,
12,
31]. Heparin, a widely used anticoagulant drug, has also been identified as a potential anti-angiogenesis agent [
32,
33]. However, heparin has been shown to both enhance and inhibit the activity of various angiogenic proteins such as fibroblast growth factors, platelet derived growth factor, and VEGF, based on dose, timing, and mode of administration [
32,
34,
35]. Consequently, the complexity of heparin’s mode of action has limited its clinical utility in anti-angiogenic therapy. Recently, we discovered that heparin enhances VEGF binding to Avastin, especially at low pH where Avastin binding to VEGF becomes destabilized [
6]. These results suggest that one might be able to utilize heparin by specifically linking it to Avastin, so as to target this activity while limiting the complex systemic actions of heparin.
In an attempt to link Avastin and heparin to soluble streptavidin, an order of addition protocol was used where Avastin was first conjugated to streptavidin and then bound to biotin–heparin (
Figure 4). Complexes containing Avastin were isolated by protein A affinity. Complexes that contained heparin showed an increase in VEGF binding, indicating a promising means to generate soluble conjugates with enhanced VEGF binding properties. However, the heparin-mediated increase in VEGF binding was much lower than that observed when the Avastin and heparin were bound to plate surfaces (
Figure 2,
Figure 5,
Figure 6, and
Figure 7 ), suggesting that this approach remains far from optimized. The effect of heparin on VEGF–Avastin binding on plate surfaces was found to be dependent on the density of streptavidin, providing additional evidence that heparin and Avastin need to be in close proximity to form a synergistic complex with VEGF (
Figure 5).
Reduced extracellular pH has long been a hallmark of hypoxic tissues, such as solid tumors, and these conditions have been demonstrated to promote the expression of pro-angiogenic signals such as VEGF [
36]. Thus, our observation that Avastin binding to VEGF is destabilized at acidic pH suggests an important limitation to Avastin therapy. Interestingly, VEGF binding to heparin is enhanced at acidic pH [
19], and the presence of heparin was found to compensate for the reduced binding of Avastin to VEGF at acidic pH (
Figure 6) [
6]. Indeed, the combination of Avastin and heparin showed significantly higher VEGF binding than with either Avastin or heparin alone, such that binding was similar at both pHs when Avastin and heparin were both present. These results suggest that the co-localization of these two drugs might provide a means for maximal anti-VEGF activity under a range of environmental conditions that might be encountered in vivo.
A critical step in VEGF-mediated activation of angiogenesis is the stimulation of endothelial cell migration [
37]. Endothelial migration is a complex process that involves changes in cell–cell and cell–ECM contacts, as well as rearrangements of the cytoskeleton [
38]. While there is no perfect way to model this complex process, the migration of cells through a microporous membrane has long been used to gain insight [
39]. Thus, we observed that colocalization of heparin and Avastin on the transwell culture plate surface showed dramatically enhanced inhibition of VEGF-stimulated HUVEC migration compared to that observed with either drug alone (
Figure 8). These findings indicate that the combination of Avastin and heparin not only binds more VEGF, but also impacts biological activity of VEGF in a similar manner. This supports the concept that an optimized approach to conjugating Avastin and heparin to one another might lead to a more effective VEGF binding drug for antiangiogenesis therapy.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







