
Cell Senescence and the DNA Single-Strand Break Damage Repair Pathway


Abstract
1. Introduction
2. Morphological and Functional Characteristics of Senescent Cells

3. Cellular Senescence and Anti-Cancer Therapies
4. DNA Damage and Radiotherapy

| Type of Damage | Radioinduced Damage per Cell per Gy | Endogenous Damage per Cell per Day |
|---|---|---|
| Single strand breaks | 1000 [80] | >10,000 [81] |
| Base damage | 2000 | 3200 |
| Abasic sites | 250 | 12,600 |
| Double strand breaks | 39 [80] | ≈50 [82] |
| DNA-protein cross-links | 150 | ? |
| Non-DSB clustered lesions | 122 | ? |
| Complex DSB | ? | ? |
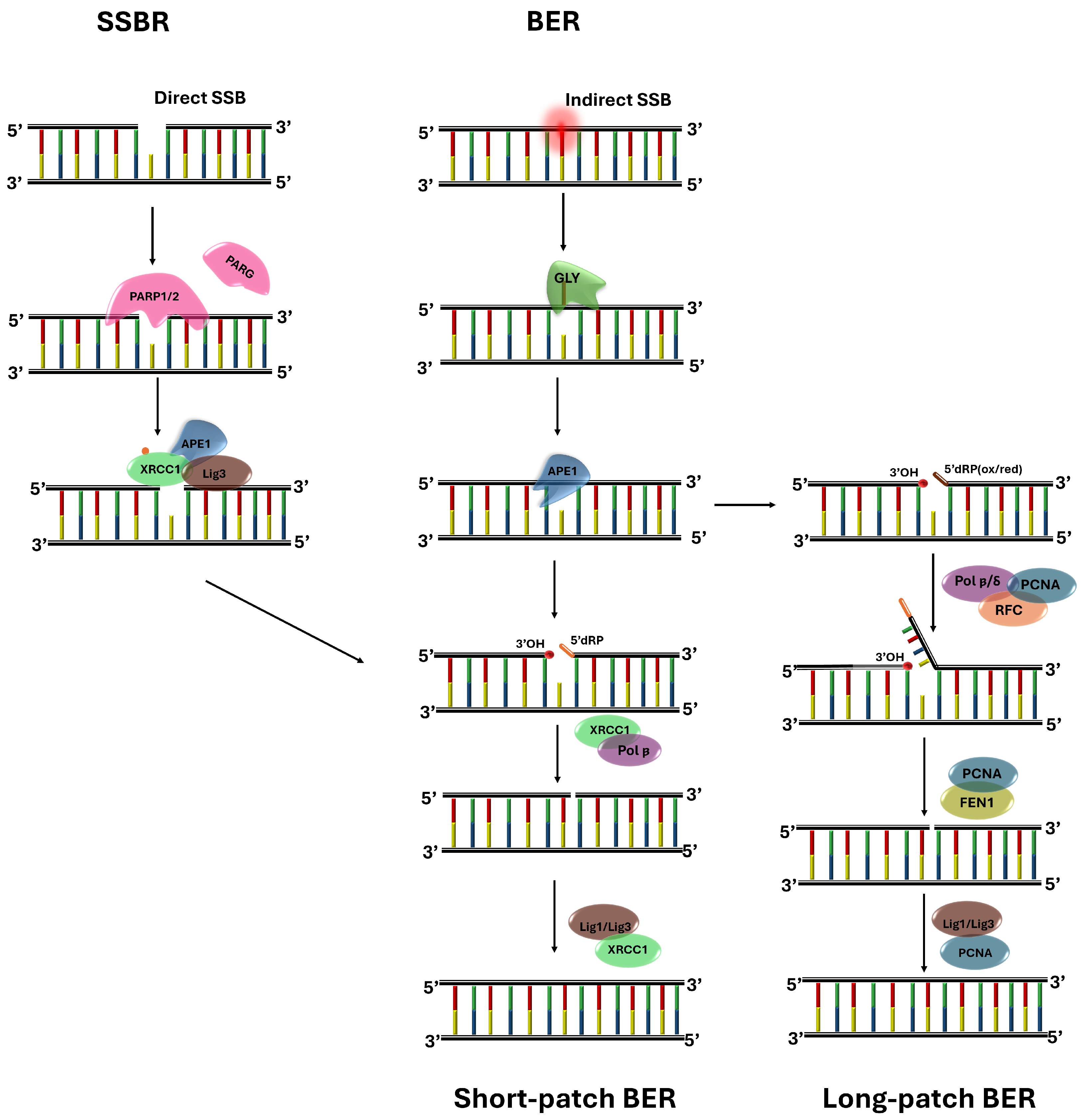
5. DNA Damage Repair Pathways
- Recognizing DNA damage: This is performed by DNA damage sensor proteins such as Poly(ADP)Ribose-1 (PARP-1) protein, Ku70/80, etc. (see [86]).
- Recruiting repair proteins and excising the damaged segment: This is performed by transducer proteins, which typically include a repair protein bound to a scaffold protein to form a complex at the lesion site. This complex removes the damaged segment and restores the correct -hydroxyl and -phosphate termini.
- Re-synthesizing the missing parts of the DNA sequence: This is done by different effector synthetases, which add new nucleotides.
- Finally, ligation of the clean broken ends by ligase enzymes.

5.1. Base-Excision Repair Pathway
5.2. Single-Strand Break Repair Pathway
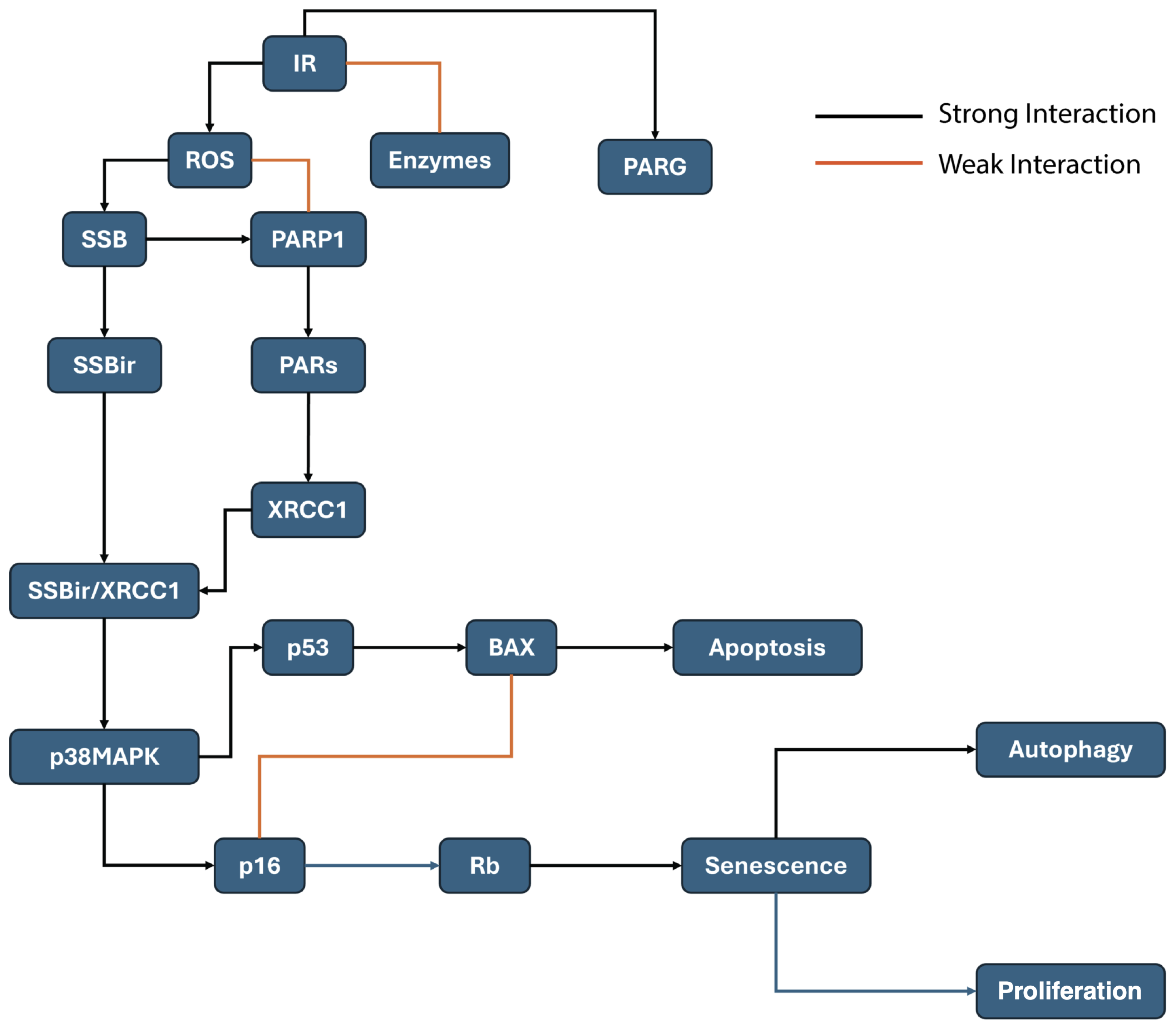
6. Radiation-Induced Single-Strand Breaks and Cellular Senescence
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. How telomeres solve the end-protection problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef]
- Hezel, A.F.; Bardeesy, N.; Maser, R.S. Telomere induced senescence: End game signaling. Curr. Mol. Med. 2005, 5, 145–152. [Google Scholar] [CrossRef]
- Deng, Y.; Chan, S.S.; Chang, S. Telomere dysfunction and tumour suppression: The senescence connection. Nat. Rev. Cancer 2008, 8, 450–458. [Google Scholar] [CrossRef]
- Mikuła-Pietrasik, J.; Niklas, A.; Uruski, P.; Tykarski, A.; Książek, K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell. Mol. Life Sci. 2020, 77, 213–229. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Marin, I.; Boix, O.; Garcia-Garijo, A.; Sirois, I.; Caballe, A.; Zarzuela, E.; Ruano, I.; Attolini, C.S.O.; Prats, N.; López-Domínguez, J.A.; et al. Cellular Senescence Is Immunogenic and Promotes Antitumor Immunity. Cancer Discov. 2022, 13, 410–431. [Google Scholar] [CrossRef]
- Sikora, E.; Mosieniak, G.; Alicja Sliwinska, M. Morphological and functional characteristic of senescent cancer cells. Curr. Drug Targets 2016, 17, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.; Idelchik, M.d.P.S.; Melendez, J.A. Redox control of senescence and age-related disease. Redox Biol. 2017, 11, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Veronesi, F.; Contartese, D.; Di Sarno, L.; Borsari, V.; Fini, M.; Giavaresi, G. In vitro models of cell senescence: A systematic review on musculoskeletal tissues and cells. Int. J. Mol. Sci. 2023, 24, 15617. [Google Scholar] [CrossRef]
- Naka, K.; Tachibana, A.; Ikeda, K.; Motoyama, N. Stress-induced premature senescence in hTERT-expressing ataxia telangiectasia fibroblasts. J. Biol. Chem. 2004, 279, 2030–2037. [Google Scholar] [CrossRef]
- Parrinello, S.; Samper, E.; Krtolica, A.; Goldstein, J.; Melov, S.; Campisi, J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 2003, 5, 741–747. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Criscione, S.W.; Teo, Y.V.; Neretti, N. The chromatin landscape of cellular senescence. Trends Genet. 2016, 32, 751–761. [Google Scholar] [CrossRef]
- Kuwano, K.; Araya, J.; Hara, H.; Minagawa, S.; Takasaka, N.; Ito, S.; Kobayashi, K.; Nakayama, K. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir. Investig. 2016, 54, 397–406. [Google Scholar] [CrossRef]
- Parry, A.J.; Narita, M. Old cells, new tricks: Chromatin structure in senescence. Mamm. Genome 2016, 27, 320–331. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Buratta, S.; Sagini, K.; Tancini, B.; Emiliani, C. Extracellular vesicles as new players in cellular senescence. Int. J. Mol. Sci. 2016, 17, 1408. [Google Scholar] [CrossRef] [PubMed]
- Micco, R.D.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef] [PubMed]
- Nassour, J.; Martien, S.; Martin, N.; Deruy, E.; Tomellini, E.; Malaquin, N.; Bouali, F.; Sabatier, L.; Wernert, N.; Pinte, S.; et al. Defective DNA single-strand break repair is responsible for senescence and neoplastic escape of epithelial cells. Nat. Commun. 2016, 7, 10399. [Google Scholar] [CrossRef] [PubMed]
- Goy, E.; Tomezak, M.; Facchin, C.; Martin, N.; Bouchaert, E.; Benoit, J.; de Schutter, C.; Nassour, J.; Saas, L.; Drullion, C.; et al. The out-of-field dose in radiation therapy induces delayed tumorigenesis by senescence evasion. eLife 2022, 11, e67190. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Qin, D.; Hou, X.; Tian, L.; Yu, Y.; Zhang, R.; Lyu, H.; Guo, D.; Chen, X.Z.; Zhou, C.; et al. Cellular senescence: A double-edged sword in cancer therapy. Front. Oncol. 2023, 13, 1189015. [Google Scholar] [CrossRef]
- Varela-Eirín, M.; Demaria, M. Cellular senescence. Curr. Biol. 2022, 32, R448–R452. [Google Scholar] [CrossRef]
- Cho, S.; Hwang, E.S. Fluorescence-based detection and quantification of features of cellular senescence. Methods Cell Biol. 2011, 103, 149–188. [Google Scholar]
- Zhao, H.; Darzynkiewicz, Z. Biomarkers of cell senescence assessed by imaging cytometry. In Cell Senescence: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2013; pp. 83–92. [Google Scholar]
- Chondrogianni, N.; Stratford, F.L.; Trougakos, I.P.; Friguet, B.; Rivett, A.J.; Gonos, E.S. Central role of the proteasome in senescence and survival of human fibroblasts: Induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 2003, 278, 28026–28037. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Noz, D.P.M.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Prata, L.G.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Gosselin, K.; Monte, D.; Vercamer, C.; Bouali, F.; Pourtier, A.; Vandenbunder, B.; Abbadie, C. Involvement of Rel/Nuclear Factor-κB Transcription Factors in Keratinocyte Senescence. Cancer Res. 2004, 64, 472–481. [Google Scholar] [CrossRef]
- Gosselin, K.; Deruy, E.; Martien, S.; Vercamer, C.; Bouali, F.; Dujardin, T.; Slomianny, C.; Houel-Renault, L.; Chelli, F.; Launoit, Y.D.; et al. Senescent keratinocytes die by autophagic programmed cell death. Am. J. Pathol. 2009, 174, 423–435. [Google Scholar] [CrossRef]
- Nayak, M.G.; George, A.; Vidyasagar, M.; Mathew, S.; Nayak, S.; Nayak, B.S.; Shashidhara, Y.; Kamath, A. Quality of life among cancer patients. Indian J. Palliat. Care 2017, 23, 445. [Google Scholar] [CrossRef]
- Debela, D.T.; Muzazu, S.G.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. Sage Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef]
- Bidram, E.; Esmaeili, Y.; Ranji-Burachaloo, H.; Al-Zaubai, N.; Zarrabi, A.; Stewart, A.; Dunstan, D.E. A concise review on cancer treatment methods and delivery systems. J. Drug Deliv. Sci. Technol. 2019, 54, 101350. [Google Scholar] [CrossRef]
- de Gonzalez, A.B.; Gilbert, E.; Curtis, R.; Inskip, P.; Kleinerman, R.; Morton, L.; Rajaraman, P.; Little, M.P. Second solid cancers after radiation therapy: A systematic review of the epidemiologic studies of the radiation dose-response relationship. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 224–233. [Google Scholar] [CrossRef]
- Dracham, C.B.; Shankar, A.; Madan, R. Radiation induced secondary malignancies: A review article. Radiat. Oncol. J. 2018, 36, 85–94. [Google Scholar] [CrossRef]
- Khanna, L.; Prasad, S.R.; Yedururi, S.; Parameswaran, A.M.; Marcal, L.P.; Sandrasegaran, K.; Tirumani, S.H.; Menias, C.O.; Katabathina, V.S. Second Malignancies after Radiation Therapy: Update on Pathogenesis and Cross-sectional Imaging Findings. Radiogr. Radiat. Oncol. 2021, 41, 876–894. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, W.; Stenman, G.; Sager, R. Suppression of tumor growth by senescence in virally transformed human fibroblasts. Proc. Natl. Acad. Sci. USA 1986, 83, 8659–8663. [Google Scholar] [CrossRef]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef]
- Han, Z.; Wei, W.; Dunaway, S.; Darnowski, J.W.; Calabresi, P.; Sedivy, J.; Hendrickson, E.A.; Balan, K.V.; Pantazis, P.; Wyche, J.H. Role of p21 in apoptosis and senescence of human colon cancer cells treated with camptothecin. J. Biol. Chem. 2002, 277, 17154–17160. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Sotgia, F.; Lisanti, M.P. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog-GLI signalling in breast cancer cells. Oncotarget 2015, 6, 10728. [Google Scholar] [CrossRef]
- Gureghian, V.; Herbst, H.; Kozar, I.; Mihajlovic, K.; Malod-Dognin, N.; Ceddia, G.; Angeli, C.; Margue, C.; Randic, T.; Philippidou, D.; et al. A multi-omics integrative approach unravels novel genes and pathways associated with senescence escape after targeted therapy in NRAS mutant melanoma. Cancer Gene Ther. 2023, 30, 1330–1345. [Google Scholar] [CrossRef]
- McDermott, M.S.; Conlon, N.; Browne, B.C.; Szabo, A.; Synnott, N.C.; O’Brien, N.A.; Duffy, M.J.; Crown, J.; O’Donovan, N. HER2-targeted tyrosine kinase inhibitors cause therapy-induced-senescence in breast cancer cells. Cancers 2019, 11, 197. [Google Scholar] [CrossRef]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; Deursen, J.V.; Goronzy, J.; et al. Therapy-induced senescence: Opportunities to improve anticancer therapy. JNCI J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef]
- Bousset, L.; Gil, J. Targeting senescence as an anticancer therapy. Mol. Oncol. 2022, 16, 3855–3880. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, R.; Caroprese, M.; Palma, G.; Oliviero, C.; Clemente, S.; Cella, L.; Conson, M. Technological evolution of radiation treatment: Implications for clinical applications. Semin. Oncol. 2019, 46, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and cancer—Role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Brown, S.L.; Gordon, M.N. Radiation-induced senescence: Therapeutic opportunities. Radiat. Oncol. 2023, 18, 10. [Google Scholar] [CrossRef]
- Arruebo, M.; Vilaboa, N.; Sáez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; González-Fernández, Á. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef]
- Melía, E.; Parsons, J.L. DNA damage and repair dependencies of ionising radiation modalities. Biosci. Rep. 2023, 43, BSR20222586. [Google Scholar] [CrossRef]
- Mitin, N.; Nyrop, K.A.; Strum, S.L.; Knecht, A.; Carey, L.A.; Reeder-Hayes, K.E.; Claire Dees, E.; Jolly, T.A.; Kimmick, G.G.; Karuturi, M.S.; et al. A biomarker of aging, p16, predicts peripheral neuropathy in women receiving adjuvant taxanes for breast cancer. NPJ Breast Cancer 2022, 8, 103. [Google Scholar] [CrossRef]
- Peng, X.; Wu, Y.; Brouwer, U.; van Vliet, T.; Wang, B.; Demaria, M.; Barazzuol, L.; Coppes, R.P. Cellular senescence contributes to radiation-induced hyposalivation by affecting the stem/progenitor cell niche. Cell Death Dis. 2020, 11, 854. [Google Scholar] [CrossRef]
- Rovillain, E.; Mansfield, L.; Lord, C.J.; Ashworth, A.; Jat, P.S. An RNA interference screen for identifying downstream effectors of the p53 and pRB tumour suppressor pathways involved in senescence. BMC Genom. 2011, 12, 355. [Google Scholar] [CrossRef]
- Domen, A.; Deben, C.; Verswyvel, J.; Flieswasser, T.; Prenen, H.; Peeters, M.; Lardon, F.; Wouters, A. Cellular senescence in cancer: Clinical detection and prognostic implications. J. Exp. Clin. Cancer Res. 2022, 41, 360. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.; Dellambra, E.; Paterna, P.; Golisano, O.; Traverso, C.E.; Rama, P.; Lacal, P.; De Luca, M. Telomerase activity is sufficient to bypass replicative senescence in human limbal and conjunctival but not corneal keratinocytes. Eur. J. Cell Biol. 2004, 83, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–15568. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988, 35, 95–125. [Google Scholar] [PubMed]
- Cohen-Jonathan, E.; Bernhard, E.J.; McKenna, W.G. How does radiation kill cells? Curr. Opin. Chem. Biol. 1999, 3, 77–83. [Google Scholar] [CrossRef]
- IARC Working Group. Ionizing radiation, part 1: X- and gamma-radiation, and neutrons. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2000; Volume 75, pp. 1–508. [Google Scholar]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 61–72. [Google Scholar] [CrossRef]
- Grün, R.; Friedrich, T.; Krämer, M.; Scholz, M. Systematics of relative biological effectiveness measurements for proton radiation along the spread-out Bragg peak: Experimental validation of the local effect model. Phys. Med. Biol. 2017, 62, 890. [Google Scholar] [CrossRef]
- Gu, B.; noz Santiburcio, D.M.; Pieve, F.D.; Cleri, F.; Artacho, E.; Kohanoff, J. Bragg’s additivity rule and core and bond model studied by real-time TDDFT electronic stopping simulations: The case of water vapor. Radiat. Phys. Chem. 2022, 193, 109961. [Google Scholar] [CrossRef]
- Wang, S.; Gonzalez, G.; Sun, L.; Xu, Y.; Pandey, P.; Chen, Y.; Xiang, S.L. Real-time tracking of the Bragg peak during proton therapy via 3D protoacoustic Imaging in a clinical scenario. NPJ Imaging 2024, 2, 34. [Google Scholar] [CrossRef]
- von Sonntag, C. Recent Trends in Radiation Chemistry; World Scientific: Wishart, JF, USA, 2010; p. 543. [Google Scholar]
- Ravanat, J.L. Endogenous natural and radiation-induced DNA lesions: Differences and similarities and possible implications for human health and radiological protection. Radioprotection 2018, 53, 241–248. [Google Scholar] [CrossRef]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef]
- Porro, M.L.T.; Greenberg, M.M. Double-Strand Breaks from a Radical Commonly Produced by DNA-Damaging Agents. Chem. Res. Toxycol. 2015, 28, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006; pp. 379–390. [Google Scholar]
- Weinfeld, M.; Soderling, K.J. 32P-postlabeling detection of radiation-induced DNA damage: Identification and estimation of thymine glycols and phosphoglycolate termini. Biochemistry 1999, 30, 1091–1097. [Google Scholar] [CrossRef]
- Zhou, T.; Akopiants, K.; Mohapatra, S.; Lin, P.S.; Valerie, K.; Ramsden, D.A.; Lees-Miller, S.P.; Povirk, L.F. Tyrosyl-DNA phosphodiesterase and the repair of 3’-phosphoglycolate-terminated DNA double-strand breaks. DNA Repair 2009, 8, 901–911. [Google Scholar] [CrossRef]
- Goodhead, D.T. The initial physical damage produced by ionizing radiations. Int. J. Radiat. Biol. 1989, 56, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kacmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- Branze, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Chalmers, A.; Carruthers, R. Radiobiology summaries: DNA damage and repair. Clin. Oncol. 2021, 33, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Schipler, A.; Iliakis, G. DNA double-strand–break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-strand DNA Break Formation in Chromatin. J. Cell Physiol. 2007, 231, 3–14. [Google Scholar] [CrossRef]
- Tounekti, O.; Kenani, A.; Foray, N.A.; Orlowski, S.; Mir, L.M. The ratio of single-to double-strand DNA breaks and their absolute values determine cell death pathway. Br. J. Cancer 2001, 84, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef]
- Kouzminova, E.A.; Kuzminov, A. Fragmentation of replicating chromosomes triggered by uracil in DNA. J. Mol. Biol. 2006, 355, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Mammalian DNA single-strand break repair: An X-ra (y) ted affair. Bioessays 2001, 23, 447–455. [Google Scholar] [CrossRef]
- Venkadakrishnan, J.; Lahane, G.; Dhar, A.; Xiao, W.; Bhat, K.M.; Pandita, T.K.; Bhat, A. Implications of translesion DNA synthesis polymerases on genomic stability and human health. Mol. Cell. Biol. 2023, 43, 401–425. [Google Scholar] [CrossRef]
- Ma, W.; Halweg, C.J.; Menendez, D.; Resnick, M.A. Differential effects of poly (ADP-ribose) polymerase inhibition on DNA break repair in human cells are revealed with Epstein–Barr virus. Proc. Natl. Acad. Sci. USA 2012, 109, 6590–6595. [Google Scholar] [CrossRef] [PubMed]
- Mokari, M.; Alamatsaz, M.H.; Moeini, H.; Taleei, R. A simulation approach for determining the spectrum of DNA damage induced by protons. Phys. Med. Biol. 2018, 63, 175003. [Google Scholar] [CrossRef] [PubMed]
- Roobol, S.J.; van den Bent, I.; van Cappellen, W.A.; Abraham, T.E.; Paul, M.W.; Kanaar, R.; Houtsmuller, A.B.; van Gent, D.C.; Essers, J. Comparison of High- and Low-LET Radiation-Induced DNA Double-Strand Break Processing in Living Cells. Int. J. Mol. Sci. 2020, 21, 6602. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. DNA single-strand break repair and human genetic disease. Trends Cell Biol. 2022, 32, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Sahadevan, M.; Lee, O.; Muzzio, M.; Phan, B.; Jacobs, L.; Khouri, N.; Wang, J.; Hu, H.; Stearns, V.; Chatterton, R.T. The relationship of single-strand breaks in DNA to breast cancer risk and to tissue concentrations of oestrogens. Biomarkers 2017, 22, 689–697. [Google Scholar] [CrossRef]
- Hossain, M.A.; Lin, Y.; Yan, S. Single-Strand Break End Resection in Genome Integrity: Mechanism and Regulation by APE2. Int. J. Mol. Sci. 2018, 19, 2389. [Google Scholar] [CrossRef]
- Gerasimova, N.S.; Akhtar, M.S.; Studitskii, V.M. Effect of Single-Strand DNA Breaks on Transcription of Nucleosomes. Mosc. Univ. Biol. Sci. Bull. 2022, 77, 216–222. [Google Scholar] [CrossRef]
- Xu, S.; Wei, J.; Sun, S.; Zhang, J.; Chan, T.F.; Li, Y. SSBlazer: A genome-wide nucleotide-resolution model for predicting single-strand break sites. Genome Biol. 2024, 25, 46. [Google Scholar] [CrossRef]
- Zhou, W.; Doetsch, P.W.E. Effects of abasic sites and DNA single-strand breaks on prokaryotic RNA polymerases. Proc. Natl. Acad. Sci. USA 1999, 90, 6601–6605. [Google Scholar] [CrossRef]
- Kathe, S.D.; Shen, G.P.; Wallace, S.S. Single stranded breaks in DNA but not oxidative DNA base damages block transcriptional elongation by RNA polymerase II in HeLa cell nuclear extracts. J. Biol. Chem. 2004, 279, 18511–18520. [Google Scholar] [CrossRef]
- Heeres, J.T.; Hergenrother, P.J.P. Poly(ADP-ribose) makes a date with death. Curr. Opin. Chem. Biol. 2007, 11, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Moroni, F. Poly(ADP-ribose polymerase 1 (PARP-1) and postischemic brain damage. Curr. Opin. Pharmacol. 2008, 8, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Khoronenkova, S.V.; Dianov, G.L. ATM prevents DSB formation by coordinating SSB repair and cell cycle progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3997–4002. [Google Scholar] [CrossRef] [PubMed]
- Fortini, P.; Dogliotti, E. Base damage and single-strand break repair: Mechanisms and functional significance of short-and long-patch repair subpathways. DNA Repair 2007, 6, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. The relationship of single-strand breaks in DNA to breast cancer risk and to tissue concentrations of oestrogens. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schär, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base Excision Repair and Cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef]
- Guan, L.; Greenberg, M.M. Irreversible Inhibition of DNA Polymerase β by an Oxidized Abasic Lesion. J. Am. Chem. Soc. 2010, 132, 5004. [Google Scholar] [CrossRef]
- Jacobs, A.C.; Kreller, C.R.; Greenberg, M.M. Long Patch Base Excision Repair Compensates for DNA Polymerase β Inactivation by the C4′-Oxidized Abasic Site. Biochemistry 2010, 50, 136–143. [Google Scholar] [CrossRef]
- Sattler, U.; Frit, P.; Salles, B.; Calsou, P. Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep. 2003, 4, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell. Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Hindi, N.N.; Elsakrmy, N.; Ramotar, D. The base excision repair process: Comparison between higher and lower eukaryotes. Cell. Mol. Life Sci. 2021, 78, 7943–7965. [Google Scholar] [CrossRef] [PubMed]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Ziegler, M.; Oei, S.L. ATP-dependent selection between single nucleotide and long patch base excision repair. DNA Repair 2003, 2, 1101–1114. [Google Scholar] [CrossRef]
- Klungland, A.; Lindahl, T. Second pathway for completion of human DNA base excision-repair: Reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 1997, 16, 3341–3348. [Google Scholar] [CrossRef] [PubMed]
- Lilyestrom, W.; van der Woerd, M.J.; Clark, N.; Luger, K. Structural and biophysical studies of human PARP-1 in complex with damaged DNA. J. Mol. Biol. 2010, 395, 983–994. [Google Scholar] [CrossRef]
- Ali, A.; Timinszky, G.; Arribas-Bosacoma, R.; Kozlowski, M.; Hassa, P.; Hassler, M.; Ladurner, A.; Pearl, L.; Oliver, A. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 2012, 19, 685–692. [Google Scholar] [CrossRef]
- Sarma, P.A.; Abbadie, C.; Cleri, F. Cooperative dynamics of PARP-1 Zinc-finger domains in the detection of DNA single-strand breaks. Sci. Rep. 2024, 14, 23257. [Google Scholar] [CrossRef]
- Langelier, M.F.; Ruhl, D.D.; Planck, J.L.; Kraus, W.L.; Pascal, J.M. The Zn3 domain of human poly(ADP-ribose) polymerase-1 (PARP-1) functions in both DNA-dependent poly(ADP-ribose) synthesis activity and chromatin compaction. J. Biol. Chem. 2010, 285, 18877–18887. [Google Scholar] [CrossRef]
- Rudolph, J.; Muthurajan, U.; Palacio, M.; Mahadevan, J.; Roberts, G.; Erbse, A.; Dyer, P.; Luger, K. The BRCT domain of PARP1 binds intact DNA and mediates intrastrand transfer. Mol. Cell 2021, 81, 4994–5006. [Google Scholar] [CrossRef] [PubMed]
- Bilokapic, S.; Suskiewicz, M.J.; Ahel, I.; Halic, M. Bridging of DNA breaks activates PARP2-HPF1 to modify chromatin. Nature 2020, 503, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Gaullier, G.; Roberts, G.; Muthurajan, U.; Bowerman, S.; Rudolph, J.; Mahadevan, J.; Jha, A.; Rae, P.; Luger, K. Bridging of nucleosome-proximal DNA double-strand breaks by PARP2 enhances its interaction with HPF1. PLoS ONE 2020, 15, e0240932. [Google Scholar] [CrossRef]
- Eustermann, S.; Wu, W.F.; Langelier, M.F.; Yang, J.C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol. Cell 2015, 60, 742–754. [Google Scholar] [CrossRef]
- Dawicki-McKenna, J.M.; Langelier, M.F.; DeNizio, J.E.; Riccio, A.A.; Cao, C.D.; Karch, K.R.; McCauley, M.; Steffen, J.D.; Black, B.E.; Pascal, J.M. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell 2015, 60, 755–768. [Google Scholar] [CrossRef]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP) – Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 2020, 7, 00191. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H.; West, M.G. XRCC1 keeps DNA from getting stranded. Mutat. Res. 2000, 459, 1–18. [Google Scholar] [CrossRef]
- Thompson, L.H.; Brookman, K.W.; Jones, N.J.; Allen, S.A.; Carrano, A.V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Mol. Cell Biol. 1990, 10, 6160–6171. [Google Scholar]
- Mani, R.S.; Karimi-Busheri, F.; Fanta, M.; Caldecott, K.W.; Cass, C.E.; Weinfeld, M. Biophysical characterization of human XRCC1 and its binding to damaged and undamaged DNA. Biochemistry 2004, 43, 16505–16514. [Google Scholar] [CrossRef]
- Caldecott, K.W.; Aoufouchi, S.; Johnson, P.; Shall, S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular “nick-sensor” in vitro. Nucleic Acids Res. 1996, 24, 4387–4394. [Google Scholar] [CrossRef]
- Schreiber, V.; Amé, J.C.; Dollé, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; de Murcia, J.M.; de Murcia, G. Poly (ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 2002, 277, 23028–23036. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lu, L.Y.; Yang, C.Y.; Wang, S.; Yu, X. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. 2013, 27, 1752–1768. [Google Scholar] [CrossRef] [PubMed]
- Mok, M.C.Y.; Campalans, A.; Pillon, M.C.; Guarné, A.; Radicella, J.P.; Junop, M.S. Identification of an XRCC1 DNA binding activity essential for retention at sites of DNA damage. Sci. Rep. 2019, 9, 3095. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W.; McKeown, C.K.; Tucker, J.D.; Ljungquist, S.; Thompson, L.H. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell Biol. 1994, 14, 68–76. [Google Scholar] [PubMed]
- Breslin, C.; Mani, R.S.; Fanta, M.; Hoch, N.; Weinfeld, M.; Caldecott, K.W. The RIR motif in the scaffold protein XRCC1 mediates a low-affinity interaction with polynucleotide kinase phosphatase (PNKP) during DNA single-strand break repair. J. Biol. Chem. 2017, 292, 16024–16031. [Google Scholar] [CrossRef]
- Loizou, J.I.; El-Khamisy, S.F.; Zlatanou, A.; Moore, D.J.; Chan, D.W.; Qin, J.; Sarno, S.; Meggio, F.; Pinna, L.A.; Caldecott, K.W. The Protein Kinase CK2 Facilitates Repair of Chromosomal DNA Single-Strand Breaks. Cell 2004, 117, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Date, H.; Igarashi, S.; Sano, Y.; Takahashi, T.; Takahashi, T.; Takano, H.; Tsuji, S.; Nishizawa, M.; Onodera, O. The FHA domain of aprataxin interacts with the C-terminal region of XRCC1. Biochem. Biophys. Res. Commun. 2004, 325, 1279–1285. [Google Scholar] [CrossRef]
- Iles, N.; Rulten, S.; El-Khamisy, S.F.; Caldecott, K.W. APLF (C2orf13) Is a Novel Human Protein Involved in the Cellular Response to Chromosomal DNA Strand Breaks. Cell 2007, 27, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Abbotts, R.; Wilson III, D.M. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006. [Google Scholar]
- Kong, F.M.; Ritter, T.; Quint, D.J.; Senan, S.; Gaspar, L.E.; Komaki, R.U.; Hurkmans, C.W.; Timmerman, R.; Bezjak, A.; Bradley, J.D.; et al. Consideration of dose limits for organs at risk of thoracic radiotherapy: Atlas for lung, proximal bronchial tree, esophagus, spinal cord, ribs, and brachial plexus. Int. J. Radiat. Oncol. Biol. Phys. 2010, 81, 1442–1457. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Loree, J.; Kutanzi, K.; Koganow, C.; Pogribny, I.; Kovalchuk, O. In vivo bystander effect: Cranial X-irradiation leads to elevated DNA damage, altered cellular proliferation and apoptosis, and increased p53 levels in shielded spleen. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 554–562. [Google Scholar] [CrossRef]
- Najafi, M.; Fardid, R.; Hadadi, G.; Fardid, M. The mechanisms of radiation-induced bystander effect. J. Biomed. Phys. Eng. 2014, 4, 163. [Google Scholar]
- Hei, T.K.; Zhou, H.; Ivanov, V.N. Mechanism of radiation-induced bystander effects: A unifying model. J. Pharmacol. Exp. Ther. 2011, 328, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Diallo, I.; Haddy, N.; Adjadj, E.; Samand, A.; Quiniou, E.; Chavaudra, J.; Alziar, I.; Perret, N.; Guérin, S.; Lefkopoulos, D.; et al. Frequency distribution of second solid cancer locations in relation to the irradiated volume among 115 patients treated for childhood cancer. J. Radiat. Oncol. Biol. Phys. 2009, 74, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Chofor, N.; Harder, D.; Willborn, K.; Poppe, B. Innovative approaches to reduce radiation-induced secondary cancer risk. Strahlenther. Und Onkol. 2012, 188, 672–677. [Google Scholar]
- Abbadie, C.; Pluquet, O.; Pourtier, A. Epithelial cell senescence: An adaptive response to pre-carcinogenic stresses? Cell. Mol. Life Sci. 2017, 74, 4471–4509. [Google Scholar] [CrossRef] [PubMed]
- Chofor, N.; Harder, D.; Poppe, B.; Rührnschopf, E.P.; Willborn, K. The impact of the scatter dose on the risk of radiation-induced second cancers in radiotherapy of the prostate. Radiat. Oncol. 2011, 6, 1–9. [Google Scholar]
- Al-Mohanna, M.A.; Al-Khalaf, H.H.; Al-Yousef, N. Apoptosis and Bax expression are inhibited by p16INK4a in breast cancer cells. Cell. Signal. 2004, 16, 681–688. [Google Scholar]
- Beyls, C.; Haustermans, K.; Deroose, C.M.; Pans, S.; Vanbeckevoort, D.; Verslype, C.; Dekervel, J. Could autoimmune disease contribute to the abscopal effect in metastatic hepatocellular carcinoma? Hepatology 2020, 72, 1152–1154. [Google Scholar] [CrossRef]
- Ahenkorah, S.; Cassells, I.; Deroose, C.M.; Cardinaels, T.; Burgoyne, A.R.; Bormans, G.; Ooms, M.; Cleeren, F. Bismuth-213 for targeted radionuclide therapy: From atom to bedside. Pharmaceutics 2021, 13, 599. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarma, P.A.; Abbadie, C.; de Launoit, Y.; Cleri, F. Cell Senescence and the DNA Single-Strand Break Damage Repair Pathway. DNA 2024, 4, 530-552. https://doi.org/10.3390/dna4040036
Sarma PA, Abbadie C, de Launoit Y, Cleri F. Cell Senescence and the DNA Single-Strand Break Damage Repair Pathway. DNA. 2024; 4(4):530-552. https://doi.org/10.3390/dna4040036
Chicago/Turabian StyleSarma, Parvathy A., Corinne Abbadie, Yvan de Launoit, and Fabrizio Cleri. 2024. "Cell Senescence and the DNA Single-Strand Break Damage Repair Pathway" DNA 4, no. 4: 530-552. https://doi.org/10.3390/dna4040036
APA StyleSarma, P. A., Abbadie, C., de Launoit, Y., & Cleri, F. (2024). Cell Senescence and the DNA Single-Strand Break Damage Repair Pathway. DNA, 4(4), 530-552. https://doi.org/10.3390/dna4040036